
The Role of Cellular Stress in Intrauterine Growth Restriction and Postnatal Dysmetabolism



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Human Studies of Intrauterine Growth Restriction and Metabolic Outcomes
3. Cellular Stress and Metabolism in IUGR Offspring
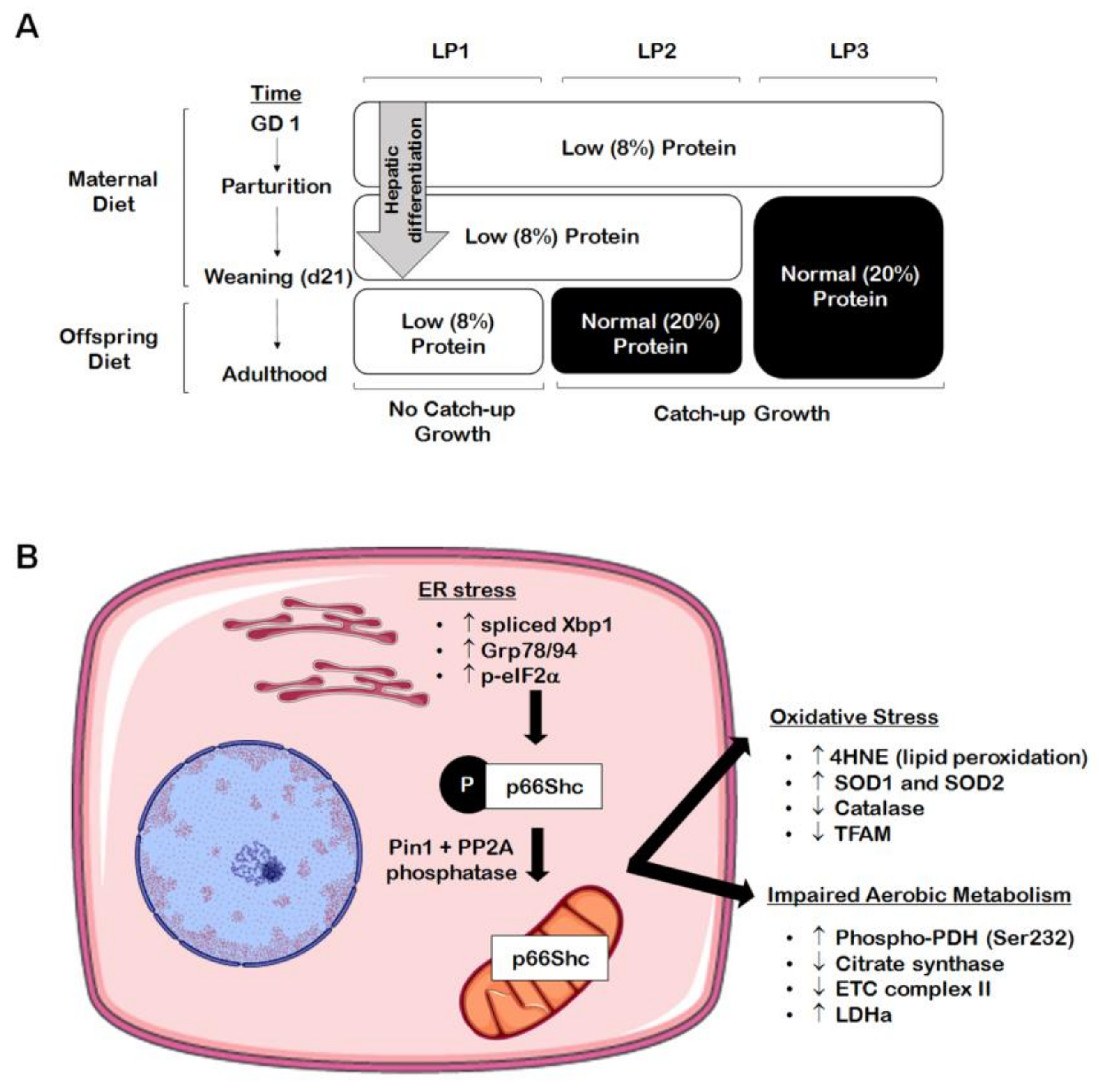
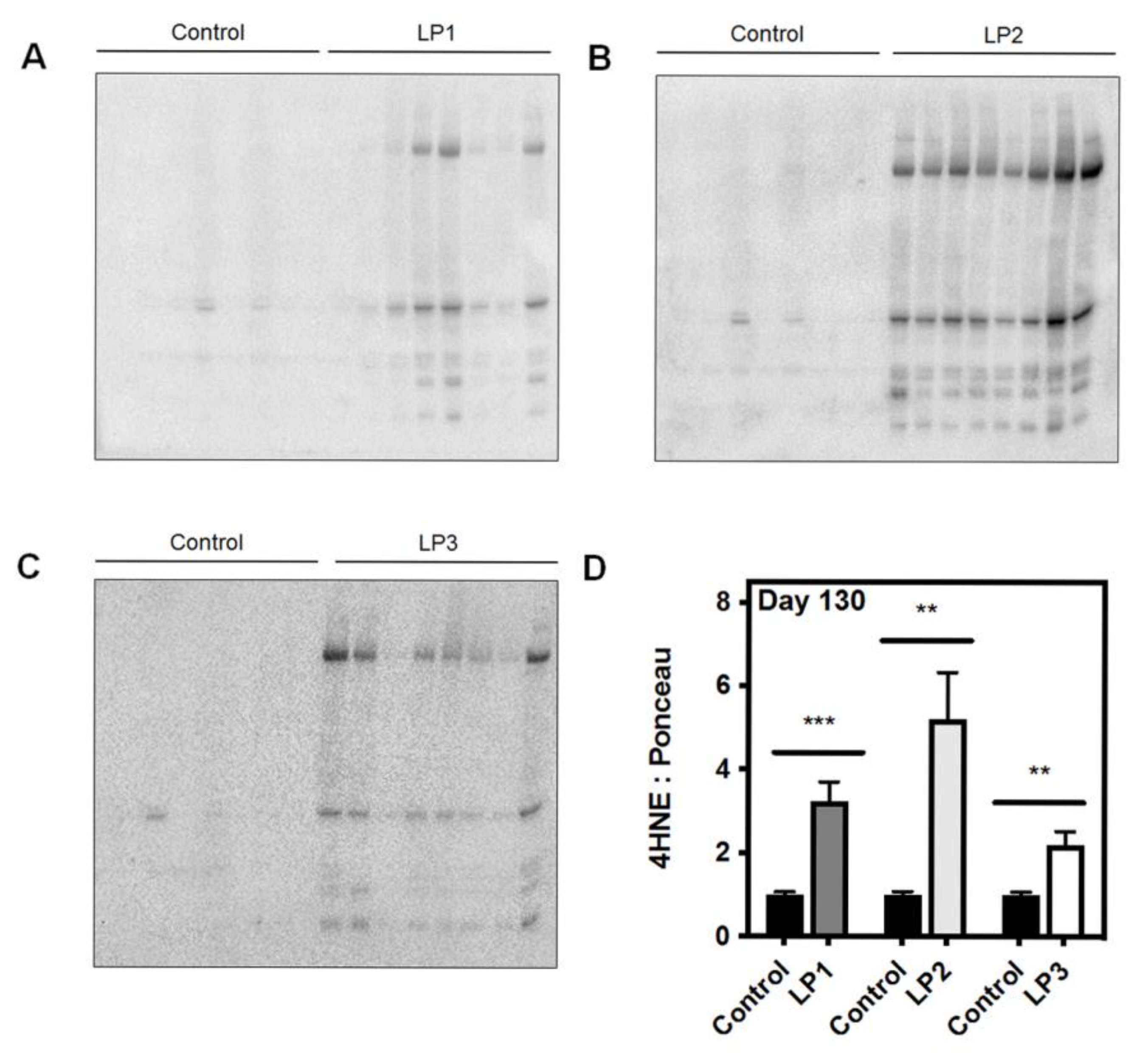
3.1. Oxidative Stress and Mitochondrial Dysfunction
3.2. Endoplasmic Reticulum (ER) Stress and the Unfolded Protein Response
3.3. Inflammation and the Immune Response
4. Programmed Cell Death and Metabolism in IUGR Offspring
4.1. Apoptosis
4.2. Autophagy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ravelli, G.P.; Stein, Z.A.; Susser, M.W. Obesity in Young Men after Famine Exposure in Utero and Early Infancy. N. Engl. J. Med. 1976, 295, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.C.; van der Meulen, J.H.; Michels, R.P.; Osmond, C.; Barker, D.J.; Hales, C.N.; Bleker, O.P. Glucose Tolerance in Adults after Prenatal Exposure to Famine. Lancet 1998, 351, 173–177. [Google Scholar] [CrossRef]
- Ravelli, A.C.; van der Meulen, J.H.; Osmond, C.; Barker, D.J.; Bleker, O.P. Obesity at the Age of 50 y in Men and Women Exposed to Famine Prenatally. Am. J. Clin. Nutr. 1999, 70, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Baschat, A.A. Pathophysiology of Fetal Growth Restriction: Implications for Diagnosis and Surveillance. Obstet. Gynecol. Surv. 2004, 59, 617–627. [Google Scholar] [CrossRef]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin. Med. Insights. Pediatrics 2016, 10, 67–83. [Google Scholar] [CrossRef]
- Barker, D.J.; Martyn, C.N.; Osmond, C.; Hales, C.N.; Fall, C.H. Growth in Utero and Serum Cholesterol Concentrations in Adult Life. BMJ 1993, 307, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J. The Thrifty Phenotype Hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Osmond, C.; Barker, D.J.P.; Winter, P.D.; Fall, C.H.D.; Simmonds, S.J. Early Growth and Death from Cardiovascular Disease in Women. Br. Med. J. 1993, 307, 1519–1524. [Google Scholar] [CrossRef]
- Barker, D.J.; Hales, C.N.; Fall, C.H.; Osmond, C.; Phipps, K.; Clark, P.M. Type 2 (Non-Insulin-Dependent) Diabetes Mellitus, Hypertension and Hyperlipidaemia (Syndrome X): Relation to Reduced Fetal Growth. Diabetologia 1993, 36, 62–67. [Google Scholar] [CrossRef]
- Fall, C.H.; Osmond, C.; Barker, D.J.; Clark, P.M.; Hales, C.N.; Stirling, Y.; Meade, T.W. Fetal and Infant Growth and Cardiovascular Risk Factors in Women. BMJ 1995, 310, 428–432. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and Infant Growth and Impaired Glucose Tolerance at Age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef]
- Cooper, C.; Fall, C.; Egger, P.; Hobbs, R.; Eastell, R.; Barker, D. Growth in Infancy and Bone Mass in Later Life. Ann. Rheum. Dis. 1997, 56, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Sayer, A.A.; Cooper, C.; Evans, J.R.; Rauf, A.; Wormald, R.P.L.; Osmond, C.; Barker, D.J.P. Are Rates of Ageing Determined in Utero? Age Ageing 1998, 27, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Victora, C.G.; Barros, F.C.; Lima, R.C.; Behague, D.P.; Gon alves, H.; Horta, B.L.; Gigante, D.P.; Vaughan, J.P. The Pelotas Birth Cohort Study, Rio Grande Do Sul, Brazil, 1982–2001. Cad. De Saúde Pública / Ministério Da Saúde Fundação Oswaldo Cruz Esc. Nac. De Saúde Pública 2003, 19, 1241–1256. [Google Scholar] [CrossRef] [PubMed]
- Musa, M.G.; Kagura, J.; Pisa, P.T.; Norris, S.A. Relationship between Early Growth and CVD Risk Factors in Adolescents. J. Dev. Orig. Health Dis. 2015, 7, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Perng, W.; Hajj, H.; Belfort, M.B.; Rifas-Shiman, S.L.; Kramer, M.S.; Gillman, M.W.; Oken, E. Birth Size, Early Life Weight Gain, and Midchildhood Cardiometabolic Health. J. Pediatrics 2016, 173, 122–130.e1. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.D.; Zybert, P.A.; Van Der Pal-De Bruin, K.; Lumey, L.H. Exposure to Famine during Gestation, Size at Birth, and Blood Pressure at Age 59 y: Evidence from the Dutch Famine. Eur. J. Epidemiol. 2006, 21, 759–765. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Han, X.; Liu, B.; Hu, H.; Wang, F.; Li, X.; Yang, K.; Yuan, J.; Yao, P.; et al. Exposure to the Chinese Famine in Childhood Increases Type 2 Diabetes Risk in Adults. J. Nutr. 2016, 146, 2289–2295. [Google Scholar] [CrossRef]
- Wang, P.-X.; Wang, J.-J.; Lei, Y.-X.; Xiao, L.; Luo, Z.-C. Impact of Fetal and Infant Exposure to the Chinese Great Famine on the Risk of Hypertension in Adulthood. PLoS ONE 2012, 7, e49720. [Google Scholar] [CrossRef]
- Huang, C.; Li, Z.; Wang, M.; Martorell, R. Early Life Exposure to the 1959-1961 Chinese Famine Has Long-Term Health Consequences. J. Nutr. 2010, 140, 1874–1878. [Google Scholar] [CrossRef]
- Eriksson, J.; Forsén, T.; Tuomilehto, J.; Osmond, C.; Barker, D. Size at Birth, Childhood Growth and Obesity in Adult Life. Int. J. Obes. 2001, 25, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, W.; Zhang, X.; Mu, R.; Zhai, Y.; Kong, L.; Chen, C. Impact of Famine during Pregnancy and Infancy on Health in Adulthood. Obes. Rev. 2008, 9, 95–99. [Google Scholar] [CrossRef]
- James, P.T.; Rigby, N.; Leach, R. The Obesity Epidemic, Metabolic Syndrome and Future Prevention Strategies. Eur. J. Cardiovasc. Prev. Rehabil. 2004, 11, 3–8. [Google Scholar] [CrossRef]
- Salgin, B.; Norris, S.A.; Prentice, P.; Pettifor, J.M.; Richter, L.M.; Ong, K.K.; Dunger, D.B. Even Transient Rapid Infancy Weight Gain Is Associated with Higher BMI in Young Adults and Earlier Menarche. Int. J. Obes. 2015, 39, 939–944. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Valsamakis, G.; Kanaka-Gantenbein, C.; Malamitsi-Puchner, A.; Mastorakos, G. Causes of Intrauterine Growth Restriction and the Postnatal Development of the Metabolic Syndrome. Ann. N. Y. Acad. Sci. 2006, 1092, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Neerhof, M.G. Causes of Intrauterine Growth Restriction. Clin. Perinatol. 1995, 22, 375–385. [Google Scholar] [CrossRef]
- Deiber, M.; Chatelain, P.; Naville, D.; Putet, G.; Salle, B. Functional hypersomatotropism in small for gestational age (sga) newborn infants. J. Clin. Endocrinol. Metab. 1989, 68, 232–234. [Google Scholar] [CrossRef]
- Singhal, A.; Cole, T.J.; Fewtrell, M.; Lucas, A. Breastmilk Feeding and Lipoprotein Profile in Adolescents Born Preterm: Follow-up of a Prospective Randomised Study. Lancet 2004, 363, 1571–1578. [Google Scholar] [CrossRef]
- Suomela, E.; Oikonen, M.; Pitkänen, N.; Ahola-Olli, A.; Virtanen, J.; Parkkola, R.; Jokinen, E.; Laitinen, T.; Hutri-Kähönen, N.; Kähönen, M.; et al. Childhood Predictors of Adult Fatty Liver. The Cardiovascular Risk in Young Finns Study. J. Hepatol. 2016, 65, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Marcellini, M.; Marchesini, G.; Vanni, E.; Manco, M.; Villani, A.; Bugianesi, E. Intrauterine Growth Retardation, Insulin Resistance, and Nonalcoholic Fatty Liver Disease in Children. Diabetes Care 2007, 30, 2638–2640. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Krumova, K.; Cosa, G. Chapter 1 Overview of Reactive Oxygen Species. In Singlet Oxygen: Applications in Biosciences and Nanosciences; Royal Society of Chemistry: London, UK, 2016; Volume 1, pp. 1–21. [Google Scholar] [CrossRef]
- Biri, A.; Bozkurt, N.; Turp, A.; Kavutcu, M.; Himmetoglu, Ö.; Durak, I. Role of Oxidative Stress in Intrauterine Growth Restriction. Gynecol. Obstet. Investig. 2007, 64, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Narang, M.; Banerjee, B.D.; Basu, S. Oxidative Stress in Term Small for Gestational Age Neonates Born to Undernourished Mothers: A Case Control Study. BMC Pediatrics 2004, 4, 14. [Google Scholar] [CrossRef]
- Karowicz-Bilinska, A.; Suzin, J.; Sieroszewski, P. Evaluation of Oxidative Stress Indices during Treatment in Pregnant Women with Intrauterine Growth Retardation. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2002, 8, CR211–CR216. [Google Scholar]
- Saker, M.; Soulimane Mokhtari, N.; Merzouk, S.A.; Merzouk, H.; Belarbi, B.; Narce, M. Oxidant and Antioxidant Status in Mothers and Their Newborns According to Birthweight. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 141, 95–99. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, P.; Ramiro-Cortijo, D.; Reyes-Hernández, C.G.; López de Pablo, A.L.; González, M.C.; Arribas, S.M. Implication of Oxidative Stress in Fetal Programming of Cardiovascular Disease. Front. Physiol. 2018, 9, 602. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial Dysfunction and Oxidative Stress in the Pathogenesis of Alcohol- and Obesity-Induced Fatty Liver Diseases. Free Radic. Biol. Med. 2008, 44, 1259–1272. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, L.; Li, D.; Yin, Y.; Wang, X.; Li, P.; Dangott, L.J.; Hu, W.; Wu, G. Intrauterine Growth Restriction Affects the Proteomes of the Small Intestine, Liver, and Skeletal Muscle in Newborn Pigs. J. Nutr. 2008, 138, 60–66. [Google Scholar] [CrossRef]
- Devarajan, A.; Rajasekaran, N.S.; Valburg, C.; Ganapathy, E.; Bindra, S.; Freije, W.A. Maternal Perinatal Calorie Restriction Temporally Regulates the Hepatic Autophagy and Redox Status in Male Rat. Free Radic. Biol. Med. 2019, 130, 592–600. [Google Scholar] [CrossRef]
- Sohi, G.; Marchand, K.; Revesz, A.; Arany, E.; Hardy, D.B. Maternal Protein Restriction Elevates Cholesterol in Adult Rat Offspring Due to Repressive Changes in Histone Modifications at the Cholesterol 7alpha-Hydroxylase Promoter. Mol. Endocrinol. 2011, 25, 785–798. [Google Scholar] [CrossRef]
- Moraes, C.; Rebelato, H.J.; Amaral, M.E.C.; Resende, T.M.; Silva, E.V.C.; Esquisatto, M.A.M.; Catisti, R. Effect of Maternal Protein Restriction on Liver Metabolism in Rat Offspring. J. Physiol. Sci. 2014, 64, 347–355. [Google Scholar] [CrossRef]
- Vo, T.X.; Revesz, A.; Ma, N.; Hardy, D.B. Maternal Protein Restriction Leads to Enhanced Hepatic Gluconeogenic Gene Expression in Adult Male Rat Offspring Due to Impaired Expression of the Liver x Receptor. J. Endocrinol. 2013, 218, 85–97. [Google Scholar] [CrossRef]
- Oke, S.L.; Sohi, G.; Hardy, D.B. Perinatal Protein Restriction with Postnatal Catch-up Growth Leads to Elevated P66Shc and Mitochondrial Dysfunction in the Adult Rat Liver. Reproduction 2020, 159, 27–39. [Google Scholar] [CrossRef]
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R.; et al. Endoplasmic Reticulum Stress Links Oxidative Stress to Impaired Pancreatic Beta-Cell Function Caused by Human Oxidized LDL. PLoS ONE 2016, 11, e0163046. [Google Scholar] [CrossRef]
- Theys, N.; Clippe, A.; Bouckenooghe, T.; Reusens, B.; Remacle, C. Early Low Protein Diet Aggravates Unbalance between Antioxidant Enzymes Leading to Islet Dysfunction. PLoS ONE 2009, 4, e6110. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Kim, S.K.; Kim, M.S.; Cho, E.Y.; Lee, J.H.; Lee, K.-U.; Pak, Y.K.; Lee, H.K. Fetal and Early Postnatal Protein Malnutrition Cause Long-Term Changes in Rat Liver and Muscle Mitochondria. J. Nutr. 2003, 133, 3085–3090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Setia, S.; Sridhar, M.G.; Bhat, V.; Chaturvedula, L.; Vinayagamoorti, R.; John, M. Insulin Sensitivity and Insulin Secretion at Birth in Intrauterine Growth Retarded Infants. Pathology 2006, 38, 236–238. [Google Scholar] [CrossRef]
- Milovanovic, I.; Njuieyon, F.; Deghmoun, S.; Chevenne, D.; Levy-Marchal, C.; Beltrand, J. SGA Children with Moderate Catch-Up Growth Are Showing the Impaired Insulin Secretion at the Age of 4. PLoS ONE 2014, 9, e100337. [Google Scholar] [CrossRef]
- Li, C.; Johnson, M.S.; Goran, M.I. Effects of Low Birth Weight on Insulin Resistance Syndrome in Caucasian and African-American Children. Diabetes Care 2001, 21, 2035–2042. [Google Scholar] [CrossRef][Green Version]
- Jensen, C.B.; Storgaard, H.; Dela, F.; Holst, J.J.; Madsbad, S.; Vaag, A.A. Early Differential Defects of Insulin Secretion and Action in 19-Year-Old Caucasian Men Who Had Low Birth Weight. Diabetes 2002, 51, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.A.; Suponitsky-Kroyter, I.; Selak, M.A. Progressive Accumulation of Mitochondrial DNA Mutations and Decline in Mitochondrial Function Lead to β-Cell Failure. J. Biol. Chem. 2005, 280, 28785–28791. [Google Scholar] [CrossRef]
- Theys, N.; Bouckenooghe, T.; Ahn, M.T.; Remacle, C.; Reusens, B. Maternal Low-Protein Diet Alters Pancreatic Islet Mitochondrial Function in a Sex-Specific Manner in the Adult Rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1516–R1525. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.H.; Chandorkar, A.K.; Flozak, A.S.; Simmons, R.A. Intrauterine Growth Retardation Alters Mitochondrial Gene Expression and Function in Fetal and Juvenile Rat Skeletal Muscle. Pediatric Res. 1998, 43, 563–570. [Google Scholar] [CrossRef]
- Selak, M.A.; Storey, B.T.; Peterside, I.; Simmons, R.A. Impaired Oxidative Phosphorylation in Skeletal Muscle of Intrauterine Growth-Retarded Rats. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E130–E137. [Google Scholar] [CrossRef]
- Liu, J.; Chen, D.; Yao, Y.; Yu, B.; Mao, X.; He, J.; Huang, Z.; Zheng, P. Intrauterine Growth Retardation Increases the Susceptibility of Pigs to High-Fat Diet-Induced Mitochondrial Dysfunction in Skeletal Muscle. PLoS ONE 2012, 7, 4835. [Google Scholar] [CrossRef]
- Beauchamp, B.; Ghosh, S.; Dysart, M.W.; Kanaan, G.N.; Chu, A.; Blais, A.; Rajamanickam, K.; Tsai, E.C.; Patti, M.-E.; Harper, M.-E. Low Birth Weight Is Associated with Adiposity, Impaired Skeletal Muscle Energetics and Weight Loss Resistance in Mice. Int. J. Obes. 2015, 39, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Tarry-Adkins, J.L.; Fernandez-Twinn, D.S.; Chen, J.H.; Hargreaves, I.P.; Neergheen, V.; Aiken, C.E.; Ozanne, S.E. Poor Maternal Nutrition and Accelerated Postnatal Growth Induces an Accelerated Aging Phenotype and Oxidative Stress in Skeletal Muscle of Male Rats. Dis. Models Mech. 2016, 9, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Barra, N.G.; VanDuzer, T.A.; Holloway, A.C.; Hardy, D.B. Maternal Nicotine Exposure Leads to Augmented Expression of the Antioxidant Adipose Tissue Triglyceride Lipase Long-Term in the White Adipose of Female Rat Offspring. Toxicol. Sci. 2018, 164, 72–84. [Google Scholar] [CrossRef]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Barra, N.G.; Lisyansky, M.; Vanduzer, T.A.; Raha, S.; Holloway, A.C.; Hardy, D.B. Maternal Nicotine Exposure Leads to Decreased Cardiac Protein Disulfide Isomerase and Impaired Mitochondrial Function in Male Rat Offspring. J. Appl. Toxicol. 2017, 37, 1517–1526. [Google Scholar] [CrossRef]
- Barnett, C.; Nnoli, O.; Abdulmahdi, W.; Nesi, L.; Shen, M.; Zullo, J.A.; Payne, D.L.; Azar, T.; Dwivedi, P.; Syed, K.; et al. Low Birth Weight Is Associated with Impaired Murine Kidney Development and Function. Pediatric Res. 2017, 82, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Rabadi, M.M.; Abdulmahdi, W.; Nesi, L.; Jules, E.; Marghani, Y.; Sheinin, E.; Tilzer, J.; Gupta, S.; Chen, S.; Cassimatis, N.D.; et al. Maternal Malnourishment Induced Upregulation of Fetuin-B Blunts Nephrogenesis in the Low Birth Weight Neonate. Dev. Biol. 2018, 443, 78–91. [Google Scholar] [CrossRef]
- Woodman, A.G.; Mah, R.; Keddie, D.; Noble, R.M.N.; Panahi, S.; Gragasin, F.S.; Lemieux, H.; Bourque, S.L. Prenatal Iron Deficiency Causes Sex-Dependent Mitochondrial Dysfunction and Oxidative Stress in Fetal Rat Kidneys and Liver. FASEB J. 2018, 32, 3254–3263. [Google Scholar] [CrossRef]
- Woodman, A.G.; Mah, R.; Keddie, D.L.; Noble, R.M.N.; Holody, C.D.; Panahi, S.; Gragasin, F.S.; Lemieux, H.; Bourque, S.L. Perinatal Iron Deficiency and a High Salt Diet Cause Long-Term Kidney Mitochondrial Dysfunction and Oxidative Stress. Cardiovasc. Res. 2020, 116, 183–192. [Google Scholar] [CrossRef]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: More than Just a Housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef]
- Michalak, M.; Gye, M.C. Endoplasmic Reticulum Stress in Periimplantation Embryos. Clin. Exp. Reprod. Med. 2015, 42, 1–7. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic Reticulum Stress: Cell Life and Death Decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.K.; Nicholson, C.J.; Holloway, A.C.; Hardy, D.B. Maternal Nicotine Exposure Leads to Impaired Disulphide Bond Formation and Augmented Endoplasmic Reticulum Stress in the Rat Placenta. PLoS ONE 2015, in press. [Google Scholar]
- Sohi, G.; Revesz, A.; Hardy, D.B. Nutritional Mismatch in Postnatal Life of Low Birth Weight Rat Offspring Leads to Increased Phosphorylation of Hepatic Eukaryotic Initiation Factor 2 Alpha in Adulthood. Metab. Clin. Exp. 2013, 62, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Hong, M.; Luo, S.; Baumeister, P.; Huang, J.M.; Gogia, R.K.; Li, M.; Lee, A.S. Underglycosylation of ATF6 as a Novel Sensing Mechanism for Activation of the Unfolded Protein Response. J. Biol. Chem. 2004, 279, 11354–11363. [Google Scholar] [CrossRef]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic Reticulum Stress, Obesity and Diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Gao, L.; Jiao, Y.; Liu, C. Maternal Protein Restriction Induces Alterations in Hepatic Unfolded Protein Response-Related Molecules in Adult Rat Offspring. Front. Endocrinol. 2018, 9, 676. [Google Scholar] [CrossRef]
- Deodati, A.; Argemí, J.; Germani, D.; Puglianiello, A.; Alisi, A.; De Stefanis, C.; Ferrero, R.; Nobili, V.; Aragón, T.; Cianfarani, S. The Exposure to Uteroplacental Insufficiency Is Associated with Activation of Unfolded Protein Response in Postnatal Life. PLoS ONE 2018, 13, e0198490. [Google Scholar] [CrossRef]
- Mounir, Z.; Krishnamoorthy, J.L.; Wang, S.; Papadopoulou, B.; Campbell, S.; Muller, W.J.; Hatzoglou, M.; Koromilas, A.E. Akt Determines Cell Fate through Inhibition of the PERK-EIF2alpha Phosphorylation Pathway. Sci. Signal. 2011, 4, ra62. [Google Scholar] [CrossRef]
- Liu, X.; Guo, Y.; Wang, J.; Zhu, L.; Gao, L. Dysregulation in the Unfolded Protein Response in the FGR Rat Pancreas. Int. J. Endocrinol. 2020, 2020, 5759182. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, D.; Li, Y.; Xin, Y. Maternal Protein Restriction Increases Autophagy in the Pancreas of Newborn Rats. J. Nutr. Sci. Vitaminol. 2020, 66, 168–175. [Google Scholar] [CrossRef]
- Sachdeva, M.M.; Claiborn, K.C.; Khoo, C.; Yang, J.; Groff, D.N.; Mirmira, R.G.; Stoffers, D.A. Pdx1 (MODY4) Regulates Pancreatic Beta Cell Susceptibility to ER Stress. Proc. Natl. Acad. Sci. USA 2009, 106, 19090–19095. [Google Scholar] [CrossRef]
- Fritz, J.M.; Dong, M.; Apsley, K.S.; Martin, E.P.; Na, C.L.; Sitaraman, S.; Weaver, T.E. Deficiency of the BiP Cochaperone ERdj4 Causes Constitutive Endoplasmic Reticulum Stress and Metabolic Defects. Mol. Biol. Cell 2014, 25, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Riddle, E.S.; Campbell, M.S.; Lang, B.Y.; Bierer, R.; Wang, Y.; Bagley, H.N.; Joss-Moore, L.A. Intrauterine Growth Restriction Increases TNF α and Activates the Unfolded Protein Response in Male Rat Pups. J. Obes. 2014, 2014, 829862. [Google Scholar] [CrossRef]
- Stojanovska, V.; Sharma, N.; Dijkstra, D.J.; Scherjon, S.A.; Jäger, A.; Schorle, H.; Plösch, T. Placental Insufficiency Contributes to Fatty Acid Metabolism Alterations in Aged Female Mouse Offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R1107–R1114. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E.; Spiegelhalter, D.J. Non-Invasive Detection of Endothelial Dysfunction in Children and Adults at Risk of Atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef]
- Mohsenzadeh, Y.; Rahmani, A.; Cheraghi, J.; Pyrani, M.; Asadollahi, K. Prenatal Exposure to Nicotine in Pregnant Rat Increased Inflammatory Marker in Newborn Rat. Mediat. Inflamm. 2014, 2014, 274048. [Google Scholar] [CrossRef]
- Bæk, O.; Sangild, P.T.; Thymann, T.; Nguyen, D.N. Growth Restriction and Systemic Immune Development in Preterm Piglets. Front. Immunol. 2019, 10, 2402. [Google Scholar] [CrossRef]
- Ferenc, K.; Pietrzak, P.; Wierzbicka, M.; Matyba, P.; Grzesiuk, E.; Gajewski, Z.; Zabielski, R. Alterations in the Liver of Intrauterine Growth Retarded Piglets May Predispose to Development of Insulin Resistance and Obesity in Later Life. J. Physiol. Pharmacol. 2018, 69, 1–8. [Google Scholar] [CrossRef]
- Liu, X.; Qi, Y.; Tian, B.; Chen, D.; Gao, H.; Xi, C.; Xing, Y.; Yuan, Z. Maternal Protein Restriction Induces Alterations in Hepatic Tumor Necrosis Factor-α/CYP7A1 Signaling and Disorders Regulation of Cholesterol Metabolism in the Adult Rat Offspring. J. Clin. Biochem. Nutr. 2014, 55, 40–47. [Google Scholar] [CrossRef]
- Cheng, K.; Jia, P.; Ji, S.; Song, Z.; Zhang, H.; Zhang, L.; Wang, T. Improvement of the Hepatic Lipid Status in Intrauterine Growth Retarded Pigs by Resveratrol Is Related to the Inhibition of Mitochondrial Dysfunction, Oxidative Stress and Inflammation. Food Funct. 2021, 12, 278–290. [Google Scholar] [CrossRef]
- Kohnert, K.-D.; Freyse, E.-J.; Salzsieder, E. Glycaemic Variability and Pancreatic SS-Cell Dysfunction. Curr. Diabetes Rev. 2012, 8, 345–354. [Google Scholar] [CrossRef]
- Quan, W.; Jo, E.-K.; Lee, M.-S. Role of Pancreatic β -Cell Death and Inflammation in Diabetes. Diabetes Obes. Metab. 2013, 15, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Pulkki, K.; Kallajoki, M.; Henriksen, K.; Parvinen, M.; Voipio-Pulkki, L.M. Apoptosis in Human Acute Myocardial Infarction. Circulation 1997, 95, 320–323. [Google Scholar] [CrossRef]
- Olivetti, G.; Quaini, F.; Sala, R.; Lagrasta, C.; Corradi, D.; Bonacina, E.; Gambert, S.R.; Cigola, E.; Anversa, P. Acute Myocardial Infarction in Humans Is Associated with Activation of Programmed Myocyte Cell Death in the Surviving Portion of the Heart. J. Mol. Cell. Cardiol. 1996, 28, 2005–2016. [Google Scholar] [CrossRef]
- Narula, J.; Haider, N.; Virmani, R.; DiSalvo, T.G.; Kolodgie, F.D.; Hajjar, R.J.; Schmidt, U.; Semigran, M.J.; Dec, G.W.; Khaw, B.-A. Apoptosis in Myocytes in End-Stage Heart Failure. N. Engl. J. Med. 1996, 335, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Aharinejad, S.; Andrukhova, O.; Lucas, T.; Zuckermann, A.; Wieselthaler, G.; Wolner, E.; Grimm, M. Programmed Cell Death in Idiopathic Dilated Cardiomyopathy Is Mediated by Suppression of the Apoptosis Inhibitor Apollon. Ann. Thorac. Surg. 2008, 86, 109–114. [Google Scholar] [CrossRef]
- Tintu, A.; Rouwet, E.; Verlohren, S.; Brinkmann, J.; Ahmad, S.; Crispi, F.; van Bilsen, M.; Carmeliet, P.; Staff, A.C.; Tjwa, M.; et al. Hypoxia Induces Dilated Cardiomyopathy in the Chick Embryo: Mechanism, Intervention, and Long-Term Consequences. PLoS ONE 2009, 4, e5155. [Google Scholar] [CrossRef]
- Botting, K.J.; Loke, X.Y.; Zhang, S.; Andersen, J.B.; Nyengaard, J.R.; L Morrison, X.J. IUGR Decreases Cardiomyocyte Endowment and Alters Cardiac Metabolism in a Sex-and Cause-of-IUGR-Specific Manner. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, 48–67. [Google Scholar] [CrossRef]
- Li, G.; Xiao, Y.; Estrella, J.L.; Ducsay, C.A.; Gilbert, R.D.; Zhang, L. Effect of Fetal Hypoxia on Heart Susceptibility to Ischemia and Reperfusion Injury in the Adult Rat. J. Soc. Gynecol. Investig. 2003, 10, 265–274. [Google Scholar] [CrossRef]
- Rueda-Clausen, C.F.; Morton, J.S.; Lopaschuk, G.D.; Davidge, S.T. Long-Term Effects of Intrauterine Growth Restriction on Cardiac Metabolism and Susceptibility to Ischaemia/Reperfusion. Cardiovasc. Res. 2011, 90, 285–294. [Google Scholar] [CrossRef]
- Boujendar, S.; Reusens, B.; Merezak, S.; Ahn, M.T.; Arany, E.; Hill, D.; Remacle, C. Taurine Supplementation to a Low Protein Diet during Foetal and Early Postnatal Life Restores a Normal Proliferation and Apoptosis of Rat Pancreatic Islets. Diabetologia 2002, 45, 856–866. [Google Scholar] [CrossRef]
- Petrik, J.; Reusens, B.; Arany, E.; Remacle, C.; Coelho, C.; Hoet, J.J.; Hill, D.J. A Low Protein Diet Alters the Balance of Islet Cell Replication and Apoptosis in the Fetal and Neonatal Rat and Is Associated with a Reduced Pancreatic Expression of Insulin-like Growth Factor-II. Endocrinology 1999, 140, 4861–4873. [Google Scholar] [CrossRef] [PubMed]
- Dahri, S.; Snoeck, A.; Reusens-Billen, B.; Remacle, C.; Hoet, J.J. Islet Function in Offspring of Mothers on Low-Protein Diet during Gestation. Diabetes 1991, 40 (Suppl. S2), 115–120. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, C.; Li, H.; Li, L.; Gao, F.; Ao, C. Effects of Intrauterine Growth Restriction during Late Pregnancy on the Cell Apoptosis and Related Gene Expression in Ovine Fetal Liver. Theriogenology 2017, 90, 204–209. [Google Scholar] [CrossRef]
- Luo, Y.; Tian, Y.; Zhao, C. Taurine Attenuates Liver Autophagy and Injury of Offspring in Gestational Diabetic Mellitus Rats. Life Sci. 2020, 257, 117889. [Google Scholar] [CrossRef]
- Lakshmi, S.; Ae, D.; Anuradha, C.V. Mitochondrial Damage, Cytotoxicity and Apoptosis in Iron-Potentiated Alcoholic Liver Fibrosis: Amelioration by Taurine. Amino Acids 2010, 38, 869–879. [Google Scholar] [CrossRef]
- Wang, Y.; Mei, X.; Yuan, J.; Lai, X.; Xu, D. Taurine Zinc Solid Dispersions Enhance Bile-Incubated L02 Cell Viability and Improve Liver Function by Inhibiting ERK2 and JNK Phosphorylation during Cholestasis. Toxicology 2016, 366–367, 10–19. [Google Scholar] [CrossRef]
- Roy, A.; Sil, P.C. Taurine Protects Murine Hepatocytes against Oxidative Stress-Induced Apoptosis by Tert-Butyl Hydroperoxide via PI3K/Akt and Mitochondrial-Dependent Pathways. Food Chem. 2012, 131, 1086–1096. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Li, C.; Nakayasu, E.S.; Casey, C.P.; Metz, T.O.; Nathanielsz, P.W.; Maloyan, A. Sexual Dimorphism in the Fetal Cardiac Response to Maternal Nutrient Restriction. J. Mol. Cell. Cardiol. 2017, 108, 181–193. [Google Scholar] [CrossRef]
- de Toro-Martín, J.; Fernández-Marcelo, T.; González-Rodríguez, Á.; Escrivá, F.; Valverde, Á.M.; Álvarez, C.; Fernández-Millán, E. Defective Liver Glycogen Autophagy Related to Hyperinsulinemia in Intrauterine Growth-Restricted Newborn Wistar Rats. Sci. Rep. 2020, 10, 17651. [Google Scholar] [CrossRef]
- Iliescu, R.; Cucchiarelli, V.E.; Yanes, L.L.; Iles, J.W.; Reckelhoff, J.F. Impact of Androgen-Induced Oxidative Stress on Hypertension in Male SHR. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R731–R735. [Google Scholar] [CrossRef] [PubMed]
- Azhary, J.M.K.; Harada, M.; Takahashi, N.; Nose, E.; Kunitomi, C.; Koike, H.; Hirata, T.; Hirota, Y.; Koga, K.; Wada-Hiraike, O.; et al. Endoplasmic Reticulum Stress Activated by Androgen Enhances Apoptosis of Granulosa Cells via Induction of Death Receptor 5 in PCOS. Endocrinology 2019, 160, 119–132. [Google Scholar] [CrossRef]
- Jazwiec, P.A.; Patterson, V.S.; Ribeiro, T.A.; Yeo, E.; Kennedy, K.M.; Mathias, P.C.F.; Petrik, J.J.; Sloboda, D.M. Paternal Obesity Results in Placental Hypoxia and Sex-Specific Impairments in Placental Vascularization and Offspring Metabolic Function. bioRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Raab, E.L.; Vuguin, P.M.; Stoffers, D.A.; Simmons, R.A. Neonatal Exendin-4 Treatment Reduces Oxidative Stress and Prevents Hepatic Insulin Resistance in Intrauterine Growth-Retarded Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1785–R1794. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oke, S.L.; Hardy, D.B. The Role of Cellular Stress in Intrauterine Growth Restriction and Postnatal Dysmetabolism. Int. J. Mol. Sci. 2021, 22, 6986. https://doi.org/10.3390/ijms22136986
Oke SL, Hardy DB. The Role of Cellular Stress in Intrauterine Growth Restriction and Postnatal Dysmetabolism. International Journal of Molecular Sciences. 2021; 22(13):6986. https://doi.org/10.3390/ijms22136986
Chicago/Turabian StyleOke, Shelby L., and Daniel B. Hardy. 2021. "The Role of Cellular Stress in Intrauterine Growth Restriction and Postnatal Dysmetabolism" International Journal of Molecular Sciences 22, no. 13: 6986. https://doi.org/10.3390/ijms22136986
APA StyleOke, S. L., & Hardy, D. B. (2021). The Role of Cellular Stress in Intrauterine Growth Restriction and Postnatal Dysmetabolism. International Journal of Molecular Sciences, 22(13), 6986. https://doi.org/10.3390/ijms22136986