
Mutagenic Analysis of the Putative ABCC6 Substrate-Binding Cavity Using a New Homology Model

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
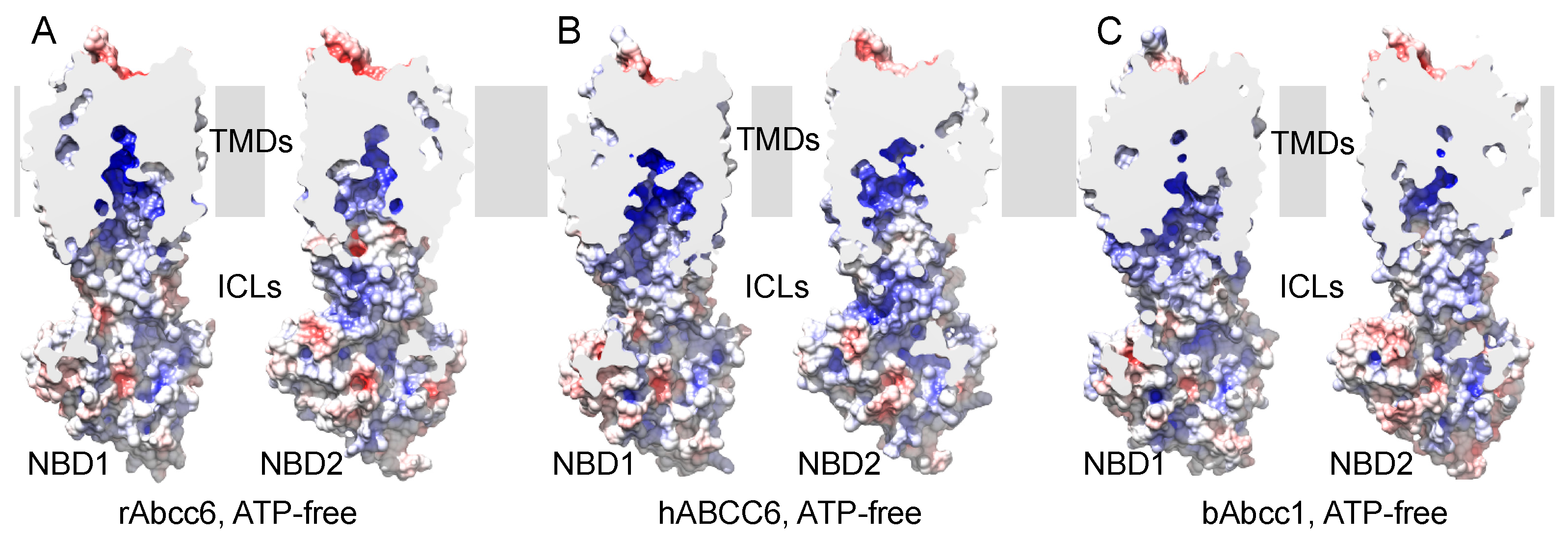
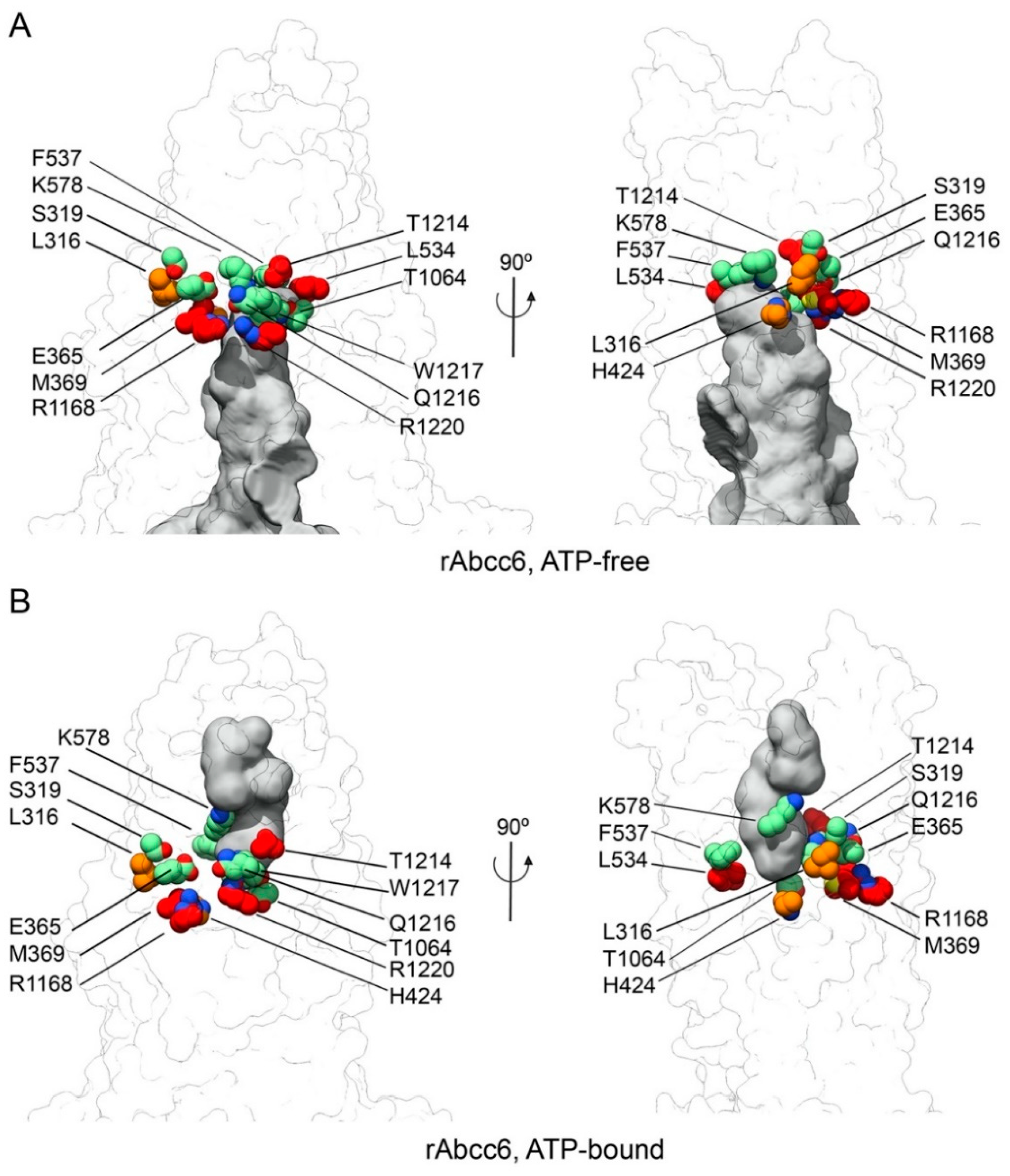
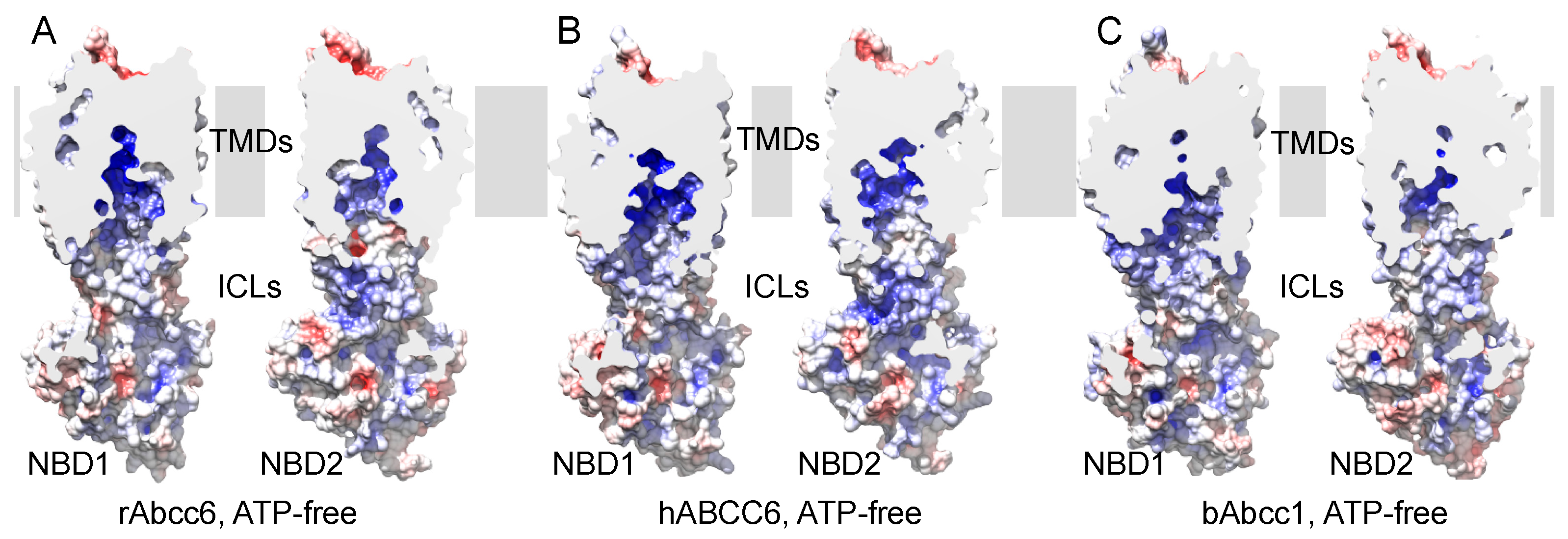
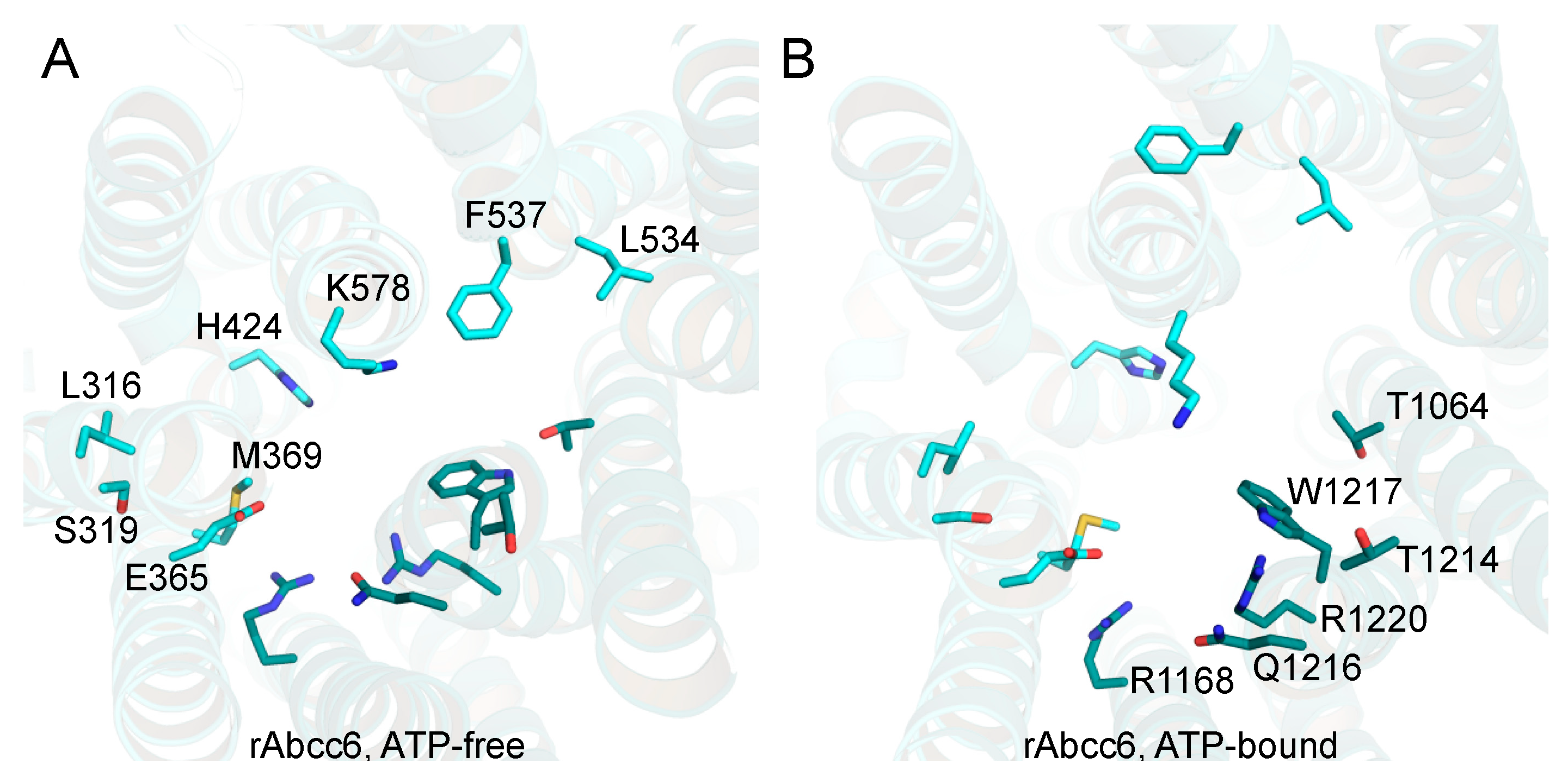
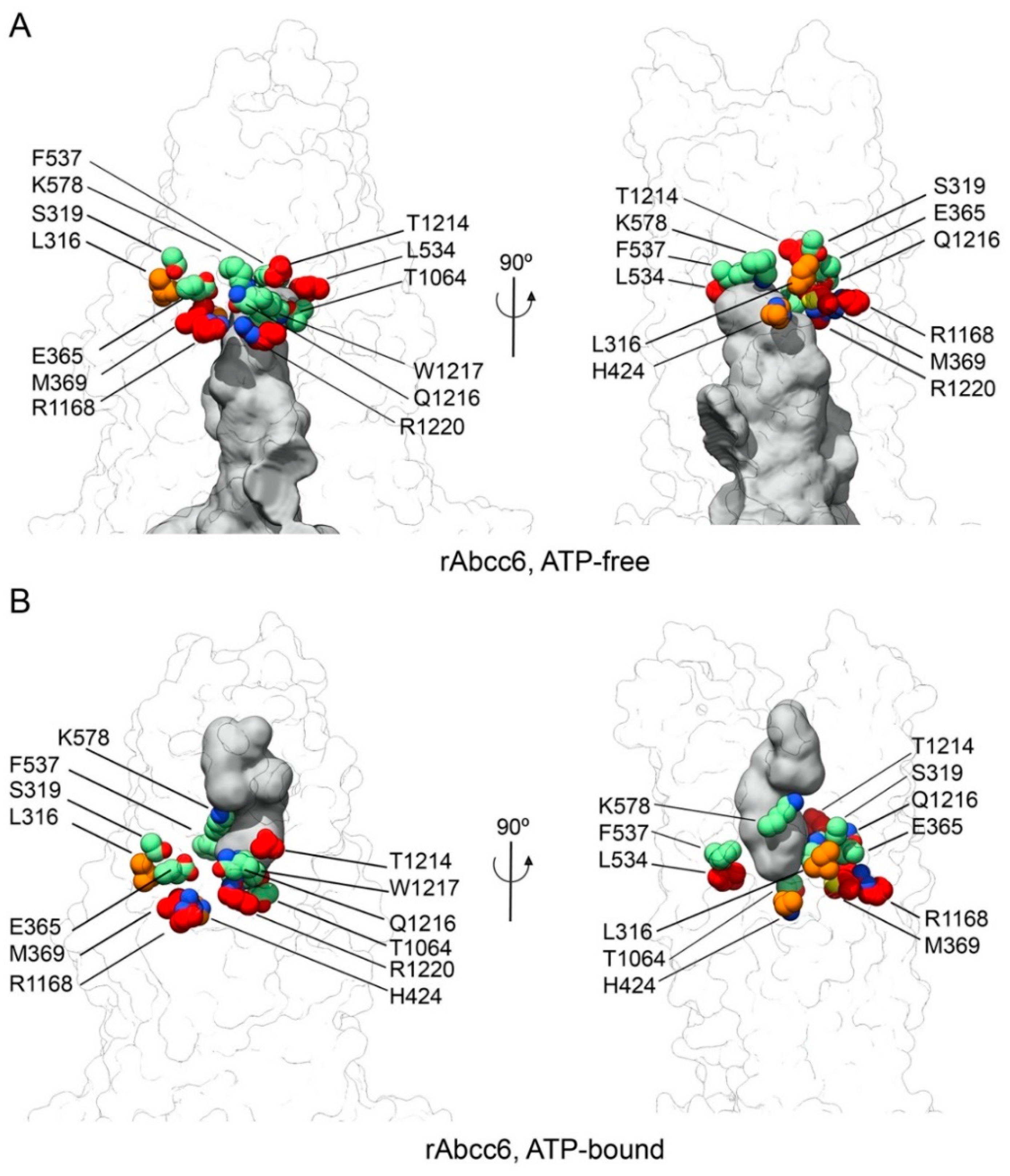
2.1. Homology Models of Human and Rat ABCC6/Abcc6
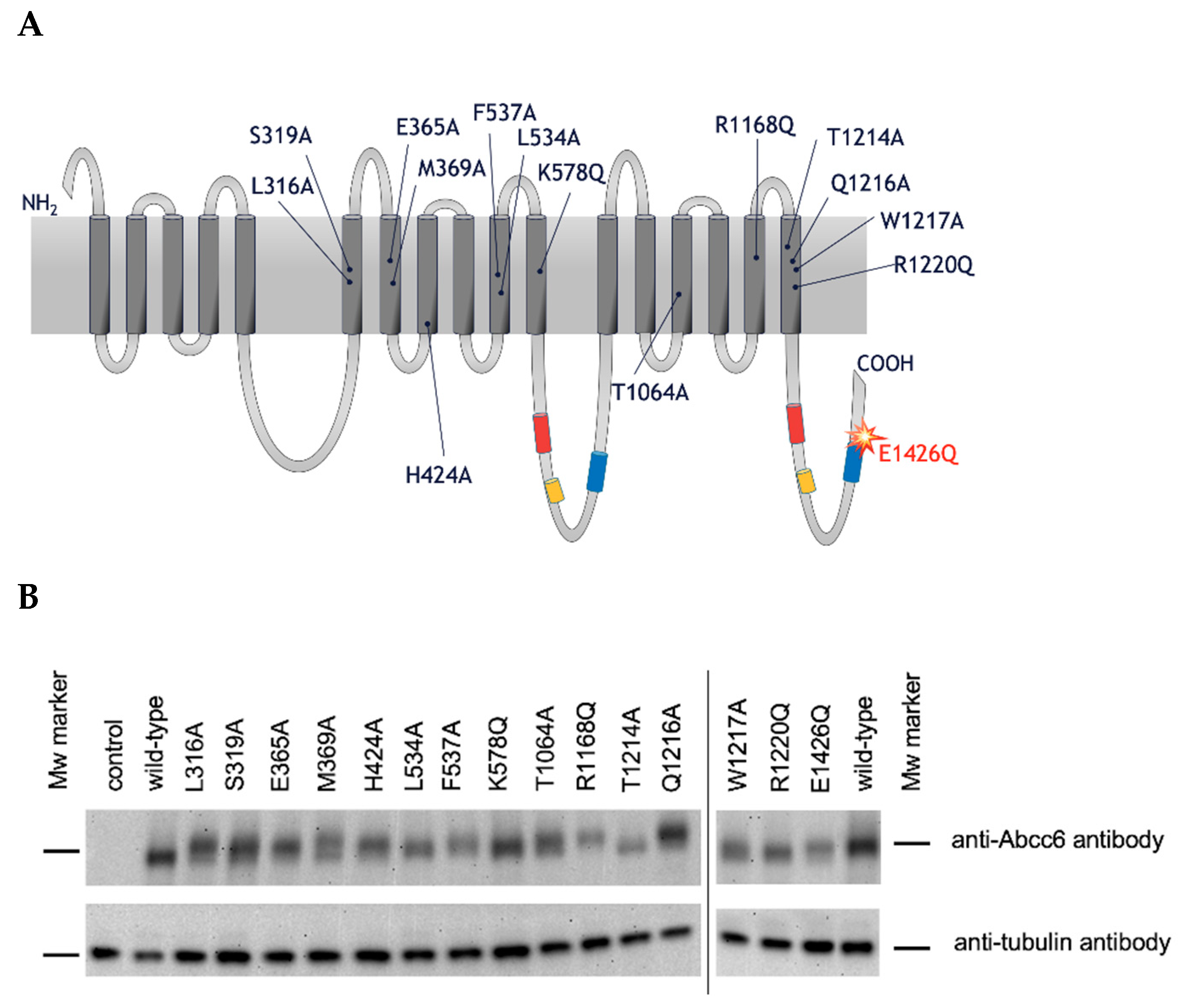
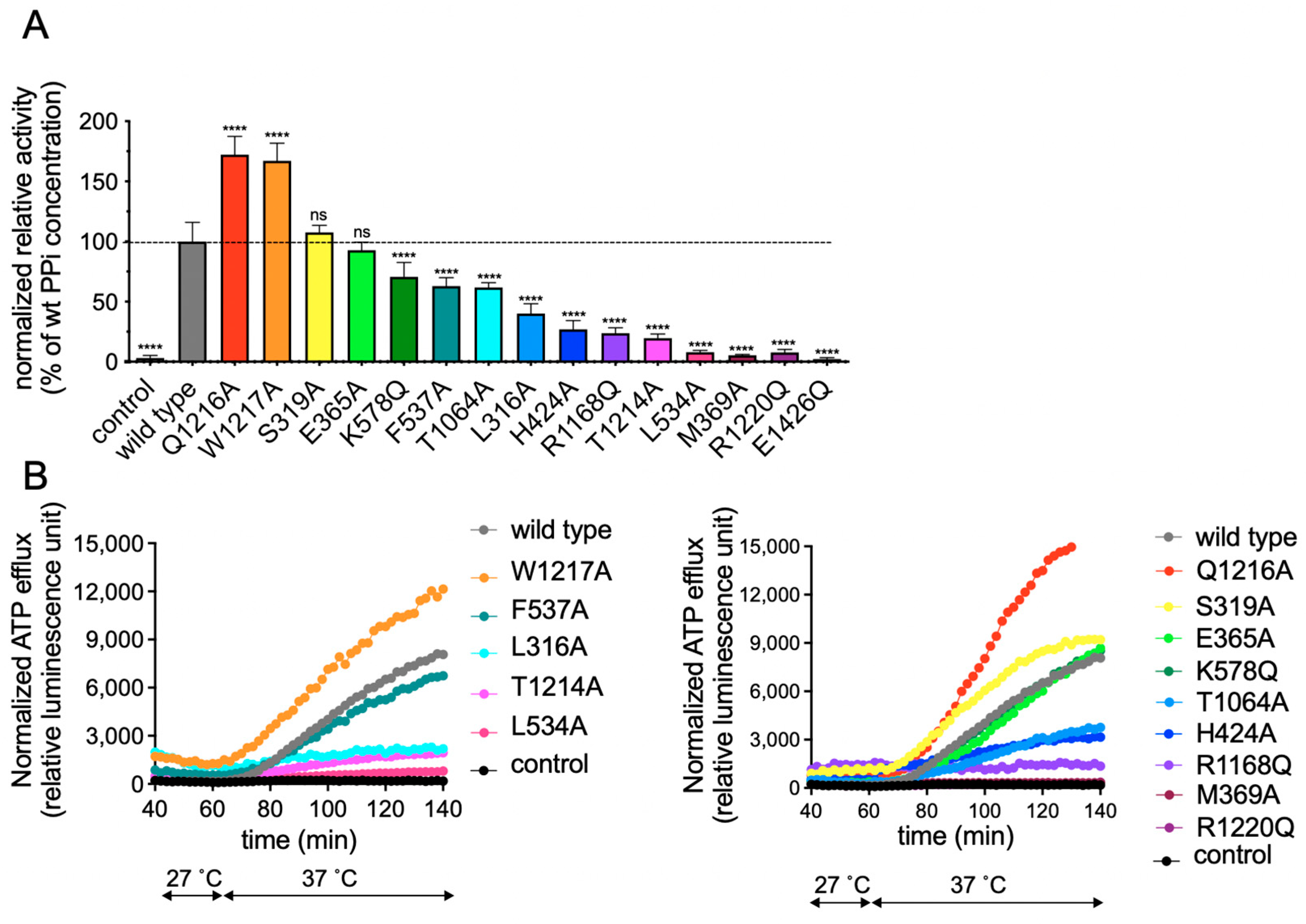
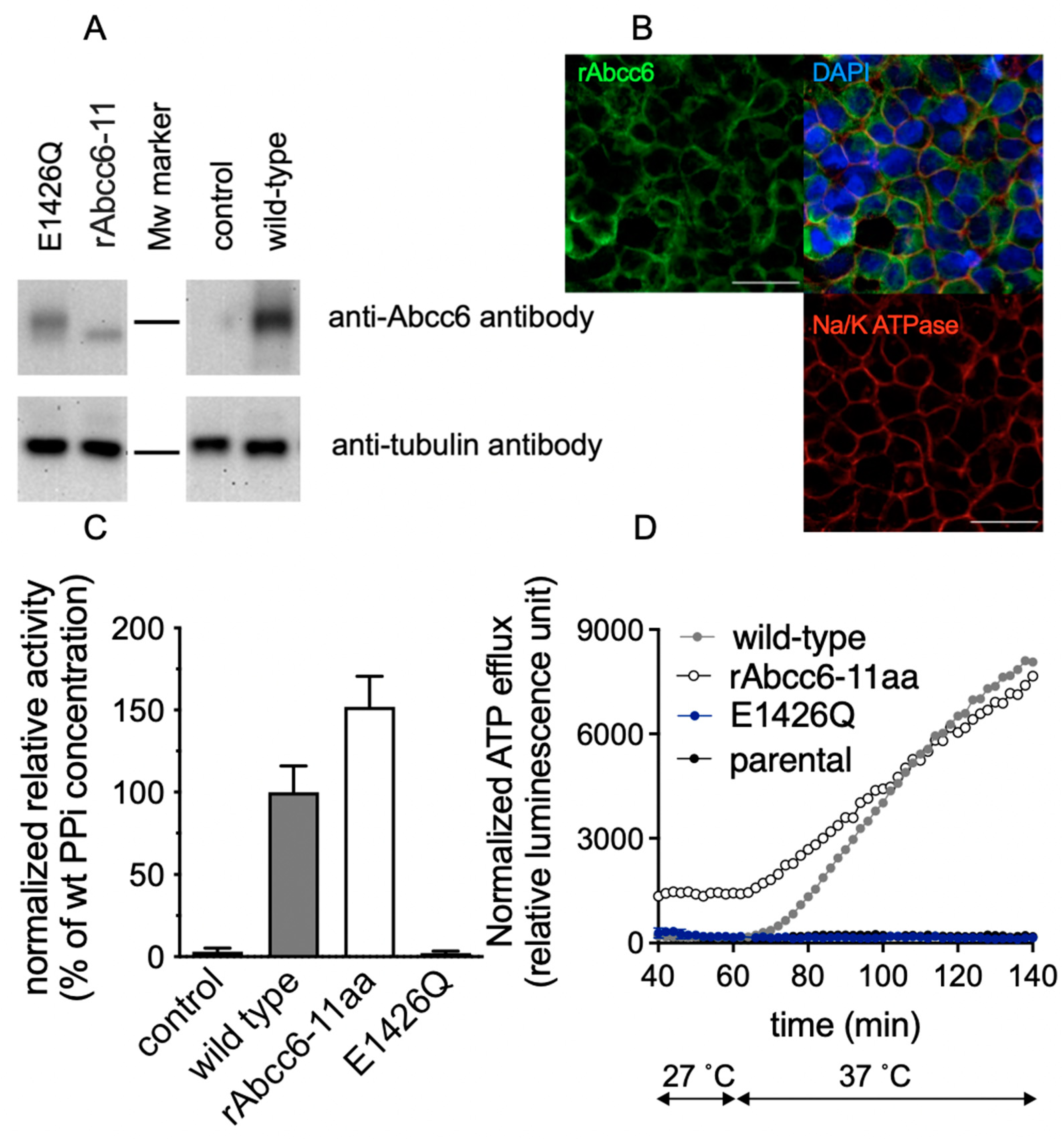
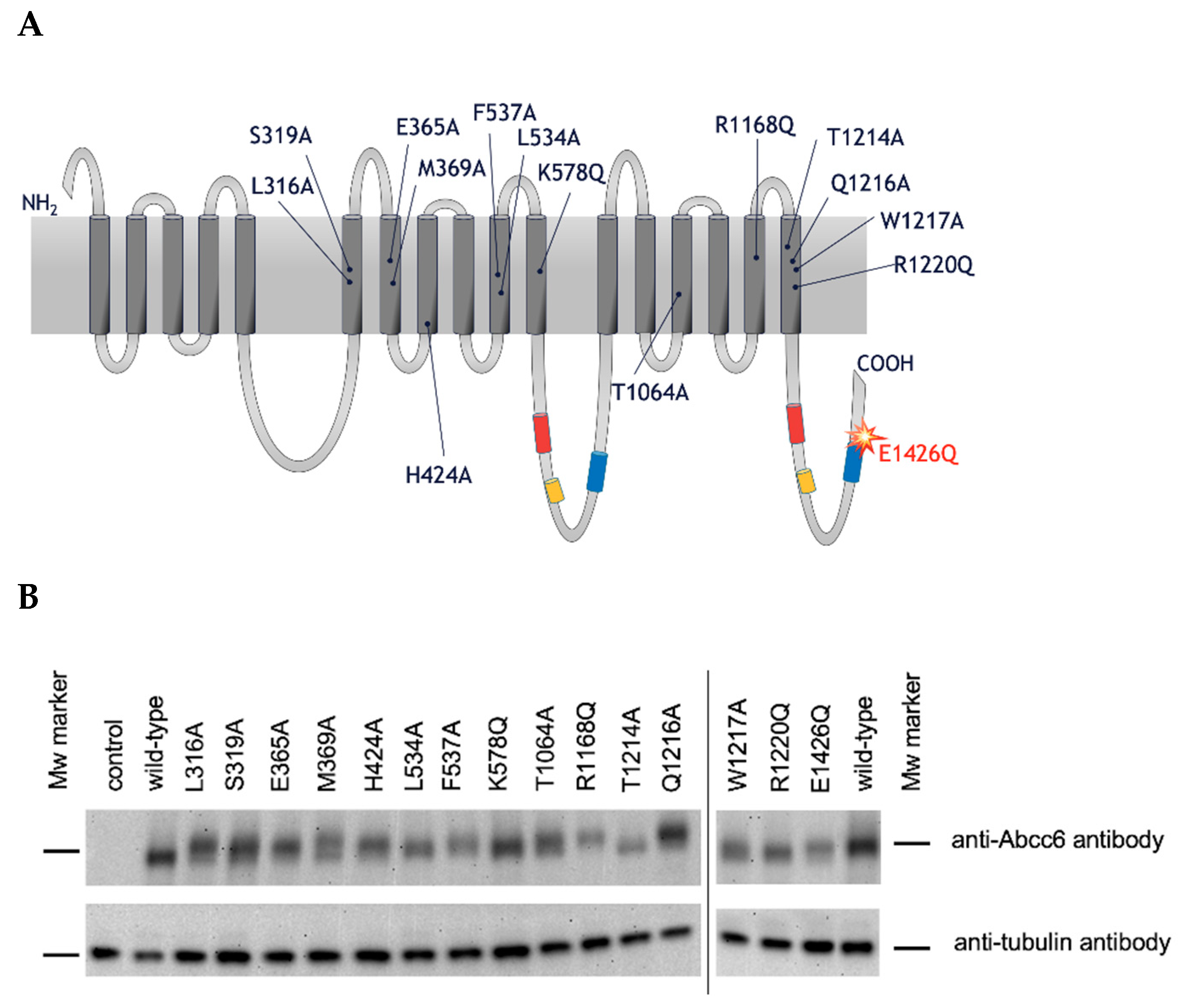
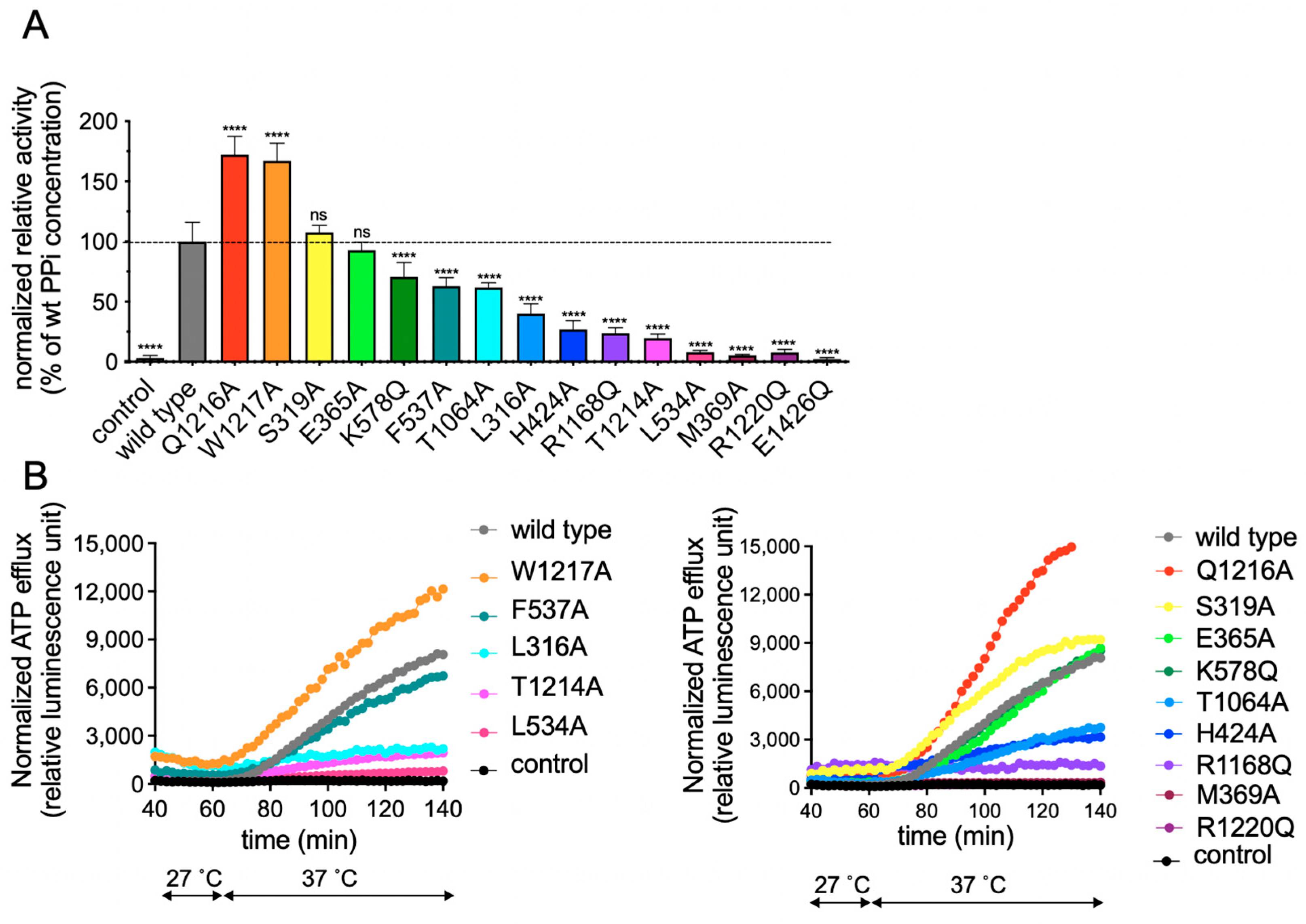
2.2. Functional Analysis of Single Amino Acid rAbcc6 Mutants
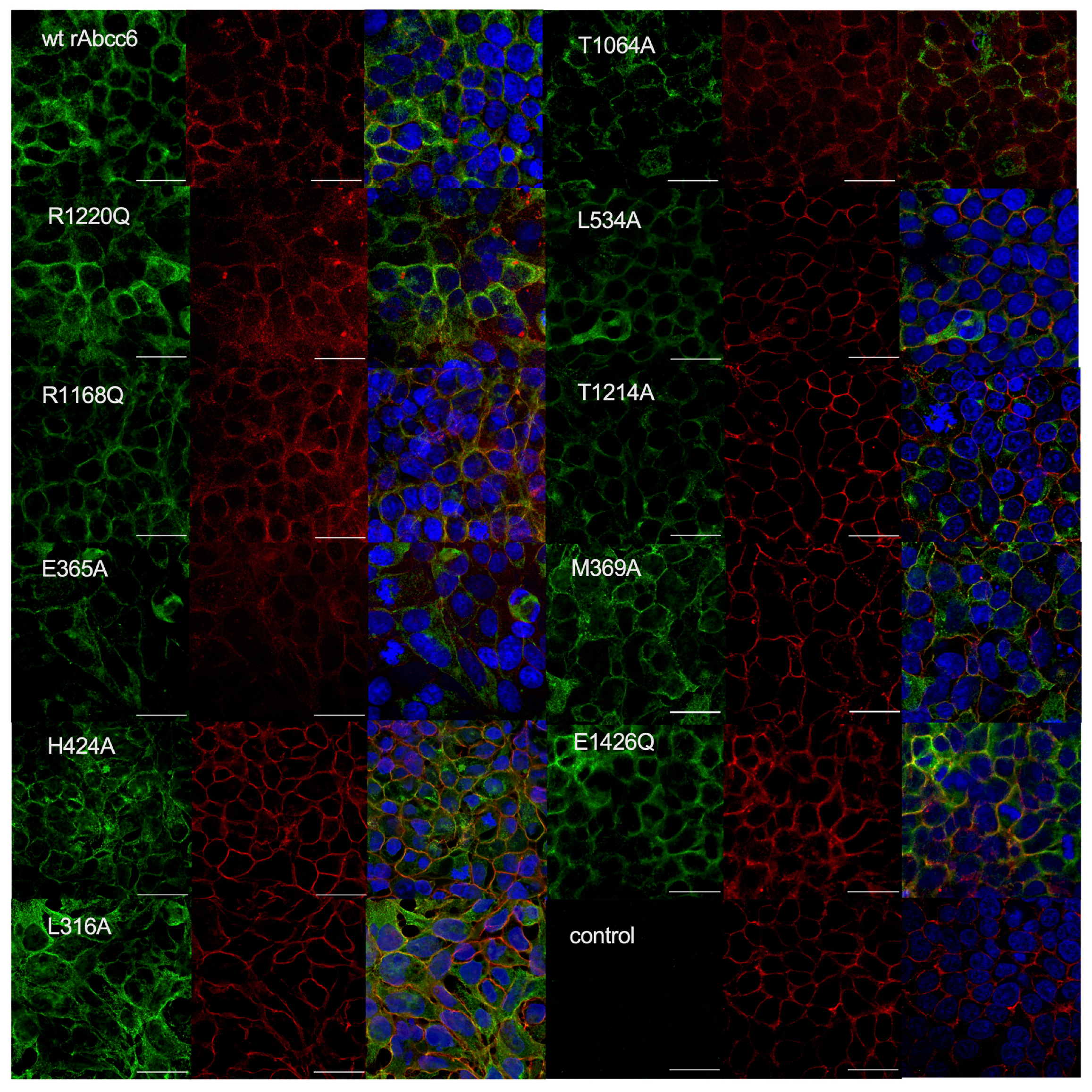
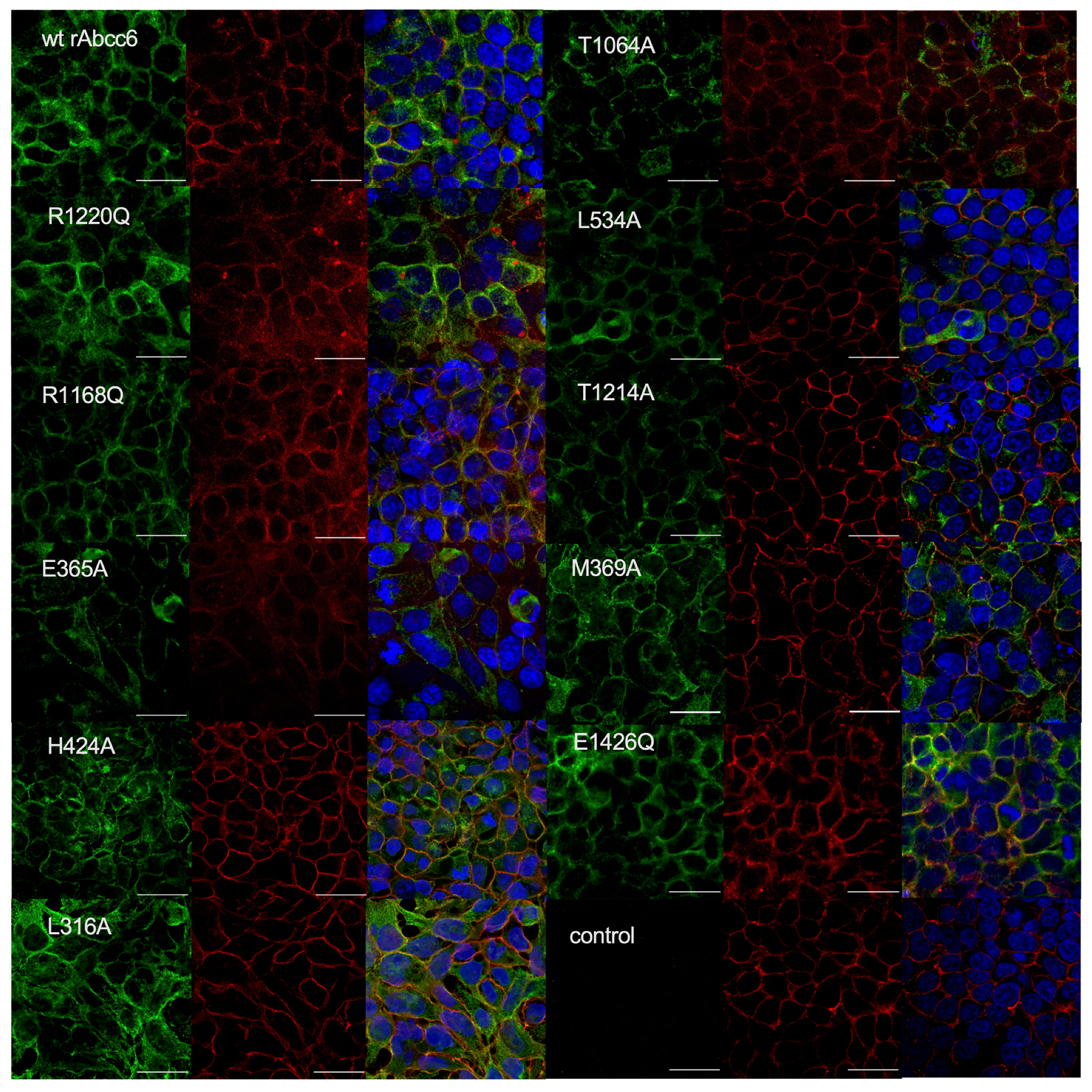
2.3. Subcellular Localization of the Single Amino Acid rAbcc6 Mutants That Showed Reduced ATP Efflux Activity
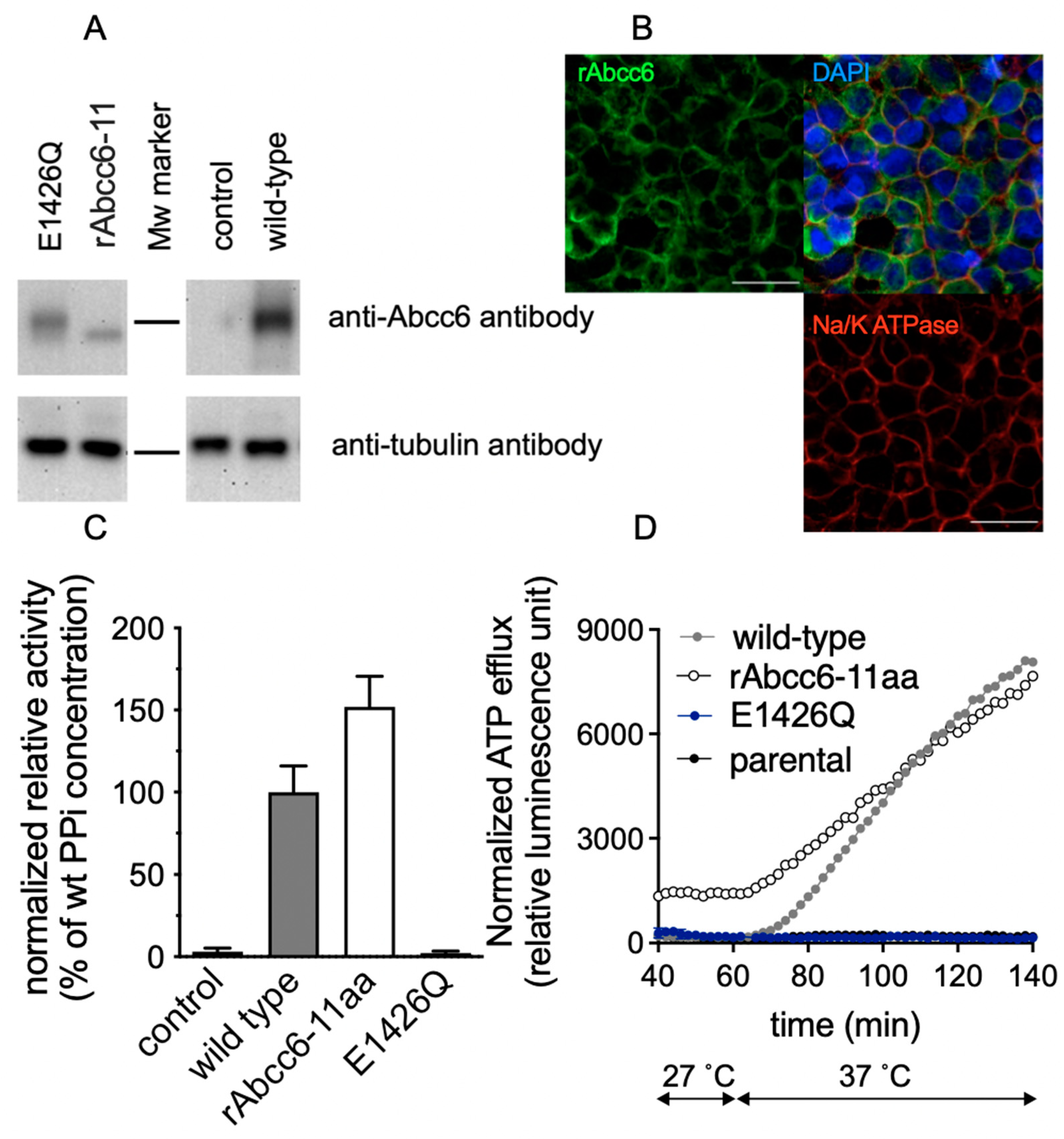
2.4. Functional Consequences of Changing All 11 Amino Acids of the Modeled rAbcc6 Substrate-Binding Site into Those That Comprise the bAbcc1 LTC4 Binding Site
3. Discussion
4. Materials and Methods
4.1. Model Building
4.2. Mutagenesis
4.3. Cell Culture and Generation of Mutant Cell Lines
4.4. Immunoblot Analysis of Wild-Type and Mutant rAbcc6
4.5. Subcellular Localization of rAbcc6 in HEK293 Cells
4.6. Quantification of PPi Levels in Medium Samples
4.7. Real-Time ATP Efflux Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.F.; Breuning, M.H.; Dauwerse, H.G.; Swart, J.; Kool, M.; Van Soest, S.; Baas, F.; et al. Mutations in ABCC6 Cause Pseudoxanthoma Elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.; et al. Mutations in a Gene Encoding an ABC Transporter Cause Pseudoxanthoma Elasticum. Nat. Genet 2000, 25, 223–227. [Google Scholar] [CrossRef]
- Ringpfeil, F.; Lebwohl, M.G.; Christiano, A.M.; Uitto, J. Pseudoxanthoma Elasticum: Mutations in the MRP6 Gene Encoding a Transmembrane ATP-Binding Cassette (ABC) Transporter. Proc. Natl. Acad. Sci. USA 2000, 97, 6001–6006. [Google Scholar] [CrossRef] [Green Version]
- Verschuere, S.; Van Gils, M.; Nollet, L.; Vanakker, O.M. From membrane to mineralization: The curious case of the ABCC6 transporter. FEBS Lett. 2020, 594, 4109–4133. [Google Scholar] [CrossRef]
- Favre, G.; Laurain, A.; Aranyi, T.; Szeri, F.; Fulop, K.; Le Saux, O.; Duranton, C.; Kauffenstein, G.; Martin, L.; Lefthériotis, G. The ABCC6 Transporter: A New Player in Biomineralization. Int. J. Mol. Sci. 2017, 18, 1941. [Google Scholar] [CrossRef]
- Borst, P.; Váradi, A.; van de Wetering, K. PXE, a Mysterious Inborn Error Clarified. Trends Biochem. Sci. 2019, 44, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, G.; Baas, A.F.; De Jong, P.A.; Asselbergs, F.W.; Visseren, F.L.J.; Spiering, W. The prevalence of pseudoxanthoma elasticum: Revised estimations based on genotyping in a high vascular risk cohort. Eur. J. Med. Genet. 2019, 62, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, Q.; Cao, Y.; Uitto, J. Therapeutics Development for Pseudoxanthoma Elasticum and Related Ectopic Mineralization Disorders: Update 2020. J. Clin. Med. 2020, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Pomozi, V.; Le Saux, O.; Brampton, C.; Apana, A.; Iliás, A.; Szeri, F.; Martin, L.; Monostory, K.; Paku, S.; Sarkadi, B.; et al. ABCC6 is a basolateral plasma membrane protein. Circ. Res. 2013, 112, e148–e151. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.S.; Kucukosmanoglu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.M.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Váradi, A.; Plomp, A.S.; Bergen, A.A.B.; Elferink, R.P.J.O.; Borst, P.; et al. ABCC6–mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation—Brief report. Arter. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef] [Green Version]
- Orriss, I.R. Extracellular pyrophosphate: The body’s water softener. Bone 2020, 134, 115243. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Sethuraman, V.; Navasiolava, N.; Makela, B.; Olomu, I.; Long, R.; Van De Wetering, K.; Martin, L.; Aranyi, T.; Szeri, F. Plasma inorganic pyrophosphate deficiency links multiparity to cardiovascular disease risk. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Szeri, F.; Lundkvist, S.; Donnelly, S.; Engelke, U.F.H.; Rhee, K.; Williams, C.J.; Sundberg, J.P.; Wevers, R.A.; Tomlinson, R.E.; Jansen, R.S.; et al. The membrane protein ANKH is crucial for bone mechanical performance by mediating cellular export of citrate and ATP. PLoS Genet. 2020, 16, e1008884. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.J.; Salm, E.; Tomita, S. ABC transporters control ATP release through cholesterol-dependent volume-regulated anion channel activity. J. Biol. Chem. 2020, 295, 5192–5203. [Google Scholar] [CrossRef] [Green Version]
- Gadsby, D.C.; Vergani, P.; Csanády, L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nat. Cell Biol. 2006, 440, 477–483. [Google Scholar] [CrossRef]
- Bryan, J.; Muñoz, A.; Zhang, X.; Düfer, M.; Drews, G.; Krippeit-Drews, P.; Aguilar-Bryan, L. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflügers Arch. Eur. J. Physiol. 2006, 453, 703–718. [Google Scholar] [CrossRef] [Green Version]
- van de Wetering, K.; Zelcer, N.; Kuil, A.; Feddema, W.; Hillebrand, M.; Vlaming, M.L.; Schinkel, A.H.; Beijnen, J.H.; Borst, P. Multidrug resistance proteins 2 and 3 provide alternative routes for hepatic excretion of morphine-glucuronides. Mole. Pharmacol. 2007, 72, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Szeri, F.; Niaziorimi, F.; Donnelly, S.; Orndorff, J.; Wetering, K. Generation of fully functional fluorescent fusion proteins to gain insights into ABCC6 biology. FEBS Lett. 2021, 595, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Ilias, A.; Urban, Z.; Seidl, T.L.; Le Saux, O.; Sinko, E.; Boyd, C.D.; Sarkadi, B.; Varadi, A. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J. Biol. Chem. 2002, 277, 16860–16867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belinsky, M.G.; Chen, Z.-S.; Shchaveleva, I.; Zeng, H.; Kruh, G.D. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res. 2002, 62, 6172–6177. [Google Scholar]
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Johnson, Z.L.; Chen, J. ATP Binding Enables Substrate Release from Multidrug Resistance Protein 1. Cell 2018, 172, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. The multidrug resistance protein family. Biochim. et Biophys. Acta BBA Biomembr. 1999, 1461, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Symmons, O.; Váradi, A.; Arányi, T. How Segmental Duplications Shape Our Genome: Recent Evolution of ABCC6 and PKD1 Mendelian Disease Genes. Mol. Biol. Evol. 2008, 25, 2601–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S. Multidrug resistance protein 1 (MRP1, ABCC1), a "multitasking" ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leier, I.; Jedlitschky, G.; Buchholz, U.; Cole, S.P.; Deeley, R.G.; Keppler, D. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J. Biol. Chem. 1994, 269, 27807–27810. [Google Scholar] [CrossRef]
- Conseil, G.; Arama-Chayoth, M.; Tsfadia, Y.; Cole, S.P.C. Structure-guided probing of the leukotriene C4binding site in human multidrug resistance protein 1 (MRP1; ABCC1). FASEB J. 2019, 33, 10692–10704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeno, K.; Nakajima, A.; Conseil, G.; Rothnie, A.; Deeley, R.G.; Cole, S.P.C. Molecular basis for reduced estrone sulfate transport and altered modulator sensitivity of transmembrane helix (TM) 6 and TM17 mutants of multidrug resistance protein 1 (ABCC1). Drug Metab. Dispos. 2009, 37, 1411–1420. [Google Scholar] [CrossRef]
- Loe, D.W.; Almquist, K.C.; Deeley, R.G.; Cole, S.P.C. Multidrug resistance protein (MRP)-Mediated transport of leukotriene C4 and chemotherapeutic agents in membrane vesicles. J. Biol. Chem. 1996, 271, 9675–9682. [Google Scholar] [CrossRef] [Green Version]
- Haimeur, A.; Deeley, R.G.; Cole, S.P.C. Charged amino acids in the sixth transmembrane helix of multidrug resistance protein 1 (MRP1/ABCC1) are critical determinants of transport activity. J. Biol. Chem. 2002, 277, 41326–41333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Situ, D.; Haimeur, A.; Conseil, G.; Sparks, K.E.; Zhang, D.; Deeley, R.G.; Cole, S.P.C. Mutational analysis of ionizable residues Proximal to the cytoplasmic interface of membrane spanning domain 3 of the multidrug resistance protein, MRP1 (ABCC1). J. Biol. Chem. 2004, 279, 38871–38880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.-Q.; Furukawa, T.; Aoki, S.; Sumizawa, T.; Haraguchi, M.; Nakajima, Y.; Ikeda, R.; Kobayashi, M.; Akiyama, S.-I. A Positively charged amino acid proximal to the C-Terminus of TM17 of MRP1 is indispensable for GSH-Dependent binding of substrates and for transport of LTC4†. Biochemistry 2002, 41, 14132–14140. [Google Scholar] [CrossRef]
- Voss, N.R.; Gerstein, M. 3V: Cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010, 38, W555–W562. [Google Scholar] [CrossRef] [Green Version]
- Madon, J.; Hagenbuch, B.; Landmann, L.; Meier, P.J.; Stieger, B. Transport function and hepatocellular localization of mrp6 in rat liver. Mol. Pharmacol. 2000, 57, 634–641. [Google Scholar] [CrossRef]
- Lone, A.M.; Taskén, K. Proinflammatory and immunoregulatory roles of eicosanoids in T cells. Front. Immunol. 2013, 4, 130. [Google Scholar] [CrossRef] [Green Version]
- Wielinga, P.R.; van der Heijden, I.; Reid, G.; Beijnen, J.H.; Wijnholds, J.; Borst, P. Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J. Biol. Chem. 2003, 278, 17664–17671. [Google Scholar] [CrossRef] [Green Version]
- Borst, P.; de Wolf, C.; van de Wetering, K. Multidrug resistance-associated proteins 3, 4, and 5. Pflügers Arch. Eur. J. Physiol. 2007, 453, 661–673. [Google Scholar] [CrossRef]
- Bäck, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceição, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement From the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2019, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Uitto, J.; Li, Q.; Jiang, Q. Pseudoxanthoma Elasticum: Molecular Genetics and Putative Pathomechanisms. J. Investig. Dermatol. 2010, 130, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Hosen, M.J.; Zubaer, A.; Thapa, S.; Khadka, B.; De Paepe, A.; Vanakker, O.M. Molecular Docking Simulations Provide Insights in the Substrate Binding Sites and Possible Substrates of the ABCC6 Transporter. PLoS ONE 2014, 9, e102779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fülöp, K.; Barna, L.; Symmons, O.; Závodszky, P.; Váradi, A. Clustering of disease-causing mutations on the domain–domain interfaces of ABCC6. Biochem. Biophys. Res. Commun. 2009, 379, 706–709. [Google Scholar] [CrossRef]
- Ito, K.-I.; Olsen, S.L.; Qiu, W.; Deeley, R.G.; Cole, S.P. Mutation of a single conserved tryptophan in multidrug resistance protein 1 (MRP1/ABCC1) results in loss of drug resistance and selective loss of organic anion transport. J. Biol. Chem. 2001, 276, 15616–15624. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Nunoya, K.; Vasa, M.; Gu, H.-M.; Cole, S.P.C.; Deeley, R.G. Mutational analysis of polar amino acid residues within predicted transmembrane helices 10 and 16 of multidrug resistance protein 1 (ABCC1): Effect on substrate specificity. Drug Metab. Dispos. 2006, 34, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.W.; Cole, S.P.; Deeley, R.G. Determinants of the substrate specificity of multidrug resistance protein 1: Role of amino acid residues with hydrogen bonding potential in predicted transmembrane helix 17. J. Biol. Chem. 2002, 277, 20934–20941. [Google Scholar] [CrossRef] [Green Version]
- Miksch, S.; Lumsden, A.; Guenther, U.P.; Foernzler, D.; Christen-Zäch, S.; Daugherty, C.; Ramesar, R.K.S.; Lebwohl, M.; Hohl, D.; Neldner, K.H.; et al. Molecular genetics of pseudoxanthoma elasticum: Type and frequency of mutations inABCC6. Hum. Mutat. 2005, 26, 235–248. [Google Scholar] [CrossRef]
- Pfendner, E.G.; Vanakker, O.M.; Terry, P.F.; Vourthis, S.E.; McAndrew, P.; McClain, M.R.; Fratta, S.; Marais, A.-S.; Hariri, S.; Coucke, P.J.; et al. Mutation Detection in the ABCC6 Gene and Genotype Phenotype Analysis in a Large International Case Series Affected by Pseudoxanthoma Elasticum. J. Med. Genet. 2007, 44, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noji, Y.; Inazu, A.; Higashikata, T.; Nohara, A.; Kawashiri, M.-A.; Yu, W.; Todo, Y.; Nozue, T.; Uno, Y.; Hifumi, S.; et al. Identification of two novel missense mutations (p.R1221C and p.R1357W) in the ABCC6 (MRP6) gene in a japanese patient with pseudoxanthoma elasticum (PXE). Intern. Med. 2004, 43, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; Du Moulin, M.; Stella, J.; Le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.-F.; et al. Generalized Arterial Calcification of Infancy and Pseudoxanthoma Elasticum Can Be Caused by Mutations in Either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.; Kawabe, T.; Nada, S.; Yamaguchi, A. Identification of basic residues involved in drug export function of human multidrug resistance-associated protein 2. J. Biol. Chem. 2000, 275, 39617–39624. [Google Scholar] [CrossRef] [Green Version]
- El-Sheikh, A.; Heuvel, J.J.M.W.V.D.; Krieger, E.; Russel, F.; Koenderink, J.B. Functional role of arginine 375 in transmembrane helix 6 of multidrug resistance protein 4 (MRP4/ABCC4). Mol. Pharmacol. 2008, 74, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Slot, A.J.; Molinski, S.; Cole, S.P. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011, 50, 179–207. [Google Scholar] [CrossRef] [Green Version]
- Zollmann, T.; Moiset, G.; Tumulka, F.; Tampé, R.; Poolman, B.; Abele, R. Single liposome analysis of peptide translocation by the ABC transporter TAPL. Proc. Natl. Acad. Sci. USA 2015, 112, 2046–2051. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic. Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Sali, A. Comparative protein modeling by satisfaction of spatial restraints. Mol. Med. Today 1995, 1, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corradi, V.; Vergani, P.; Tieleman, D. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR): Closed and open state channel models. J. Biol. Chem. 2015, 290, 22891–22906. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.; Martin, D.; Clamp, M.; Barton, G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, L. The PyMOL Molecular Graphics System. Version 2.5. Available online: https://www.schrodinger.com/products/pymol (accessed on 7 May 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| bAbcc1 | hABCC1 | hABCC6 | rAbcc6 | hABCC5 | TM Helix |
|---|---|---|---|---|---|
| K332 | K332 | L318 | L316 | L186 | TMD1, TM6 |
| H335 | H335 | S321 | S319 | T189 | TMD1, TM6 |
| L381 | L381 | E367 | E365 | L236 | TMD1, TM7 |
| F385 | F385 | M371 | M369 | W240 | TMD1, TM7 |
| Y440 | Y440 | Y426 | H424 | V293 | TMD1, TM8 |
| T550 | T550 | L536 | L534 | V403 | TMD1, TM10 |
| W553 | W553 | F539 | F537 | A406 | TMD1, TM10 |
| F594 | F594 | K579 | K578 | Q432 | TMD1, TM11 |
| M1092 | M1093 | S1065 | T1064 | M992 | TMD2, TM14 |
| R1196 | R1197 | R1169 | R1168 | R1096 | TMD2, TM16 |
| Y1242 | Y1243 | T1215 | T1214 | L1142 | TMD2, TM17 |
| N1244 | N1245 | Q1217 | Q1216 | Q1144 | TMD2, TM17 |
| W1245 | W1246 | W1218 | W1217 | F1145 | TMD2, TM17 |
| R1248 | R1249 | R1221 | R1220 | R1148 | TMD2, TM17 |
| Construct | Mutation | Forward Primer | Reverse Primer |
|---|---|---|---|
| rAbcc6-L316A | L316A | AGCGCCGUCATTAGCGATGCCTTCAGGTTTG | ACGGCGCUGAGGGTCCCCAGCAGGAAA |
| rAbcc6-S319A | S319A | ATTGCCGAUGCCTTCAGGTTTGCTGTT | ATCGGCAAUGACCAGGCTGAGGGTCCC |
| rAbcc6-E365A | E365A | ACTGTTUGCCCAGCAGTACATGTACAGA | AAACAGUGTCTGTAGGCAGGCCGACAA |
| rAbcc6-M369A | M369A | AGTACGCCUACAGAGTCAAGGTCCTGCAGATG | AGGCGTACUGCTGTTCAAACAGTGTCTG |
| rAbcc6-H424A | H424A | ATCCTCGCCCUCAACGGGCTGTGGCTGCTCTT | AGGGCGAGGAUGCTCTCGACCAGCCGCTG |
| rAbcc6-L534A | L534A | AAGTGTCUACATTTCTGGTGGCGCTGGTTGT | AGACACTUGGAAGGACACGGCAGACACAGAGAAGAGGAAG |
| rAbcc6-F537A | F537A | AAGTGTCUACATTTCTGGTGGCGCTGGTTGT | AGACACTUGGGCGGACACGAGAGACACAGA |
| rAbcc6-K578Q | K578Q | AGCCAGGCCUTCCTCCCCTTCTCTGTGC | AGGCCTGGGCUTGGTTAAGGATG |
| rAbcc6-T1064A | T1064A | AGGGCCCUGCTGACCTATGCCTTTGG | AGGGCCCUCATCTTGTCTGGGATGTCCACAT |
| rAbcc6-R1168Q | R1168Q | ACCAGTGGCUGGCTGCCAACCTGGAGCT | AGCCACTGGUCAGCCACCAGCCTCGGGA |
| rAbcc6-T1214A | T1214A | AGGCTCUGCAGTGGGTGGTCCGCAGCTG | AGAGCCUGTGTTACCTGGAGGGCAGCAGAAACCG |
| rAbcc6-Q1216A | Q1216A | ACTCTGGCCUGGGTGGTCCGCAGCTGGAC | AGGCCAGAGUCTGTGTTACCTGGAGGGC |
| rAbcc6-W1217A | W1217A | AGGCCGUGGTCCGCAGCTGGACAGATC | ACGGCCUGCAGAGTCTGTGTTACCT |
| rAbcc6-R1220Q | R1220Q | AGTGGGTGGUCCAATCTGGAGAACAG | ACCACCCACUGCAGTCTGTGTTACCT |
| rAbcc6-11AA | L316K & S319H | AGGTCATUCACGATGCCTTCAGGTTTGCTGTTCCCAAGC | AATGACCUTGCTGAGGGTCCCCAGCAGGAAAGT |
| E365L & M369F | AGCAGTACUTCTACAGAGTCAAGGTCCTGCAGATGAGGCTG | AGTACTGCUGCAGAAACAGTGTCTGTAGGCAGGCCGACAAG | |
| H424Y | ATCCTCUACCTCAACGGGCTGTGGCTGC | AGAGGAUGCTCTCGACCAGCCGCTG | |
| L534T | ACCGTGUCCTGGCAAGTGTCTACATTTCTGGTGGC | ACACGGUAGACACAGAGAAGAGGAAGGCGGAGGTCT | |
| F537W | AAGTGTCUACATTTCTGGTGGCGCTGGTTG | AGACACTUGCCAGGACACGAGAGACACAGAGAAGAGGAAGGC | |
| K578F | ATCCTTAACUTCGCCCAGGCCTTCCTCCCCTTC | AGTTAAGGAUGCTGAGCACCGTGAGCGT | |
| T1064M | AGGATGCUGCTGACCTATGCCTTTGGACTCCTGG | AGCATCCUCATCTTGTCTGGGATGTCCACATCCAC | |
| T1214Y | AGTATCUGAACTGGGTGGTCCGCAGCTGG | AGATACUGTGTTACCTGGAGGGCAGCAGAAACCG | |
| Q1216N | AACTGGGUGGTCCGCAGCTGGACAGATC | ACCCAGTUCAGAGTCTGTGTTACCTGGAGGGCAGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szeri, F.; Corradi, V.; Niaziorimi, F.; Donnelly, S.; Conseil, G.; Cole, S.P.C.; Tieleman, D.P.; van de Wetering, K. Mutagenic Analysis of the Putative ABCC6 Substrate-Binding Cavity Using a New Homology Model. Int. J. Mol. Sci. 2021, 22, 6910. https://doi.org/10.3390/ijms22136910
Szeri F, Corradi V, Niaziorimi F, Donnelly S, Conseil G, Cole SPC, Tieleman DP, van de Wetering K. Mutagenic Analysis of the Putative ABCC6 Substrate-Binding Cavity Using a New Homology Model. International Journal of Molecular Sciences. 2021; 22(13):6910. https://doi.org/10.3390/ijms22136910
Chicago/Turabian StyleSzeri, Flora, Valentina Corradi, Fatemeh Niaziorimi, Sylvia Donnelly, Gwenaëlle Conseil, Susan P. C. Cole, D. Peter Tieleman, and Koen van de Wetering. 2021. "Mutagenic Analysis of the Putative ABCC6 Substrate-Binding Cavity Using a New Homology Model" International Journal of Molecular Sciences 22, no. 13: 6910. https://doi.org/10.3390/ijms22136910
APA StyleSzeri, F., Corradi, V., Niaziorimi, F., Donnelly, S., Conseil, G., Cole, S. P. C., Tieleman, D. P., & van de Wetering, K. (2021). Mutagenic Analysis of the Putative ABCC6 Substrate-Binding Cavity Using a New Homology Model. International Journal of Molecular Sciences, 22(13), 6910. https://doi.org/10.3390/ijms22136910