
The Role of ABCG2 in the Pathogenesis of Primary Hyperuricemia and Gout—An Update


Abstract
:1. Introduction
2. Gout and Hyperuricemia
3. ABCG2 and Its Function in Renal Urate Elimination
4. Relevance of ABCG2 in Extrarenal Urate Elimination
5. ABCG2 Polymorphisms in Pediatric-Onset Hyperuricemia and Early-Onset Gout
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bardin, T.; Richette, P. Definition of hyperuricemia and gouty conditions. Curr. Opin. Rheumatol. 2014, 26, 186–191. [Google Scholar] [CrossRef]
- Masseoud, D.; Rott, K.; Liu-Bryan, R.; Agudelo, C. Overview of Hyperuricaemia and Gout. Curr. Pharm. Des. 2005, 11, 4117–4124. [Google Scholar] [CrossRef] [PubMed]
- Grassi, W.; De Angelis, R. Clinical features of gout. Reumatismo 2012, 63, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Kc, M.; Leslie, S.W. Uric Acid Nephrolithiasis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lanaspa, M.A.; Andres-Hernando, A.; Kuwabara, M. Uric acid and hypertension. Hypertens. Res. 2020, 43, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, M.; Niwa, K.; Hisatome, I.; Nakagawa, T.; Roncal-Jimenez, C.A.; Andres-Hernando, A.; Bjornstad, P.; Jensen, T.; Sato, Y.; Milagres, T.; et al. Asymptomatic Hyperuricemia Without Comorbidities Predicts Cardiometabolic Diseases: Five-Year Japanese Cohort Study. Hypertension 2017, 69, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, H.; Tian, W.; Shi, L.; Chen, L.; Li, J.; Zhao, S.; Qi, G. Association between serum uric acid levels and coronary artery disease in different age and gender: A cross-sectional study. Aging Clin. Exp. Res. 2019, 31, 1783–1790. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Hsu, H.-L.; Huang, Y.-C.; Lee, M.; Huang, W.-Y.; Huang, Y.-C.; Lee, T.-H.; Lee, J.-D. Gouty Arthritis in Acute Cerebrovascular Disease. Cerebrovasc. Dis. 2009, 28, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Thottam, G.E.; Krasnokutsky, S.; Pillinger, M.H. Gout and Metabolic Syndrome: A Tangled Web. Curr. Rheumatol. Rep. 2017, 19, 60. [Google Scholar] [CrossRef]
- Ejaz, A.A.; Nakagawa, T.; Kanbay, M.; Kuwabara, M.; Kumar, A.; Arroyo, F.E.G.; Roncal-Jimenez, C.; Sasai, F.; Kang, D.-H.; Jensen, T.; et al. Hyperuricemia in Kidney Disease: A Major Risk Factor for Cardiovascular Events, Vascular Calcification, and Renal Damage. Semin. Nephrol. 2020, 40, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Rodriguez-Iturbe, B.; Kelley, E.E.; Nakagawa, T.; Madero, M.; Feig, D.I.; Borghi, C.; Piani, F.; Cara-Fuentes, G.; Bjornstad, P.; et al. Uric Acid and Hypertension: An Update With Recommendations. Am. J. Hypertens. 2020, 33, 583–594. [Google Scholar] [CrossRef]
- Johnson, R.J.; Bakris, G.L.; Borghi, C.; Chonchol, M.B.; Feldman, D.; Lanaspa, M.A.; Merriman, T.R.; Moe, O.W.; Mount, D.B.; Lozada, L.G.S.; et al. Hyperuricemia, Acute and Chronic Kidney Disease, Hypertension, and Cardiovascular Disease: Report of a Scientific Workshop Organized by the National Kidney Foundation. Am. J. Kidney Dis. 2018, 71, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ge, J.-Y.; Zhang, Y.-Y.; Wang, F.-F.; Ji, Y.; Li, H.-Y. The relationship between elevated serum uric acid and arterial stiffness in a healthy population. Vascular 2020, 28, 494–501. [Google Scholar] [CrossRef]
- Soriano, L.C.; Rothenbacher, D.; Choi, H.K.; Rodríguez, L.A.G. Contemporary epidemiology of gout in the UK general population. Arthritis Res. Ther. 2011, 13, R39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Fang, W.; Zeng, X.; Zhang, Y.; Ma, Y.; Sheng, F.; Zhang, X. Clinical characteristics of early- and late-onset gout: A cross-sectional observational study from a Chinese gout clinic. Medicine 2016, 95, e5425. [Google Scholar] [CrossRef]
- Pascart, T.; Norberciak, L.; Ea, H.; Guggenbuhl, P.; Lioté, F. Patients With Early-Onset Gout and Development of Earlier Severe Joint Involvement and Metabolic Comorbid Conditions: Results From a Cross-Sectional Epidemiologic Survey. Arthritis Rheum. 2019, 71, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.C. Gout—An update of aetiology, genetics, co-morbidities and management. Maturitas 2018, 118, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Church, T.S.; Meriwether, R.A.; Lobelo, F.; Blair, S.N. Uric acid and the development of metabolic syndrome in women and men. Metabolism 2008, 57, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estiverne, C.; Mandal, A.K.; Mount, D.B. Molecular Pathophysiology of Uric Acid Homeostasis. Semin. Nephrol. 2020, 40, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.K.; Atkinson, K.; Karlson, E.W.; Willett, W.; Curhan, G. Purine-Rich Foods, Dairy and Protein Intake, and the Risk of Gout in Men. N. Engl. J. Med. 2004, 350, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.-H.; Mahmoud, H.H.; Wiley, J.M.; Woods, G.M.; Leverger, G.; Camitta, B.; Hastings, C.; Blaney, S.M.; Relling, M.V.; Reaman, G.H. Recombinant Urate Oxidase for the Prophylaxis or Treatment of Hyperuricemia in Patients with Leukemia or Lymphoma. J. Clin. Oncol. 2001, 19, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.S.; Kaplow, R. Tumor Lysis Syndrome: Implications for Oncology Nursing Practice. Semin. Oncol. Nurs. 2021, 37, 151136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, L.; Zhang, Y.; Zeng, C. Recent advances in fructose intake and risk of hyperuricemia. Biomed. Pharmacother. 2020, 131, 110795. [Google Scholar] [CrossRef]
- Sayehmiri, K.; Ahmadi, I.; Anvari, E. Fructose Feeding and Hyperuricemia: A Systematic Review and Meta-Analysis. Clin. Nutr. Res. 2020, 9, 122–133. [Google Scholar] [CrossRef]
- Hernández-Rubio, A.; Sanvisens, A.; Bolao, F.; Pérez-Mañá, C.; García-Marchena, N.; Fernández-Prendes, C.; Muñoz, A.; Muga, R. Association of hyperuricemia and gamma glutamyl transferase as a marker of metabolic risk in alcohol use disorder. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Choi, H.K.; McCormick, N.; Lu, N.; Rai, S.K.; Yokose, C.; Zhang, Y. Population Impact Attributable to Modifiable Risk Factors for Hyperuricemia. Arthritis Rheumatol. 2020, 72, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef]
- Nanagiri, A.; Shabbir, N. Lesch Nyhan Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Krishnan, E.; Lessov-Schlaggar, C.N.; Krasnow, R.E.; Swan, G.E. Nature Versus Nurture in Gout: A Twin Study. Am. J. Med. 2012, 125, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Langlois, V.; Noone, D. Hyperuricemia and Hypertension: Links and Risks. Integr. Blood Press. Control 2019, 12, 43–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, O.M. ABCG2: The molecular mechanisms of urate secretion and gout. Am. J. Physiol. Physiol. 2015, 309, F485–F488. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate–anion exchanger that regulates blood urate levels. Nat. Cell Biol. 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.A.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bhatnagar, V. The systems biology of uric acid transporters: The role of remote sensing and signaling. Curr. Opin. Nephrol. Hypertens. 2018, 27, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef]
- Bobulescu, I.A.; Moe, O.W. Renal Transport of Uric Acid: Evolving Concepts and Uncertainties. Adv. Chronic Kidney Dis. 2012, 19, 358–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, V.F.; Kos, I.A.; Vargas-Santos, A.B.; Pinheiro, G.D.R.C.; Paiva, E.D.S. Benzbromarone in the treatment of gout. Adv. Rheumatol. 2019, 59, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoque, K.M.; Dixon, E.E.; Lewis, R.M.; Allan, J.; Gamble, G.D.; Phipps-Green, A.J.; Kuhns, V.L.H.; Horne, A.M.; Stamp, L.K.; Merriman, T.R.; et al. The ABCG2 Q141K hyperuricemia and gout associated variant illuminates the physiology of human urate excretion. Nat. Commun. 2020, 11, 2767. [Google Scholar] [CrossRef]
- DeBosch, B.J.; Kluth, O.; Fujiwara, H.; Schurmann, A.; Moley, K.H. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat. Commun. 2014, 5, 4642. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, H.; Hruz, P.; Zimmermann, C.; Beglinger, C.; Drewe, J. Distribution of breast cancer resistance protein (BCRP/ABCG2) mRNA expression along the human GI tract. Biochem. Pharmacol. 2005, 70, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lin, H.; Niu, Y.; Liu, Y.; Wang, Z.; Song, L.; Gao, L.; Li, L. Mangiferin promotes intestinal elimination of uric acid by modulating intestinal transporters. Eur. J. Pharmacol. 2020, 888, 173490. [Google Scholar] [CrossRef]
- Chen, M.; Ye, C.; Zhu, J.; Zhang, P.; Jiang, Y.; Lu, X.; Wu, H. Bergenin as a Novel Urate-Lowering Therapeutic Strategy for Hyperuricemia. Front. Cell Dev. Biol. 2020, 8, 703. [Google Scholar] [CrossRef]
- Chen, X.; Ge, H.-Z.; Lei, S.-S.; Jiang, Z.-T.; Su, J.; He, X.; Zheng, X.; Wang, H.-Y.; Yu, Q.-X.; Li, B.; et al. Dendrobium officinalis six nostrum ameliorates urate under-excretion and protects renal dysfunction in lipid emulsion-induced hyperuricemic rats. Biomed. Pharmacother. 2020, 132, 110765. [Google Scholar] [CrossRef] [PubMed]
- Ristic, B.; Sikder, M.O.F.; Bhutia, Y.D.; Ganapathy, V. Pharmacologic inducers of the uric acid exporter ABCG2 as potential drugs for treatment of gouty arthritis. Asian J. Pharm. Sci. 2020, 15, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-H.; Chang, Y.-P.; Li, T.; Han, F.; Li, C.-J.; Li, X.-Y.; Xue, M.; Cheng, Y.; Meng, Z.-Y.; Han, Z.; et al. Empagliflozin Attenuates Hyperuricemia by Upregulation of ABCG2 via AMPK/AKT/CREB Signaling Pathway in Type 2 Diabetic Mice. Int. J. Biol. Sci. 2020, 16, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, A.; Köttgen, A.; Yang, Q.; Hwang, S.-J.; Kao, W.L.; Rivadeneira, F.; Boerwinkle, E.; Levy, D.; Hofman, A.; Astor, B.C.; et al. Association of three genetic loci with uric acid concentration and risk of gout: A genome-wide association study. Lancet 2008, 372, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Richette, P.; Bardin, T. Gout. Lancet 2010, 375, 318–328. [Google Scholar] [CrossRef]
- Woodward, O.M.; Köttgen, M.; Coresh, J.; Boerwinkle, E.; Guggino, W.B. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosomi, A.; Nakanishi, T.; Fujita, T.; Tamai, I. Extra-Renal Elimination of Uric Acid via Intestinal Efflux Transporter BCRP/ABCG2. PLoS ONE 2012, 7, e30456. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S.; et al. Common Defects of ABCG2, a High-Capacity Urate Exporter, Cause Gout: A Function-Based Genetic Analysis in a Japanese Population. Sci. Transl. Med. 2009, 1, 5ra11. [Google Scholar] [CrossRef]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef]
- Mo, W.; Zhang, J.-T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Boil. 2011, 3, 1–27. [Google Scholar]
- Polgar, O.; Robey, R.W.; Bates, S.E. ABCG2: Structure, function and role in drug response. Expert Opin. Drug Metab. Toxicol. 2007, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q. BCRP/ABCG2 in the placenta: Expression, function and regulation. Pharm. Res. 2008, 25, 1244–1255. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Mojsilovic-Petrovic, J.; Andrade, M.F.; Zhang, H.; Ball, M.; Stanimirovic, D.B. Expression and functional characterization of ABCG2 in brain endothelial cells and vessels. FASEB J. 2003, 17, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; Van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; Van De Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar] [PubMed]
- Huls, M.; Brown, C.; Windass, A.; Sayer, R.; Heuvel, J.V.D.; Heemskerk, S.; Russel, F.; Masereeuw, R.; Huls, M.; Brown, C.; et al. The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 2008, 73, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Sarkadi, B.; Ozvegy-Laczka, C.; Német, K.; Váradi, A. ABCG2—A transporter for all seasons. FEBS Lett. 2004, 567, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.; Ross, D.D. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003, 22, 7340–7358. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Polgar, O.; Deeken, J.; To, K.; Bates, S.E. ABCG2: Determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007, 26, 39–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Singh, R.R.; Cho-Vega, J.H.; Drakos, E.; Davuluri, Y.; Khokhar, F.A.; Fayad, L.; Medeiros, L.J.; Vega, F. Sonic hedgehog signaling proteins and ATP-binding cassette G2 are aberrantly expressed in diffuse large B-Cell lymphoma. Mod. Pathol. 2009, 22, 1312–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiani, D.; Tiribelli, M.; Calistri, E.; Geromin, A.; Chiarvesio, A.; Michelutti, A.; Cavallin, M.; Fanin, R. The prognostic value of P-glycoprotein (ABCB) and breast cancer resistance protein (ABCG2) in adults with de novo acute myeloid leukemia with normal karyotype. Haematologica 2006, 91, 825–828. [Google Scholar] [PubMed]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nat. Cell Biol. 2017, 546, 504–509. [Google Scholar] [CrossRef]
- Kowal, J.; Ni, D.; Jackson, S.M.; Manolaridis, I.; Stahlberg, H.; Locher, K.P. Structural Basis of Drug Recognition by the Multidrug Transporter ABCG2. J. Mol. Biol. 2021, 433, 166980. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nat. Cell Biol. 2018, 563, 426–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckenstaler, R.; Benndorf, R.A. 3D structure of the transporter ABCG2-What’s new? Br. J. Pharmacol. 2020, 177, 1485–1496. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, P.; Horsey, A.J.; Cox, M.H.; Kerr, I.D. ABCG2: Does resolving its structure elucidate the mechanism? Biochem. Soc. Trans. 2018, 46, 1485–1494. [Google Scholar] [CrossRef]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Guo, S.; Yang, Y.; Wu, J.; Guan, M.; Zou, H.; Jin, L.; Wang, J. Association between ABCG2 Q141K polymorphism and gout risk affected by ethnicity and gender: A systematic review and meta-analysis. Int. J. Rheum. Dis. 2015, 18, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Kannangara, D.; Phipps-Green, A.J.; Dalbeth, N.; Stamp, L.K.; Williams, K.M.; Graham, G.G.; Day, R.O.; Merriman, T.R. Hyperuricaemia: Contributions of urate transporter ABCG2 and the fractional renal clearance of urate. Ann. Rheum. Dis. 2015, 75, 1363–1366. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Miao, L.; Qin, L.; Xiang, Y.; Zhang, X.; Peng, H.; Mailamuguli; Sun, Y.; Yao, H. A meta-analysis of the associations between the Q141K and Q126X ABCG2 gene variants and gout risk. Int. J. Clin. Exp. Pathol. 2015, 8, 9812–9823. [Google Scholar] [PubMed]
- Deppe, S.; Ripperger, A.; Weiss, J.; Ergün, S.; Benndorf, R.A. Impact of genetic variability in the ABCG2 gene on ABCG2 expression, function, and interaction with AT1 receptor antagonist telmisartan. Biochem. Biophys. Res. Commun. 2014, 443, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, A.; Benndorf, R.A. The C421A (Q141K) polymorphism enhances the 3′-untranslated region (3′-UTR)-dependent regulation of ATP-binding cassette transporter ABCG2. Biochem. Pharmacol. 2016, 104, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Tukaye, D.N.; Cui, J.; Greenwell, P.; Constantoulakis, L.M.; Parker, B.S.; Rao, A.; Köttgen, M.; Maloney, P.C.; Guggino, W.B. Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, 5223–5228. [Google Scholar] [CrossRef] [Green Version]
- László, L.; Sarkadi, B.; Hegedűs, T. Jump into a New Fold—A Homology Based Model for the ABCG2/BCRP Multidrug Transporter. PLoS ONE 2016, 11, e0164426. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Wakabayashi, K.; Tamura, A.; Nakagawa, H.; Morishima, Y.; Osawa, Y.; Ishikawa, T. Major SNP (Q141K) Variant of Human ABC Transporter ABCG2 Undergoes Lysosomal and Proteasomal Degradations. Pharm. Res. 2008, 26, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Bartos, Z.; Homolya, L. Identification of Specific Trafficking Defects of Naturally Occurring Variants of the Human ABCG2 Transporter. Front. Cell Dev. Biol. 2021, 9, 615729. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Nakayama, A.; Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Ikebuchi, Y.; Ito, K.; Hosoya, T.; Kanai, Y.; Suzuki, H.; et al. ABCG2 is a High-Capacity Urate Transporter and its Genetic Impairment Increases Serum Uric Acid Levels in Humans. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1091–1097. [Google Scholar] [CrossRef]
- Varela-Echavarría, A.; Oca-Luna, R.M.; Barrera-Saldaña, H.A. Uricase protein sequences: Conserved during vertebrate evolution but absent in humans. FASEB J. 1988, 2, 3092–3096. [Google Scholar] [CrossRef]
- Morimoto, C.; Tamura, Y.; Asakawa, S.; Kuribayashi-Okuma, E.; Nemoto, Y.; Li, J.; Murase, T.; Nakamura, T.; Hosoyamada, M.; Uchida, S.; et al. ABCG2 expression and uric acid metabolism of the intestine in hyperuricemia model rat. Nucleosides Nucleotides Nucleic Acids 2020, 39, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Takada, T.; Nakayama, A.; Shimizu, T.; Sakiyama, M.; Shimizu, S.; Chiba, T.; Nakashima, H.; Nakamura, T.; Takada, Y.; et al. ABCG2 Dysfunction Increases the Risk of Renal Overload Hyperuricemia. Nucleosides Nucleotides Nucleic Acids 2014, 33, 266–274. [Google Scholar] [CrossRef]
- Matsuo, H.; Nakayama, A.; Sakiyama, M.; Chiba, T.; Shimizu, S.; Kawamura, Y.; Nakashima, H.; Nakamura, T.; Takada, Y.; Oikawa, Y.; et al. ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci. Rep. 2014, 4, 3755. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Marten, J.; Halperin Kuhns, V.L.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; Yu, Z.; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukkunaprasit, T.; Rattanasiri, S.; Turongkaravee, S.; Suvannang, N.; Ingsathit, A.; Attia, J.; Thakkinstian, A. The association between genetic polymorphisms in ABCG2 and SLC2A9 and urate: An updated systematic review and meta-analysis. BMC Med. Genet. 2020, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Pavelcová, K.; Bohatá, J.; Ješina, P.; Kubota, Y.; Suzuki, H.; Takada, T.; Stiburkova, B. Identification of Two Dysfunctional Variants in the ABCG2 Urate Transporter Associated with Pediatric-Onset of Familial Hyperuricemia and Early-onset Gout. Int. J. Mol. Sci. 2021, 22, 1935. [Google Scholar] [CrossRef] [PubMed]
- Stiburkova, B.; Pavelcova, K.; Pavlikova, M.; Ješina, P.; Pavelka, K. The impact of dysfunctional variants of ABCG2 on hyperuricemia and gout in pediatric-onset patients. Arthritis Res. 2019, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, F.; Narang, R.K.; Phipps-Green, A.; Gamble, G.G.; Tausche, A.-K.; So, A.; Riches, P.; Andres, M.; Perez-Ruiz, F.; Doherty, M.; et al. Systematic genetic analysis of early-onset gout: ABCG2 is the only associated locus. Rheumatology 2020, 59, 2544–2549. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Ichida, K.; Takada, T.; Nakayama, A.; Nakashima, H.; Nakamura, T.; Kawamura, Y.; Takada, Y.; Yamamoto, K.; Inoue, H.; et al. Common dysfunctional variants in ABCG2 are a major cause of early-onset gout. Sci. Rep. 2013, 3, srep02014. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, Y.; Pavelcová, K.; Klein, M.; Suzuki, H.; Takada, T.; Stiburkova, B. Familial early-onset hyperuricemia and gout associated with a newly identified dysfunctional variant in urate transporter ABCG2. Arthritis Res. 2019, 21, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Gulley, M.L.; Dunnen, J.T.D.; Wilson, R.B. Standard Mutation Nomenclature in Molecular Diagnostics: Practical and Educational Challenges. J. Mol. Diagn. 2007, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Homolya, L. Medically Important Alterations in Transport Function and Trafficking of ABCG2. Int. J. Mol. Sci. 2021, 22, 2786. [Google Scholar] [CrossRef] [PubMed]
- Zámbó, B.; Mózner, O.; Bartos, Z.; Török, G.; Várady, G.; Telbisz, Á.; Homolya, L.; Orbán, T.I.; Sarkadi, B. Cellular expression and function of naturally occurring variants of the human ABCG2 multidrug transporter. Cell. Mol. Life Sci. 2020, 77, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Heyes, N.; Kapoor, P.; Kerr, I.D. Polymorphisms of the Multidrug Pump ABCG2: A Systematic Review of Their Effect on Protein Expression, Function, and Drug Pharmacokinetics. Drug Metab. Dispos. 2018, 46, 1886–1899. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Ichida, K.; Ohkido, I.; Yokoyama, K.; Matsuo, H.; Ohashi, Y.; Takada, T.; Nakayama, A.; Suzuki, H.; Shinomiya, N.; et al. Dysfunctional ABCG2 gene polymorphisms are associated with serum uric acid levels and all-cause mortality in hemodialysis patients. Hum. Cell 2020, 33, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Sjöstedt, N.; Heuvel, J.J.; Koenderink, J.B.; Kidron, H. Transmembrane Domain Single-Nucleotide Polymorphisms Impair Expression and Transport Activity of ABC Transporter ABCG2. Pharm. Res. 2017, 34, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, Y.; Mančíková, A.; Krylov, V.; Morimoto, K.; Pavelcová, K.; Bohatá, J.; Pavelka, K.; Pavlikova, M.; Suzuki, H.; Matsuo, H.; et al. Functional Characterization of Clinically-Relevant Rare Variants in ABCG2 Identified in a Gout and Hyperuricemia Cohort. Cells 2019, 8, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Liu, G.; Liu, Z.; Gong-Lu, L.; Wu, Z. Identification and functional characterization of two missense mutations in NDRG1 associated with Charcot-Marie-Tooth disease type 4D. Hum. Mutat. 2017, 38, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Gabrikova, D.; Mistrik, M.; Bernasovska, J.; Bozikova, A.; Behulova, R.; Tothova, I.; Macekova, S. Founder mutations in NDRG1 and HK1 genes are common causes of inherited neuropathies among Roma/Gypsies in Slovakia. J. Appl. Genet. 2013, 54, 455–460. [Google Scholar] [CrossRef]
- Lassuthova, P.; Sišková, D.; Haberlová, J.; Sakmaryová, I.; Filouš, A.; Seeman, P. Congenital cataract, facial dysmorphism and demyelinating neuropathy (CCFDN) in 10 Czech Gypsy children—Frequent and underestimated cause of disability among Czech Gypsies. Orphanet J. Rare. Dis. 2014, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Peña, H.; Coto, E.; Santos, F.; Espino, M.; Cea Crespo, J.M.; Chantzopoulos, G.; Komianou, F.; Gómez, J.; Alonso, B.; Iglesias, S.; et al. A new SLC12A3 founder mutation (p.Val647Met) in Gitelman’s syndrome patients of Roma ancestry. Nefrologia 2017, 37, 423–428. [Google Scholar] [CrossRef]
- Schulpis, K.H.; Thodi, G.; Iakovou, K.; Chatzidaki, M.; Dotsikas, Y.; Molou, E.; Triantafylli, O.; Loukas, Y.L. Clinical evaluation and mutational analysis of GALK and GALE genes in patients with galactosemia in Greece: One novel mutation and two rare cases. J. Pediatr. Endocrinol. Metab. 2017, 30, 775–779. [Google Scholar] [CrossRef]
- Butler, F.; Alghubayshi, A.; Roman, Y. The Epidemiology and Genetics of Hyperuricemia and Gout across Major Racial Groups: A Literature Review and Population Genetics Secondary Database Analysis. J. Pers. Med. 2021, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Piranavan, P.; Sundaresan, D.; Yood, R. Clinical Characteristics of Early-Onset Gout in Outpatient Setting. ACR Open Rheumatol. 2019, 1, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Singer, R.F.; Walters, G. Uric acid lowering therapies for preventing or delaying the progression of chronic kidney disease. Cochrane Database Syst. Rev. 2011, 10, 10. [Google Scholar] [CrossRef]
- Ying, H.; Yuan, H.; Tang, X.; Guo, W.; Jiang, R.; Jiang, C. Impact of Serum Uric Acid Lowering and Contemporary Uric Acid-Lowering Therapies on Cardiovascular Outcomes: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 641062. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.J.; Anoopkumar, K.; Krishnan, V. Asymptomatic hyperuricemia: Is it time to intervene? Clin. Rheumatol. 2017, 36, 2637–2644. [Google Scholar] [CrossRef]
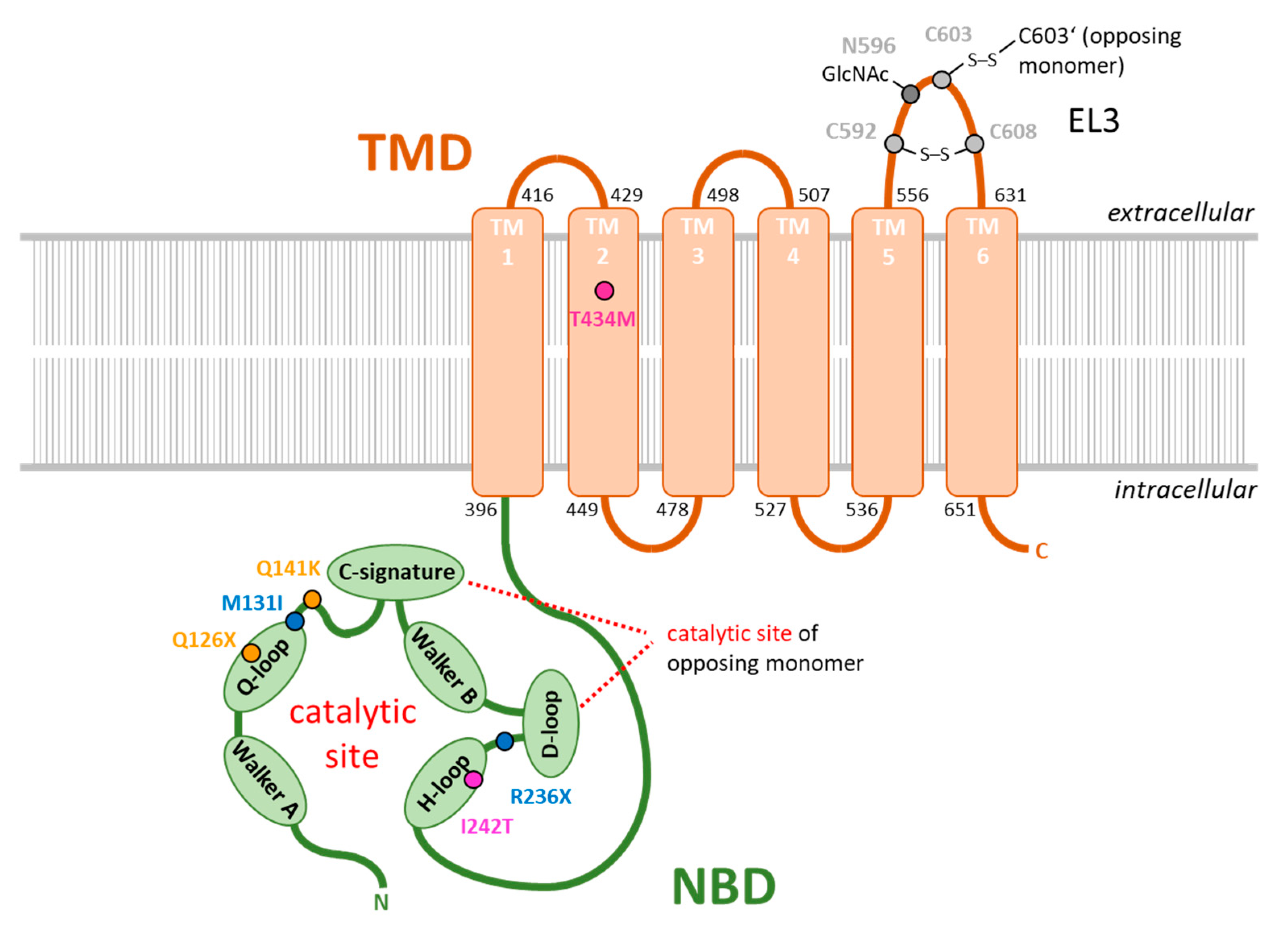

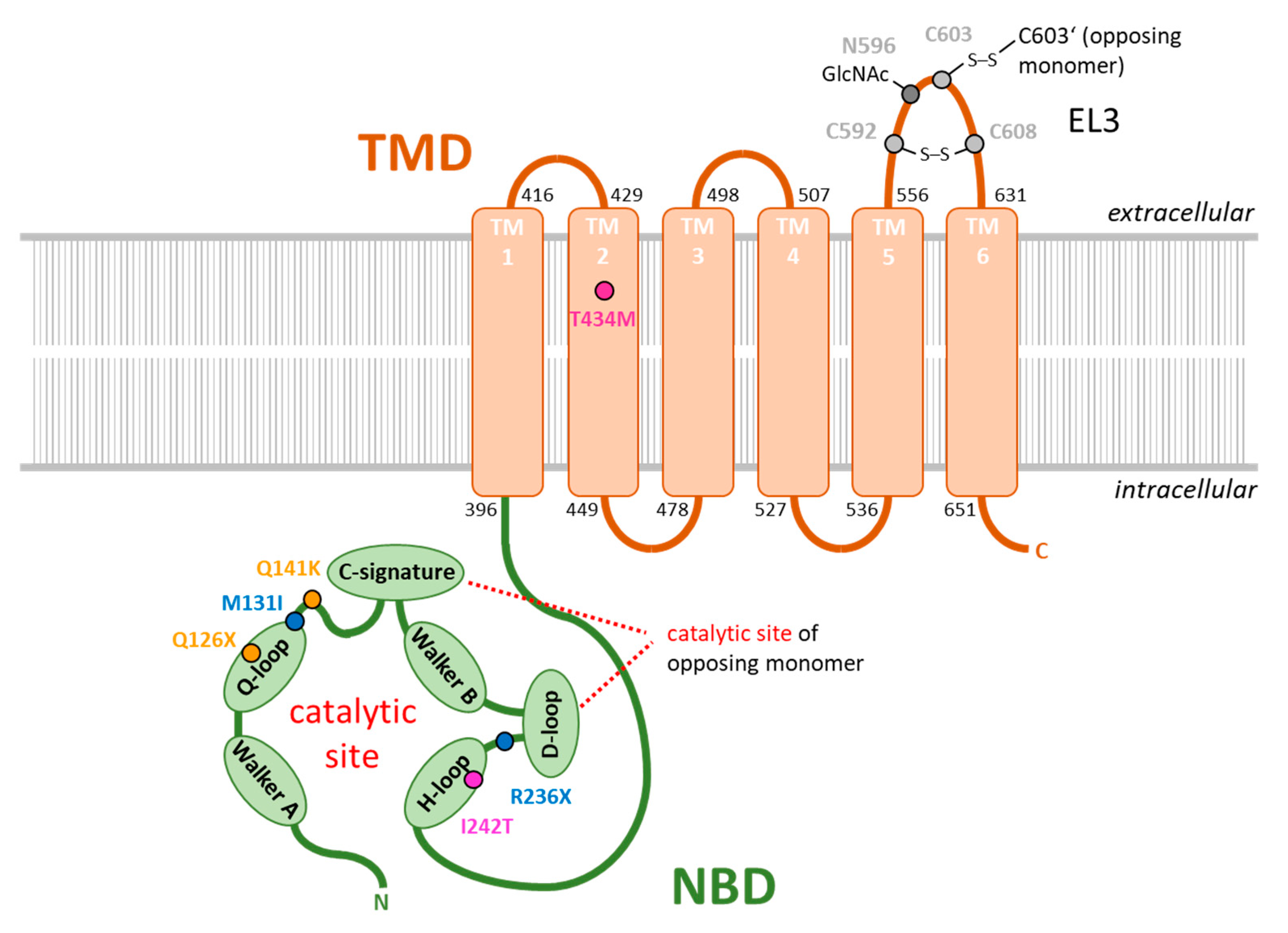{kind=link}
| rs ID | Coding Sequence | Protein Sequence | Citation |
|---|---|---|---|
| rs72552713 | c.376C > T | p.Q126X | [92] |
| rs759726272 | c.393G > T | p.M131I | [89] |
| rs2231142 | c.C421 > A | p.Q141K | [91,92] |
| rs140207606 | c.706C > T | p.R236X | [89] |
| not annotated | c.725T > C | p.I242T | [93] |
| rs769734146 | c.1301C > T | p.T434M | [90,100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckenstaler, R.; Benndorf, R.A. The Role of ABCG2 in the Pathogenesis of Primary Hyperuricemia and Gout—An Update. Int. J. Mol. Sci. 2021, 22, 6678. https://doi.org/10.3390/ijms22136678
Eckenstaler R, Benndorf RA. The Role of ABCG2 in the Pathogenesis of Primary Hyperuricemia and Gout—An Update. International Journal of Molecular Sciences. 2021; 22(13):6678. https://doi.org/10.3390/ijms22136678
Chicago/Turabian StyleEckenstaler, Robert, and Ralf A. Benndorf. 2021. "The Role of ABCG2 in the Pathogenesis of Primary Hyperuricemia and Gout—An Update" International Journal of Molecular Sciences 22, no. 13: 6678. https://doi.org/10.3390/ijms22136678
APA StyleEckenstaler, R., & Benndorf, R. A. (2021). The Role of ABCG2 in the Pathogenesis of Primary Hyperuricemia and Gout—An Update. International Journal of Molecular Sciences, 22(13), 6678. https://doi.org/10.3390/ijms22136678