
Regulation of Melatonin and Neurotransmission in Alzheimer’s Disease



Abstract
:1. Introduction
2. Major Pathologies Involved in AD
2.1. Role of Aβ Plaques and Neurofibrillary Tangles
2.2. Role of Aging
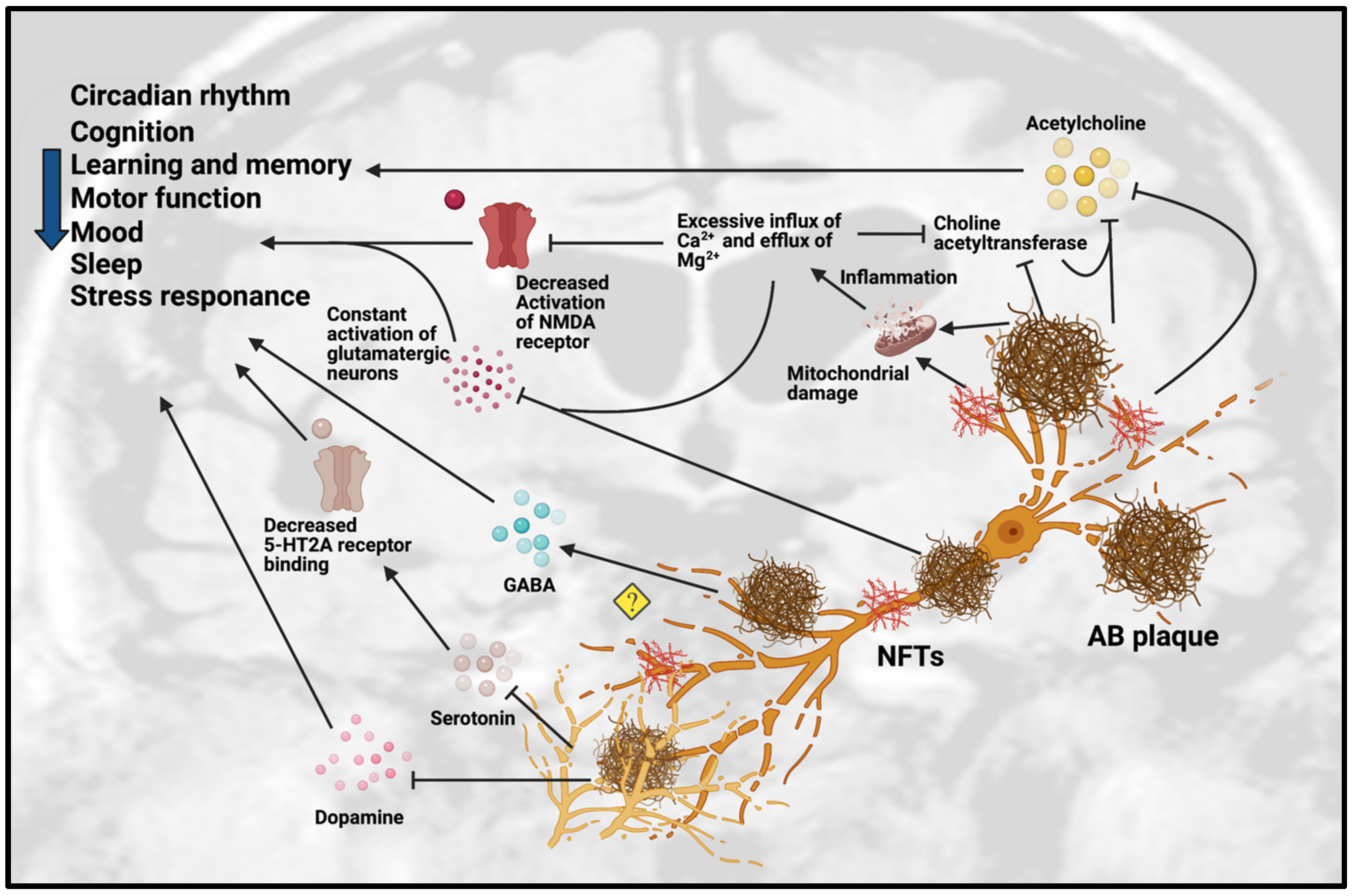
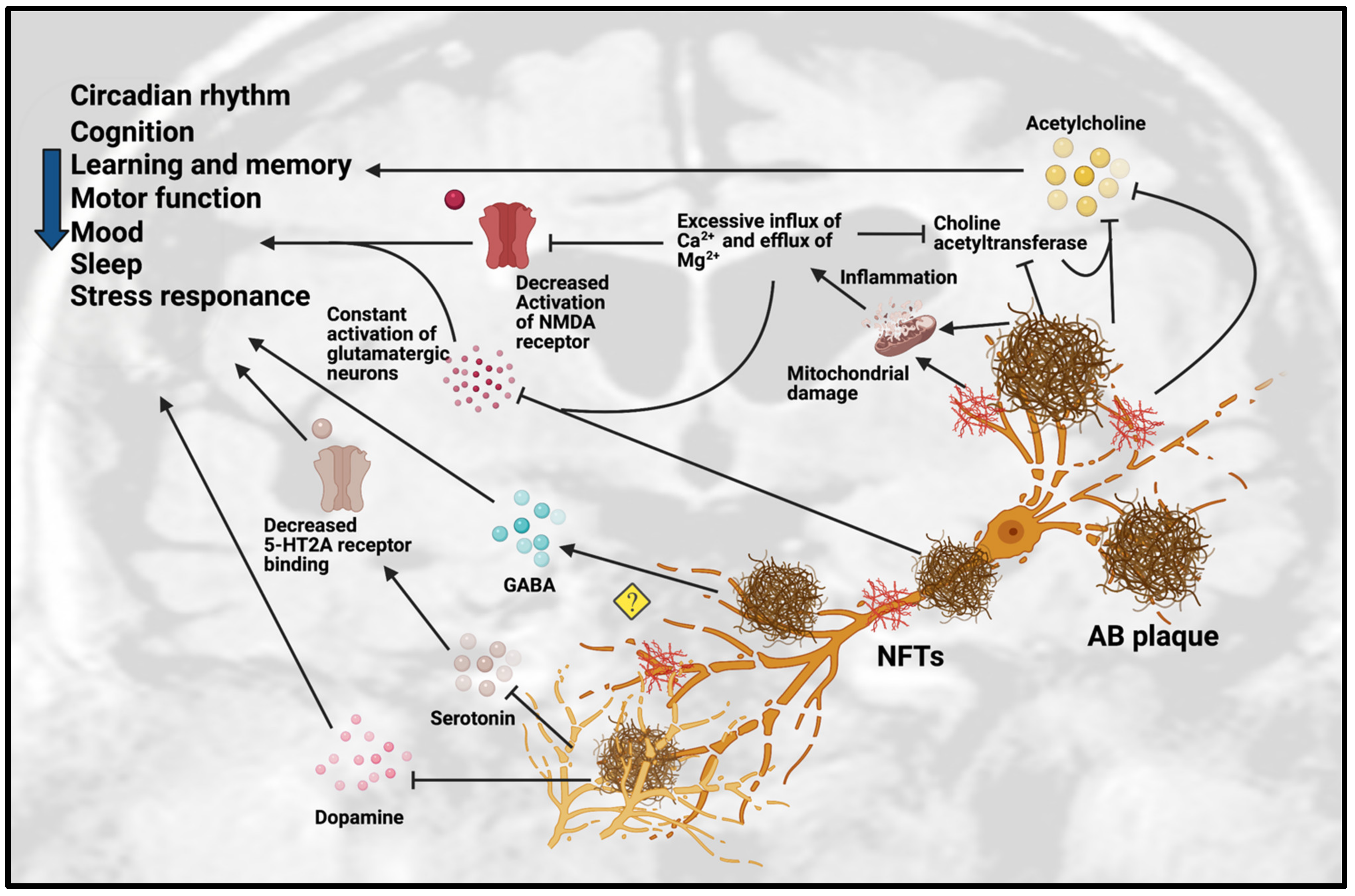
3. Dysfunction of Neurotransmission in AD
3.1. Dysfunction of Cholinergic Neurotransmission
3.2. Dysfunction of Monoaminergic Neurotransmission

{kind=link}
{kind=link}
| Animal Model. | Gender | Age | Pathology Involved | Neurotransmission Dysfunction | Behavioral Effects | References |
|---|---|---|---|---|---|---|
| APPswe/PS1dE9 mice | N/A | 4–18 months old | Degeneration and loss of forebrain 5-HT and NA axons after Aβ deposits | Monoaminergic neurodegeneration | Anxiety-related behaviors in 18 months | [57] |
| Swiss mice treated with AβO | N/A | 3 months old | Development of Aβ plaques | AβO disrupts 5-HT homeostasis | Depressive-like behavior | [58] |
| APPswe/PS1dE9 mice | Male | 4, 8, 11 months old | Progressive accumulation of Aβ protein. | Significant decrease in 5-HT2A receptor binding | Memory impairment | [59] |
| 5xFAD mice | Male | 6 months old | Significant decrease of both TH+ and TH- cells in DA-producing areas | SN-VTA networks are enhanced to the synchronization of neuronal firing activity in DA-producing nuclei |
| [65] |
| Tg2576 mice | Male | 2 and 6 months old | Degeneration of VTA DAergic neurons | Reduced noradrenergic transmission in dorsal subiculum | Age-related impairment of memory and non-cognitive functions | [66] |
| Tg2576 mice | N/A | 4–6 and 9–11 months old | Aβ were prominent in 20-month-old mice | Reduced ACh release from hippocampus in 9- to 11-month-old mice | Memory impairment present in 9- to 11-month-old mice | [30] |
| APP/PS1 mice | N/A | 3 and 7 months old | Aβ plaques deposition after cholinergic degeneration |
|
| [34] |
| APP/PS1 and 5xFAD mice | N/A | 8 and 13 months old | Aβ plaques deposition and reactive astrocytes | Aberrant increase in GABA release from reactive astrocytes | Impaired learning and memory | [36] |
| AβPP/PS mice | Male | 2–4 months old | Abnormal glutamate release precedes cognitive decline | Significantly increased potassium-evoked glutamate release in CA1 | Cognitive decline | [51] |
| AβPPswe-PS1dE9 mice | N/A | 6 months old | Deposition of Aβ plaques |
| Impairment of cognitive function and memory | [37] |
| TgAPP23 mice | Male and female | 24 months old | Deposition of Aβ plaques and cholinergic degeneration |
| N/A | [26] |
| PS2APP mice | Female | 20 or 24 months old | Deposition of Aβ plaques | Significant reduction of glutamate level in frontal cortex | N/A | [52] |
| TgAPP23 mice | N/A | 7–8 months old | Dysfunction of cholinergic and monoaminergic systems |
| N/A | [27] |
| PDAPP mice | Male and female | 4–6 months old | Deposition of Aβ plaques | Reduced basal and evoked ACh release from hippocampus | Hyper-locomotor function | [33] |
| 3xTg-AD mice | Male and female | 2–4, 13–15 and 18–20 months old | Aβ plaques deposition with cholinergic degeneration and alteration of neurotrophic factors |
| N/A | [28] |
| hAPP-J20 mice | N/A | 6 months old | Altered synaptic plasticity and cognitive function | Significantly decreased phospho GluN2B levels and hippocampal LTP | Impaired learning and memory | [42] |
| TgCRND8 mice | N/A | 2 and 7 months old | Aβ plaques deposition, oxidative stress, reactive glial cells and neurodegeneration | Reduced ChAT-positive neurons and ACh levels. | Cognitive impairment | [31] |
| PS2APP mice | Male | 5, 9, 13 and 17 months old | Deposition of Aβ plaques | Significant loss of mGlu2 receptors in entorhinal cortex and lacunosum moleculare regions | N/A | [53] |
| PS2APP mice | Male | 3–4 months old | Altered synaptic plasticity | Aberrant GluN2B-NMDAR function | N/A | [54] |
| PDAPP mice | Male | 2, 4, 12 and 24 months old | Aβ plaques deposition with cholinergic degeneration |
| N/A | [32] |
| 3xTg-AD mice | N/A | 9–23 months old | Deposition of Aβ plaques | Reduced ChAT and AChE-positive neurons | N/A | [29] |
| TgCRND8 mice | Male | 3 months old | Deposition of Aβ plaques and neuronal degeneration |
| Cognitive impairment | [55] |
| TgCRND8 mice | Male and female | 2–3 and 12–13 months old | Deposition of Aβ plaques |
| N/A | [38] |
| TgCRND8 mice | Male | 3 months old | Dysfunction of dopaminergic system |
| Cognitive impairment | [64] |
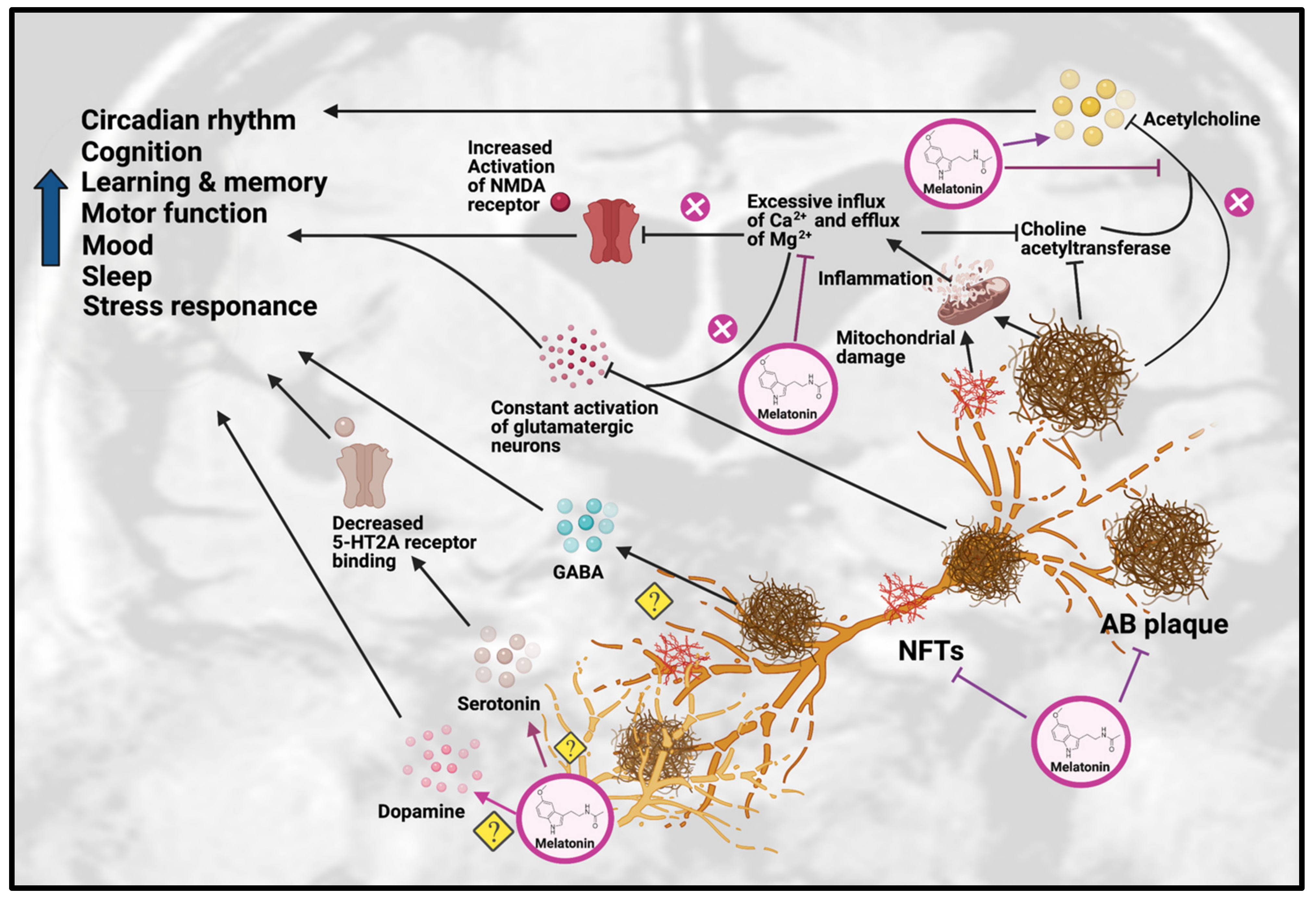
4. Role of Melatonin against AD Hallmarks
4.1. Anti-Amyloidogenic Effects
4.2. Inhibition of Tau Hyperphosphorylation
| Study Model | Effects on AD Pathology | References |
|---|---|---|
| Multiple cell types (SK-N-SH, SHSY5Y, U-138, SV770, C6, PC12, N1E-115) | Decrease in soluble APP secretion in PC12, SV770, U-138, HeLa, N1E-115 | [1] |
| N2a neuroblastoma cell | Protective effects against tau hyperphosphorylation induced by wortmannin | [77] |
| N2a neuroblastoma cell | Protective effects against tau hyperphosphorylation induced by calyculin-A | [76] |
| Animal Model | Gender | Age | Treatment Dosage and Duration | AD Pathology Involved | Effects on AD Pathology | Effects on Neurotransmission | References |
|---|---|---|---|---|---|---|---|
| Tg2576 mice | N/A | 8, 9.5, 11, 12.5 months old | 0.5 mg/mL, 4, 5.5, 7, 8.5 months | Plaque-like deposits of amyloid-beta |
| N/A | [72] |
| ICR mice treated with Aβ1-42 | Male | N/A | 10 mg/kg, 5 mg/kg, 2.5 mg/kg, 14 days | Affected cognitive functions |
| Improved neuron viability | [79] |
| AβPP/PS mice | N/A | 4 months old | 100 µg/mL,0.5 mg/day | Amyloid plaques, behavioral deficits |
| N/A | [44] |
| APP/PS1 mice | N/A | 2–2.5 months old | 100 mg/L | Aβ plaques |
| N/A | [71] |
| 3xTg-AD mice | Male | 6 months old | 10 mg/kg body weight | AβO, hyper-phosphorylated tau |
| N/A | [78] |
| SD rats treated with LPS | N/A | N/A | 5 and 10 mg/kg | Inflammation, oxidation, increased AChE activity | Lowered levels of induced inflammation and oxidation | Inhibited increase in AchE activity | [13] |
| APP695 mice | N/A | 4 months old | 10 mg/kg/day | Aβ plaques, decreased ChAT levels | Long-term treatment significantly reduced Aβ plaque levels | Increased ChAT activity in frontal cortex and hippocampus | [81] |
| Swiss mice treated with AlCl3 and d-galactose | Male | N/A | 80 mg/kg/day | Affected cognitive functions, decreased BDNF, CREB and AChE levels |
| Increased AChE level | [82] |
5. Role of Melatonin on Neurotransmission
6. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lahiri, D.K. Melatonin affects the metabolism of the β-amyloid precursor protein in different cell types. J. Pineal Res. 1999, 26, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International. World Alzheimer Report 2019; Alzheimer’s Disease International: London, UK, 2019; p. 13. [Google Scholar]
- Mayuri, S.; Piyarat, G.; Parichart, B.; Russel, J.R.; Jutamaad, S. Mechanisms of Melatonin in Alleviating Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 1010–1031. [Google Scholar]
- Bature, F.; Guinn, B.-A.; Pang, D.; Pappas, Y. Signs and symptoms preceding the diagnosis of Alzheimer’s disease: A systematic scoping review of literature from 1937 to 2016. BMJ Open 2017, 7, e015746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H. A Critical Assessment of Research on Neurotransmitters in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 969–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- McEnery, M.W.; Siegel, R.E. Neurotransmitter Receptors, 2nd ed.; Elsevier Inc.: Oxford, UK, 2014; pp. 552–564. [Google Scholar]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.Y.; Roy, J.; Fung, M.L.; Heng, B.C.; Zhang, C.; Lim, L.W. Relationships between mitochondrial dysfunction and neurotransmission failure in Alzheimer’s disease. Aging Dis. 2020, 11, 1291. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef] [Green Version]
- Pandiperumal, S.; Trakht, I.; Srinivasan, V.; Spence, D.; Maestroni, G.; Zisapel, N.; Cardinali, D. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef]
- Tyagi, E.; Agrawal, R.; Nath, C.; Shukla, R. Effect of melatonin on neuroinflammation and acetylcholinesterase activity induced by LPS in rat brain. Eur. J. Pharm. 2010, 640, 206–210. [Google Scholar] [CrossRef]
- Escames, G.; Macías, M.; León, J.; García, J.; Khaldy, H.; Martín, M.; Vives, F.; Acuña-Castroviejo, D. Calcium-Dependent Effects of Melatonin Inhibition of Glutamatergic Response in Rat Striatum. J. Neuroendocr. 2001, 13, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [Green Version]
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Rivera, R.M.C.; Simon, M.J. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. eLife 2018, 7, e40070. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta (BBA)-Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef]
- Herrup, K. Reimagining Alzheimer’s Disease--An Age-Based Hypothesis. J. Neurosci. 2010, 30, 16755–16762. [Google Scholar] [CrossRef] [Green Version]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Mufson, E.J.; Herrup, K. Neuronal Cell Death Is Preceded by Cell Cycle Events at All Stages of Alzheimer’s Disease. J. Neurosci. 2003, 23, 2557–2563. [Google Scholar] [CrossRef] [Green Version]
- Talita, H.F.-V.; Isabella, M.G.; Flavia, R.S.; Fabiola, M.R. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar]
- Perry, E.K.; Perry, R.H.; Blessed, G.; Tomlinson, B.E. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol. Appl. Neurobiol. 1978, 4, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Boncristiano, S.; Calhoun, M.E.; Kelly, P.H.; Pfeifer, M.; Bondolfi, L.; Stalder, M.; Phinney, A.L.; Abramowski, D.; Sturchler-Pierrat, C.; Enz, A. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J. Neurosci. 2002, 22, 3234–3243. [Google Scholar] [CrossRef] [Green Version]
- Van Dam, D.; Marescau, B.; Engelborghs, S.; Cremers, T.; Mulder, J.; Staufenbiel, M.; De Deyn, P.P. Analysis of cholinergic markers, biogenic amines, and amino acids in the CNS of two APP overexpression mouse models. Neurochem. Int. 2005, 46, 409–422. [Google Scholar] [CrossRef]
- Perez, S.E.; He, B.; Muhammad, N.; Oh, K.-J.; Fahnestock, M.; Ikonomovic, M.D.; Mufson, E.J. Cholinotrophic basal forebrain system alterations in 3xTg-AD transgenic mice. Neurobiol. Dis. 2011, 41, 338–352. [Google Scholar] [CrossRef] [Green Version]
- Robertson, R.T.; Baratta, J.; Yu, J.; LaFerla, F.M. Amyloid-β expression in retrosplenial cortex of triple transgenic mice: Relationship to cholinergic axonal afferents from medial septum. Neuroscience 2009, 164, 1334–1346. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Yamagata, N.; Takasaki, K.; Sano, K.; Hayakawa, K.; Katsurabayashi, S.; Egashira, N.; Mishima, K.; Iwasaki, K.; Fujiwara, M. Decreased acetylcholine release is correlated to memory impairment in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res. 2008, 1249, 222–228. [Google Scholar] [CrossRef]
- Bellucci, A.; Luccarini, I.; Scali, C.; Prosperi, C.; Giovannini, M.G.; Pepeu, G.; Casamenti, F. Cholinergic dysfunction, neuronal damage and axonal loss in TgCRND8 mice. Neurobiol. Dis. 2006, 23, 260–272. [Google Scholar] [CrossRef]
- German, D.C.; Yazdani, U.; Speciale, S.G.; Pasbakhsh, P.; Games, D.; Liang, C.L. Cholinergic neuropathology in a mouse model of Alzheimer’s disease. J. Comp. Neurol. 2003, 462, 371–381. [Google Scholar] [CrossRef]
- Bales, K.R.; Tzavara, E.T.; Wu, S.; Wade, M.R.; Bymaster, F.P.; Paul, S.M.; Nomikos, G.G. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-Aβ antibody. J. Clin. Investig. 2006, 116, 825–832. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-Amyloid Deposition and Cognitive Impairment After Cholinergic Denervation in APP/PS1 Mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285. [Google Scholar] [CrossRef]
- Govindpani, K.; Calvo-Flores Guzmán, B.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1813. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Tiwari, V.; Patel, A.B. Impaired glutamatergic and GABAergic function at early age in AβPPswe-PS1dE9 mice: Implications for Alzheimer’s disease. J. Alzheimers Dis. 2012, 28, 765–769. [Google Scholar] [CrossRef]
- Salek, R.M.; Xia, J.; Innes, A.; Sweatman, B.C.; Adalbert, R.; Randle, S.; McGowan, E.; Emson, P.C.; Griffin, J.L. A metabolomic study of the CRND8 transgenic mouse model of Alzheimer’s disease. Neurochem. Int. 2010, 56, 937–947. [Google Scholar] [CrossRef]
- Mandal, P.K.; Kansara, K.; Dabas, A. The GABA-Working Memory Relationship in Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2017, 1, 43–45. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Pocernich, C.B. The Glutamatergic System and Alzheimer’s Disease: Therapeutic Implications. CNS Drugs 2003, 17, 641–652. [Google Scholar] [CrossRef]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease: Potential targets for treatment. J. Clin. Psychiatry 2006, 67 (Suppl. 3), 3–7. [Google Scholar]
- Zhang, L.; Qin, Z.; Sharmin, F.; Lin, W.; Ricke, K.M.; Zasloff, M.; Stewart, A.F.; Chen, H.-H. Tyrosine phosphatase PTP1B impairs presynaptic NMDA receptor-mediated plasticity in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 156, 105402. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Ferreiro, E.; Schmidt, J.; Cardoso, J.M.; Pereira, C.M.; Carvalho, A.L.; Oliveira, C.R.; Rego, A.C. Aβ and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiol. Aging 2015, 36, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corlew, R.; Brasier, D.J.; Feldman, D.E.; Philpot, B.D. Presynaptic NMDA receptors: Newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist 2008, 14, 609–625. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Tomé, P.; Brera, B.; Arévalo, M.A.-A.; de Ceballos, M.A.L. β-Amyloid25-35 inhibits glutamate uptake in cultured neurons and astrocytes: Modulation of uptake as a survival mechanism. Neurobiol. Dis. 2004, 15, 580–589. [Google Scholar] [CrossRef]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.-I.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Hascup, K.N.; Hascup, E.R. Altered neurotransmission prior to cognitive decline in AβPP/PS1 mice, a model of Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 771–776. [Google Scholar] [CrossRef] [Green Version]
- von Kienlin, M.; Künnecke, B.; Metzger, F.; Steiner, G.; Richards, J.G.; Ozmen, L.; Jacobsen, H.; Loetscher, H. Altered metabolic profile in the frontal cortex of PS2APP transgenic mice, monitored throughout their life span. Neurobiol. Dis. 2005, 18, 32–39. [Google Scholar] [CrossRef]
- Richards, G.; Messer, J.; Faull, R.L.; Stadler, H.; Wichmann, J.; Huguenin, P.; Bohrmann, B.; Mutel, V. Altered distribution of mGlu2 receptors in β-amyloid-affected brain regions of Alzheimer cases and aged PS2APP mice. Brain Res. 2010, 1363, 180–190. [Google Scholar] [CrossRef]
- Hanson, J.E.; Pare, J.-F.; Deng, L.; Smith, Y.; Zhou, Q. Altered GluN2B NMDA receptor function and synaptic plasticity during early pathology in the PS2APP mouse model of Alzheimer’s disease. Neurobiol. Dis. 2015, 74, 254–262. [Google Scholar] [CrossRef] [Green Version]
- Steele, J.W.; Brautigam, H.; Short, J.A.; Sowa, A.; Shi, M.; Yadav, A.; Weaver, C.M.; Westaway, D.; Fraser, P.E.; St George-Hyslop, P.H. Early fear memory defects are associated with altered synaptic plasticity and molecular architecture in the TgCRND8 Alzheimer’s disease mouse model. J. Comp. Neurol. 2014, 522, 2319–2335. [Google Scholar] [CrossRef] [Green Version]
- Borroni, B.; Costanzi, C.; Padovani, A. Genetic susceptibility to behavioural and psychological symptoms in Alzheimer disease. Curr. Alzheimer Res 2010, 7, 158–164. [Google Scholar] [CrossRef]
- Liu, Y.; Yoo, M.-J.; Savonenko, A.; Stirling, W.; Price, D.L.; Borchelt, D.R.; Mamounas, L.; Lyons, W.E.; Blue, M.E.; Lee, M.K. Amyloid Pathology Is Associated with Progressive Monoaminergic Neurodegeneration in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2008, 28, 13805–13814. [Google Scholar] [CrossRef]
- Ledo, J.H.; Azevedo, E.P.; Beckman, D.; Ribeiro, F.C.; Santos, L.E.; Razolli, D.S.; Kincheski, G.C.; Melo, H.M.; Bellio, M.; Teixeira, A.L.; et al. Cross Talk Between Brain Innate Immunity and Serotonin Signaling Underlies Depressive-Like Behavior Induced by Alzheimer’s Amyloid- Oligomers in Mice. J. Neurosci. 2016, 36, 12106–12116. [Google Scholar] [CrossRef]
- Holm, P.; Ettrup, A.; Klein, A.B.; Santini, M.A.; El-Sayed, M.; Elvang, A.B.; Stensbøl, T.B.; Mikkelsen, J.D.; Knudsen, G.M.; Aznar, S. Plaque Deposition Dependent Decrease in 5-HT2A Serotonin Receptor in AβPPswe/PS1dE9 Amyloid Overexpressing Mice. J. Alzheimers Dis. 2010, 20, 1201–1213. [Google Scholar] [CrossRef]
- Palmer, A.M.; Wilcock, G.K.; Esiri, M.M.; Francis, P.T.; Bowen, D.M. Monoaminergic innervation of the frontal and temporal lobes in Alzheimer’s disease. Brain Res. 1987, 401, 231–238. [Google Scholar] [CrossRef]
- Roh, J.H.; Huang, Y.; Bero, A.W.; Kasten, T.; Stewart, F.R.; Bateman, R.J.; Holtzman, D.M. Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med. 2012, 4, 150ra122. [Google Scholar] [CrossRef] [Green Version]
- Dahlstroem, A.; Fuxe, K. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol. Scand. Suppl. 1964, 232, 1–55. [Google Scholar]
- Storga, D.; Vrecko, K.; Birkmayer, J.G.D.; Reibnegger, G. Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci. Lett. 1996, 203, 29–32. [Google Scholar] [CrossRef]
- Ambrée, O.; Richter, H.; Sachser, N.; Lewejohann, L.; Dere, E.; de Souza Silva, M.A.; Herring, A.; Keyvani, K.; Paulus, W.; Schäbitz, W.-R. Levodopa ameliorates learning and memory deficits in a murine model of Alzheimer’s disease. Neurobiol. Aging 2009, 30, 1192–1204. [Google Scholar] [CrossRef]
- Vorobyov, V.; Bakharev, B.; Medvinskaya, N.; Nesterova, I.; Samokhin, A.; Deev, A.; Tatarnikova, O.; Ustyugov, A.A.; Sengpiel, F.; Bobkova, N. Loss of Midbrain Dopamine Neurons and Altered Apomorphine EEG Effects in the 5xFAD Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 70, 241–256. [Google Scholar] [CrossRef] [Green Version]
- Cordella, A.; Krashia, P.; Nobili, A.; Pignataro, A.; La Barbera, L.; Viscomi, M.T.; Valzania, A.; Keller, F.; Ammassari-Teule, M.; Mercuri, N.B.; et al. Dopamine loss alters the hippocampus-nucleus accumbens synaptic transmission in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2018, 116, 142–154. [Google Scholar] [CrossRef]
- Mihardja, M.; Roy, J.; Wong, K.Y.; Aquili, L.; Heng, B.C.; Chan, Y.S.; Fung, M.L.; Lim, L.W. Therapeutic potential of neurogenesis and melatonin regulation in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2020, 1478, 43–62. [Google Scholar] [CrossRef]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer β-Fibrillogenesis by Melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef] [Green Version]
- Rosales-Corral, S.A.; Acuña-Castroviejo, D.; Coto-Montes, A.; Boga, J.A.; Manchester, L.C.; Fuentes-Broto, L.; Korkmaz, A.; Ma, S.; Tan, D.-X.; Reiter, R.J. Alzheimer’s disease: Pathological mechanisms and the beneficial role of melatonin. J. Pineal Res. 2012, 52, 167–202. [Google Scholar] [CrossRef]
- O’Neal-Moffitt, G.; Delic, V.; Bradshaw, P.C.; Olcese, J. Prophylactic melatonin significantly reduces Alzheimer’s neuropathology and associated cognitive deficits independent of antioxidant pathways in AβPPswe/PS1 mice. Mol. Neurodegener. 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Olcese, J.M.; Cao, C.; Mori, T.; Mamcarz, M.B.; Maxwell, A.; Runfeldt, M.J.; Wang, L.; Zhang, C.; Lin, X.; Zhang, G.; et al. Protection against cognitive deficits and markers of neurodegeneration by long-term oral administration of melatonin in a transgenic model of Alzheimer disease. J. Pineal Res. 2009, 47, 82–96. [Google Scholar] [CrossRef]
- Matsubara, E.; Bryant-Thomas, T.; Pacheco Quinto, J.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.-J.; Smith, M.A.; Perry, G.; et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef]
- Quinn, J.; Kulhanek, D.; Nowlin, J.; Jones, R.; Praticò, D.; Rokach, J.; Stackman, R. Chronic melatonin therapy fails to alter amyloid burden or oxidative damage in old Tg2576 mice: Implications for clinical trials. Brain Res. 2005, 1037, 209–213. [Google Scholar] [CrossRef]
- Cardinali, D.P. Melatonin: Clinical perspectives in neurodegeneration. Front. Endocrinol. 2019, 10, 480. [Google Scholar] [CrossRef]
- Pappolla, M.; Matsubara, E.; Vidal, R.; Pacheco-Quinto, J.; Poeggeler, B.; Zagorski, M.; Sambamurti, K. Melatonin treatment enhances Aβ lymphatic clearance in a transgenic mouse model of amyloidosis. Curr. Alzheimer Res. 2018, 15, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-C.; Wang, Z.-F.; Zhang, J.-X.; Wang, Q.; Wang, J.-Z. Effect of melatonin on calyculin A-induced tau hyperphosphorylation. Eur. J. Pharm. 2005, 510, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-Q.; Xu, G.-G.; Duan, P.; Zhang, Q.; Wang, J.-Z. Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharm. Sin. 2005, 26, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Mesa, Y.; Giménez-Llort, L.; López, L.C.; Venegas, C.; Cristòfol, R.; Escames, G.; Acuña-Castroviejo, D.; Sanfeliu, C. Melatonin plus physical exercise are highly neuroprotective in the 3xTg-AD mouse. Neurobiol. Aging 2012, 33, 1124.e1113–1124.e1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.H.; Hua, N.; Zang, X.; Huang, T.; He, L. Melatonin ameliorates Aβ1-42-induced Alzheimer’s cognitive deficits in mouse model. J. Pharm. Pharmacol. 2018, 70, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.Q.; Wang, S.H.; Ling, Z.Q.; Wang, D.L.; Wang, J.Z. Effect of inhibiting melatonin biosynthesis on spatial memory retention and tau phosphorylation in rat. J. Pineal Res. 2004, 37, 71–77. [Google Scholar] [CrossRef]
- Feng, Z.; Chang, Y.; Cheng, Y.; Zhang, B.-L.; Qu, Z.-W.; Qin, C.; Zhang, J.-T. Melatonin alleviates behavioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer’s disease. J. Pineal Res. 2004, 37, 129–136. [Google Scholar] [CrossRef]
- Labban, S.; Alghamdi, B.S.; Alshehri, F.S.; Kurdi, M. Effects of melatonin and resveratrol on recognition memory and passive avoidance performance in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2021, 402, 113100. [Google Scholar] [CrossRef]
- Shi, Y.; Fang, Y.-Y.; Wei, Y.-P.; Jiang, Q.; Zeng, P.; Tang, N.; Lu, Y.; Tian, Q. Melatonin in Synaptic Impairments of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 911–926. [Google Scholar] [CrossRef]
- Rong, K.; Zheng, H.; Yang, R.; Liu, X.; Li, L.; Chen, N.; Zhao, G.; Gong, C.; Deng, Y. Melatonin and its metabolite N (1)-acetyl-N (1)-formyl-5-methoxykynuramine improve learning and memory impairment related to Alzheimer’s disease in rats. J. Biochem. Mol. Toxicol. 2020, 34, e22430. [Google Scholar] [CrossRef]
- Luo, X.-T.; Wang, C.-M.; Liu, Y.; Huang, Z.-G. New multifunctional melatonin-derived benzylpyridinium bromides with potent cholinergic, antioxidant, and neuroprotective properties as innovative drugs for Alzheimer’s disease. Eur. J. Med. Chem. 2015, 103, 302–311. [Google Scholar] [CrossRef]
- Wang, X.-C.; Zhang, Y.-C.; Chatterjie, N.; Grundke-Iqbal, I.; Iqbal, K.; Wang, J.-Z. Effect of melatonin and melatonylvalpromide on β-amyloid and neurofilaments in N2a cells. Neurochem. Res. 2008, 33, 1138–1144. [Google Scholar] [CrossRef]
- Buendia, I.; Egea, J.; Parada, E.; Navarro, E.; León, R.; Rodríguez-Franco, M.I.; López, M.G. The Melatonin–N, N-Dibenzyl (N-methyl) amine Hybrid ITH91/IQM157 Affords Neuroprotection in an in Vitro Alzheimer’s Model via Hemo-oxygenase-1 Induction. ACS Chem. Neurosci. 2015, 6, 288–296. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Ouyang, X.; Zhou, S.; Yin, W.; Tang, C.; Laudon, M.; Tian, S. A novel melatonin agonist Neu-P11 facilitates memory performance and improves cognitive impairment in a rat model of Alzheimer’disease. Horm. Behav. 2013, 64, 1–7. [Google Scholar] [CrossRef]
- Yao, K.; Zhao, Y.-F.; Zu, H.-B. Melatonin receptor stimulation by agomelatine prevents Aβ-induced tau phosphorylation and oxidative damage in PC12 cells. Drug Des. Dev. Ther. 2019, 13, 387. [Google Scholar] [CrossRef] [Green Version]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, J.; Tsui, K.C.; Ng, J.; Fung, M.-L.; Lim, L.W. Regulation of Melatonin and Neurotransmission in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6841. https://doi.org/10.3390/ijms22136841
Roy J, Tsui KC, Ng J, Fung M-L, Lim LW. Regulation of Melatonin and Neurotransmission in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(13):6841. https://doi.org/10.3390/ijms22136841
Chicago/Turabian StyleRoy, Jaydeep, Ka Chun Tsui, Jonah Ng, Man-Lung Fung, and Lee Wei Lim. 2021. "Regulation of Melatonin and Neurotransmission in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 13: 6841. https://doi.org/10.3390/ijms22136841
APA StyleRoy, J., Tsui, K. C., Ng, J., Fung, M.-L., & Lim, L. W. (2021). Regulation of Melatonin and Neurotransmission in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(13), 6841. https://doi.org/10.3390/ijms22136841