
Cohesin Mutations in Cancer: Emerging Therapeutic Targets



Abstract
1. Introduction
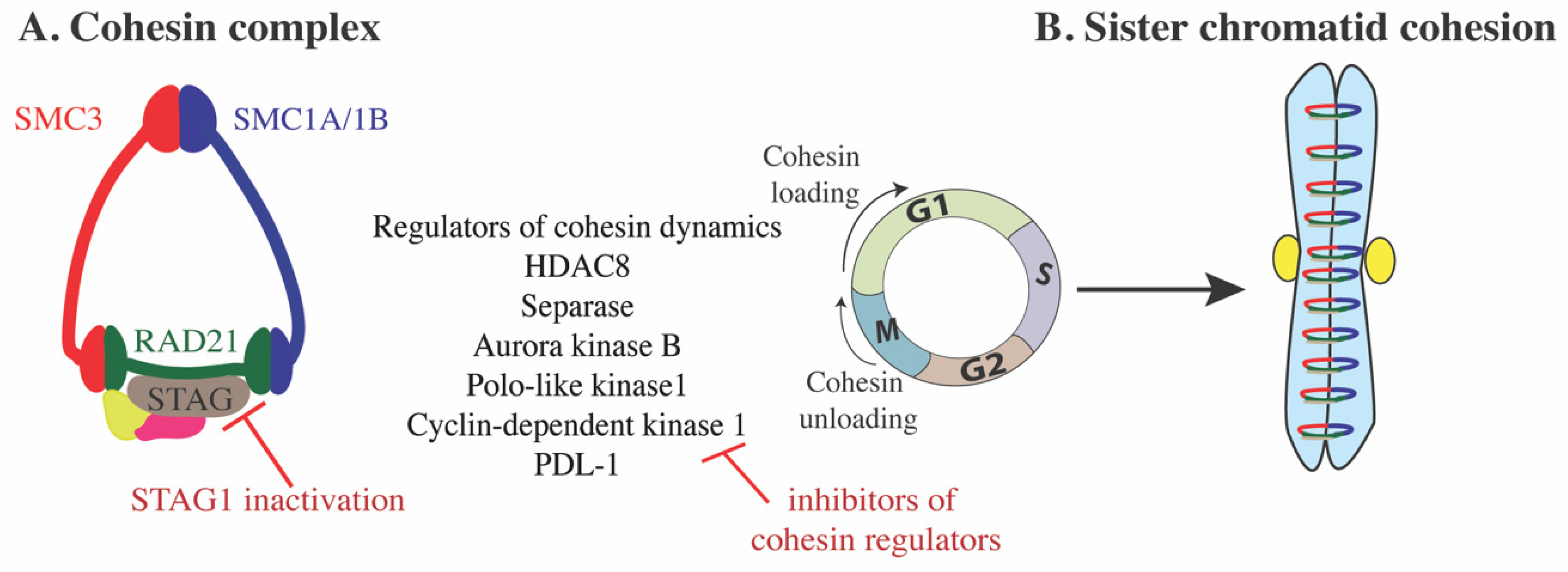
2. Cohesin Structure and Dynamics
3. Cohesin Function
4. Cohesin in Developmental Disorders
5. Cohesin in Cancer
6. Overexpression of Cohesin in Cancers
7. Therapeutic Targeting in Cohesin Mutant Cancers
7.1. Targeting Cohesin Complex Assembly
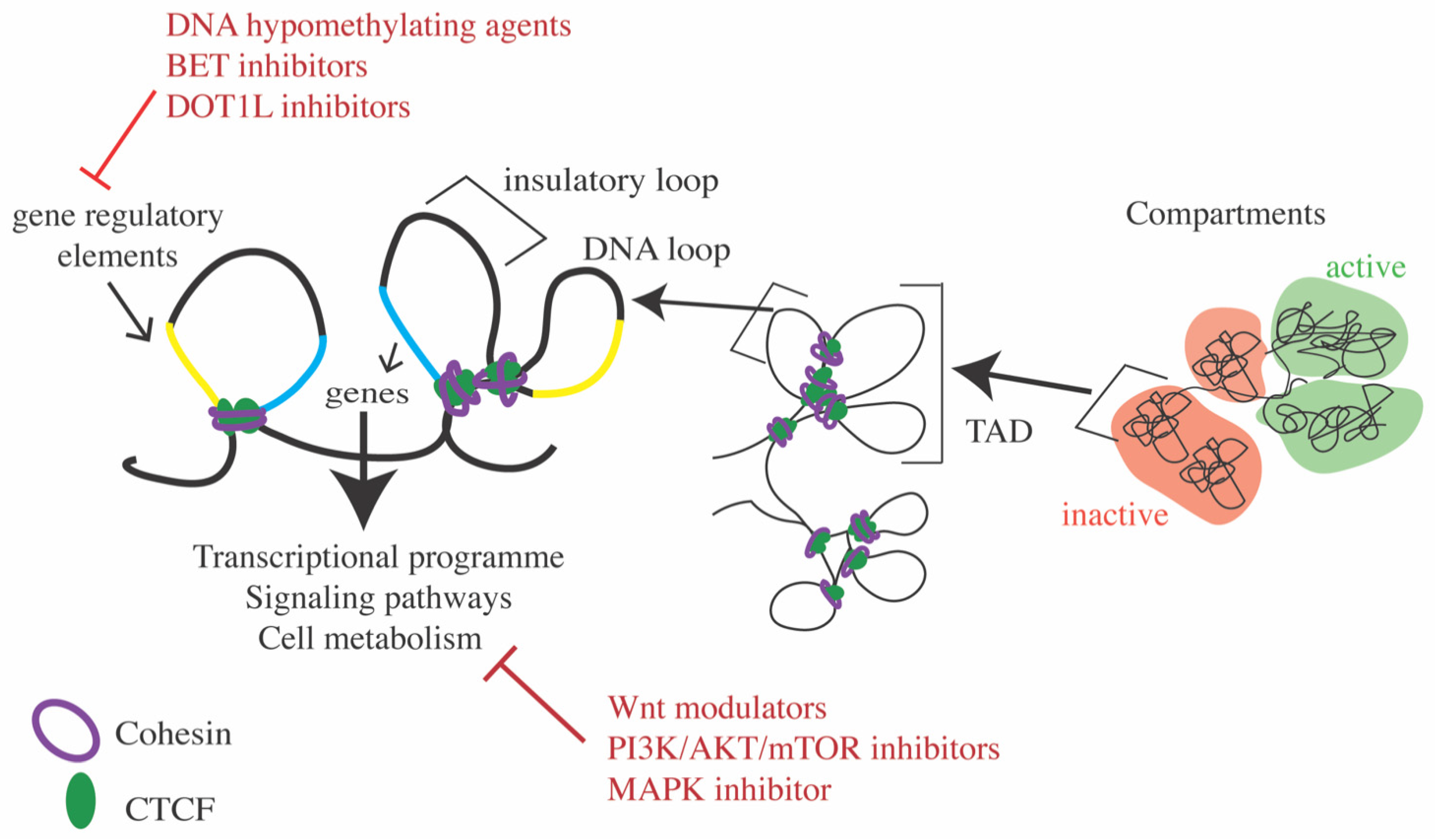
7.2. Modulating Transcription Using Inhibitors to Epigenetic Targets
7.3. Targeting Signaling Pathways
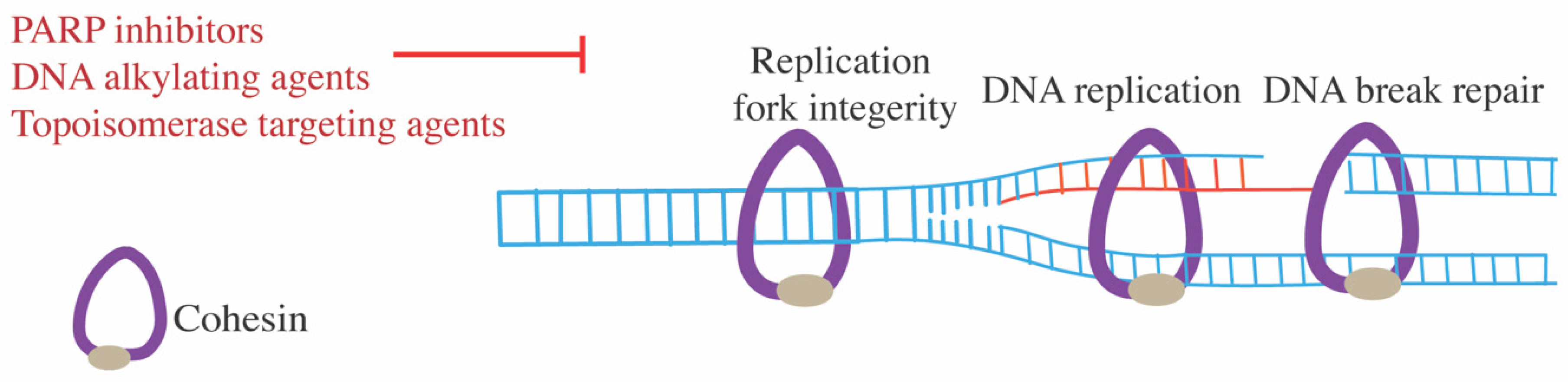
7.4. DNA-Damaging Agents
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PLK1 | Polo-like kinase 1 |
| CDK1 | Cyclin-dependent kinase 1 |
| SGO1 | Shugoshin 1 |
| PP2A | Protein phosphatase 2A |
| TADs | Topologically associated domains |
| PcG | Polycomb group |
| hTERT | Human telomerase reverse transcriptase |
| rDNA | Ribosomal DNA |
| rRNA | Ribosomal RNA |
| CdLS | Cornelia de Lange syndrome |
| RBS | Roberts syndrome |
| AML | Acute myeloid leukemia |
| ES | Embryonic stem |
| HSC | Hematopoietic stem cells |
| PROTAC | Proteolysis targeting chimera |
| GA | Glycyrrhizic acid |
| HMA | Hypomethylating agents |
| BET | Bromodomain and extra-terminal |
| GSK3 | Glycogen synthase kinase 3 |
| LPS | Lipopolysaccharide |
| PARP | Poly(ADP-ribose) |
| PD1 | Programmed death 1 |
| PDL1 | Programmed death ligand 1 |
References
- Minchell, N.E.; Keszthelyi, A.; Baxter, J. Cohesin Causes Replicative DNA Damage by Trapping DNA Topological Stress. Mol. Cell 2020, 78, 739–751. [Google Scholar] [CrossRef]
- Horsfield, J.A.; Print, C.G.; Monnich, M. Diverse developmental disorders from the one ring: Distinct molecular pathways underlie the cohesinopathies. Front. Genet. 2012, 3, 171. [Google Scholar] [CrossRef]
- Kim, Y.; Shi, Z.; Zhang, H.; Finkelstein, I.J.; Yu, H. Human cohesin compacts DNA by loop extrusion. Science 2019, 366, 1345–1349. [Google Scholar] [CrossRef]
- Nasmyth, K.; Haering, C.H. Cohesin: Its Roles and Mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef]
- Wendt, K.S. Resolving the Genomic Localization of the Kollerin Cohesin-Loader Complex. Methods Mol. Biol. 2017, 1515, 115–123. [Google Scholar] [CrossRef]
- Murayama, Y.; Uhlmann, F. DNA Entry into and Exit out of the Cohesin Ring by an Interlocking Gate Mechanism. Cell 2015, 163, 1628–1640. [Google Scholar] [CrossRef]
- Wutz, G.; Várnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef]
- Haarhuis, J.H.I.; van der Weide, R.H.; Blomen, V.A.; Yáñez-Cuna, J.O.; Amendola, M.; van Ruiten, M.S.; Krijger, P.H.L.; Teunissen, H.; Medema, R.H.; van Steensel, B.; et al. The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 2017, 169, 693–707. [Google Scholar] [CrossRef]
- Kriz, A.J.; Colognori, D.; Sunwoo, H.; Nabet, B.; Lee, J.T. Balancing cohesin eviction and retention prevents aberrant chromosomal interactions, Polycomb-mediated repression, and X-inactivation. Mol. Cell 2021, 81, 1970–1987. [Google Scholar] [CrossRef]
- Liu, N.Q.; Maresca, M.; van den Brand, T.; Braccioli, L.; Schijns, M.M.G.A.; Teunissen, H.; Bruneau, B.G.; Nora, E.P.; de Wit, E. WAPL maintains a cohesin loading cycle to preserve cell-type-specific distal gene regulation. Nat. Genet. 2021, 53, 100–109. [Google Scholar] [CrossRef]
- Rolef Ben-Shahar, T.; Heeger, S.; Lehane, C.; East, P.; Flynn, H.; Skehel, M.; Uhlmann, F. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 2008, 321, 563–566. [Google Scholar] [CrossRef]
- Unal, E.; Heidinger-Pauli, J.M.; Kim, W.; Guacci, V.; Onn, I.; Gygi, S.P.; Koshland, D.E. A molecular determinant for the establishment of sister chromatid cohesion. Science 2008, 321, 566–569. [Google Scholar] [CrossRef] [PubMed]
- van der Lelij, P.; Godthelp, B.C.; van Zon, W.; van Gosliga, D.; Oostra, A.B.; Steltenpool, J.; de Groot, J.; Scheper, R.J.; Wolthuis, R.M.; Waisfisz, Q.; et al. The cellular phenotype of Roberts syndrome fibroblasts as revealed by ectopic expression of ESCO2. PLoS ONE 2009, 4, e6936. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, X.; Li, Y.; Kim, B.J.; Jia, J.; Huang, Z.; Yang, T.; Fu, X.; Jung, S.Y.; Wang, Y.; et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell 2008, 31, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Mondal, G.; Stevers, M.; Goode, B.; Ashworth, A.; Solomon, D.A. A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat. Commun. 2019, 10, 1686. [Google Scholar] [CrossRef]
- Ladurner, R.; Kreidl, E.; Ivanov, M.P.; Ekker, H.; Idarraga-Amado, M.H.; Busslinger, G.A.; Wutz, G.; Cisneros, D.A.; Peters, J.-M. Sororin actively maintains sister chromatid cohesion. EMBO J. 2016, 35, 635–653. [Google Scholar] [CrossRef]
- Nishiyama, T.; Ladurner, R.; Schmitz, J.; Kreidl, E.; Schleiffer, A.; Bhaskara, V.; Bando, M.; Shirahige, K.; Hyman, A.A.; Mechtler, K.; et al. Sororin Mediates Sister Chromatid Cohesion by Antagonizing Wapl. Cell 2010, 143, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Losada, A.; Hirano, M.; Hirano, T. Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev. 2002, 16, 3004–3016. [Google Scholar] [CrossRef] [PubMed]
- Sumara, I.; Vorlaufer, E.; Stukenberg, P.T.; Kelm, O.; Redemann, N.; Nigg, E.A.; Peters, J.M. The dissociation of cohesin from chromosomes in prophase is regulated by polo-like kinase. Mol. Cell 2002, 9, 515–525. [Google Scholar] [CrossRef]
- Borton, M.T.; Rashid, M.S.; Dreier, M.R.; Taylor, W.R. Multiple Levels of Regulation of Sororin by Cdk1 and Aurora B. J Cell Biochem 2016, 117, 351–360. [Google Scholar] [CrossRef]
- Hara, K.; Zheng, G.; Qui, Q.; Liu, H.; Ouyang, Z.; Chen, Z.; Tomchick, D.R.; Yu, H. Structure of cohesin subcomplex pinpoints direct shugoshin-Wapl antagonism in centromeric cohesion. Nat. Struct. Mol. Biol. 2014, 21, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jia, L.; Yu, H. Phospho-H2A and Cohesin Specify Distinct Tension-Regulated Sgo1 Pools at Kinetochores and Inner Centromeres. Curr. Biol. 2013, 23, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rankin, S.; Yu, H. Phosphorylation-enabled binding of SGO1-PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat. Cell Biol. 2013, 15, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Shindo, N.; Kumada, K.; Hirota, T. Separase Sensor Reveals Dual Roles for Separase Coordinating Cohesin Cleavage and Cdk1 Inhibition. Dev. Cell 2012, 23, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef]
- De Koninck, M.; Losada, A. Cohesin Mutations in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Cuadrado, A.; Remeseiro, S.; Gomez-Lopez, G.; Pisano, D.G.; Losada, A. The specific contributions of cohesin-SA1 to cohesion and gene expression Implications for cancer and development. Cell Cycle 2012, 11, 2233–2238. [Google Scholar] [CrossRef]
- Remeseiro, S.; Cuadrado, A.; Carretero, M.; Martinez, P.; Drosopoulos, W.C.; Canamero, M.; Schildkraut, C.L.; Blasco, M.A.; Losada, A. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 2012, 31, 2076–2089. [Google Scholar] [CrossRef]
- Couturier, A.M.; Fleury, H.; Patenaude, A.M.; Bentley, V.L.; Rodrigue, A.; Coulombe, Y.; Niraj, J.; Pauty, J.; Berman, J.N.; Dellaire, G.; et al. Roles for APRIN (PDS5B) in homologous recombination and in ovarian cancer prediction. Nucleic Acids Res. 2016, 44, 10879–10897. [Google Scholar] [CrossRef]
- Misulovin, Z.; Pherson, M.; Gause, M.; Dorsett, D. Brca2, Pds5 and Wapl differentially control cohesin chromosome association and function. PLoS Genet. 2018, 14, e1007225. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Tu, X.; Dong, H.; Boughey, J.C.; Goetz, M.P.; Weinshilboum, R.; Lou, Z.; et al. Regulation of sister chromatid cohesion by nuclear PD-L1. Cell Res. 2020, 30, 590–601. [Google Scholar] [CrossRef]
- Sakuno, T.; Tada, K.; Watanabe, Y. Kinetochore geometry defined by cohesion within the centromere. Nature 2009, 458, 852–858. [Google Scholar] [CrossRef]
- Kong, X.; Ball, A.R., Jr.; Sonoda, E.; Feng, J.; Takeda, S.; Fukagawa, T.; Yen, T.J.; Yokomori, K. Cohesin associates with spindle poles in a mitosis-specific manner and functions in spindle assembly in vertebrate cells. Mol. Biol. Cell 2009, 20, 1289–1301. [Google Scholar] [CrossRef]
- Yan, J.; Enge, M.; Whitington, T.; Dave, K.; Liu, J.; Sur, I.; Schmierer, B.; Jolma, A.; Kivioja, T.; Taipale, M.; et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 2013, 154. [Google Scholar] [CrossRef] [PubMed]
- Heidinger-Pauli, J.M.; Mert, O.; Davenport, C.; Guacci, V.; Koshland, D. Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr. Biol. 2010, 20, 957–963. [Google Scholar] [CrossRef]
- Davidson, I.F.; Bauer, B.; Goetz, D.; Tang, W.; Wutz, G.; Peters, J.M. DNA loop extrusion by human cohesin. Science 2019, 366, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 2016, 167, 1188–1200. [Google Scholar] [CrossRef]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huang, S.-C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.-R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320. [Google Scholar] [CrossRef]
- Fudenberg, G.; Getz, G.; Meyerson, M.; Mirny, L.A. High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nat. Biotechnol. 2011, 29, 1109–1113. [Google Scholar] [CrossRef]
- Seitan, V.C.; Faure, A.J.; Zhan, Y.; McCord, R.P.; Lajoie, B.R.; Ing-Simmons, E.; Lenhard, B.; Giorgetti, L.; Heard, E.; Fisher, A.G.; et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013, 23, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Sofueva, S.; Yaffe, E.; Chan, W.-C.; Georgopoulou, D.; Vietri Rudan, M.; Mira-Bontenbal, H.; Pollard, S.M.; Schroth, G.P.; Tanay, A.; Hadjur, S. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013, 32. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Wutz, G.; Ladurner, R.; St Hilaire, B.G.; Stocsits, R.R.; Nagasaka, K.; Pignard, B.; Sanborn, A.; Tang, W.; Várnai, C.; Ivanov, M.P.; et al. ESCO1 and CTCF enable formation of long chromatin loops by protecting cohesinSTAG1 from WAPL. eLife 2020, 9, e52091. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Giménez-Llorente, D.; Kojic, A.; Rodríguez-Corsino, M.; Cuartero, Y.; Martín-Serrano, G.; Gómez-López, G.; Marti-Renom, M.A.; Losada, A. Specific Contributions of Cohesin-SA1 and Cohesin-SA2 to TADs and Polycomb Domains in Embryonic Stem Cells. Cell Rep. 2019, 27, 3500–3510. [Google Scholar] [CrossRef]
- Cunningham, M.D.; Gause, M.; Cheng, Y.; Noyes, A.; Dorsett, D.; Kennison, J.A.; Kassis, J.A. Wapl antagonizes cohesin binding and promotes Polycomb-group silencing in Drosophila. Development 2012, 139, 4172–4179. [Google Scholar] [CrossRef]
- Rhodes, J.D.P.; Feldmann, A.; Hernández-Rodríguez, B.; Díaz, N.; Brown, J.M.; Fursova, N.A.; Blackledge, N.P.; Prathapan, P.; Dobrinic, P.; Huseyin, M.K.; et al. Cohesin Disrupts Polycomb-Dependent Chromosome Interactions in Embryonic Stem Cells. Cell Rep. 2020, 30, 820–835. [Google Scholar] [CrossRef]
- Arruda, N.L.; Carico, Z.M.; Justice, M.; Liu, Y.F.; Zhou, J.; Stefan, H.C.; Dowen, J.M. Distinct and overlapping roles of STAG1 and STAG2 in cohesin localization and gene expression in embryonic stem cells. Epigenetics Chromatin 2020, 13, 32. [Google Scholar] [CrossRef]
- Casa, V.; Moronta Gines, M.; Gade Gusmao, E.; Slotman, J.A.; Zirkel, A.; Josipovic, N.; Oole, E.; van IJcken, W.F.J.; Houtsmuller, A.B.; Papantonis, A.; et al. Redundant and specific roles of cohesin STAG subunits in chromatin looping and transcriptional control. Genome Res. 2020, 30, 515–527. [Google Scholar] [CrossRef]
- Ketharnathan, S.; Labudina, A.; Horsfield, J.A. Cohesin Components Stag1 and Stag2 Differentially Influence Haematopoietic Mesoderm Development in Zebrafish Embryos. Front. Cell Dev. Biol. 2020, 8, 7545. [Google Scholar] [CrossRef]
- Kojic, A.; Cuadrado, A.; De Koninck, M.; Giménez-Llorente, D.; Rodríguez-Corsino, M.; Gómez-López, G.; Le Dily, F.; Marti-Renom, M.A.; Losada, A. Distinct roles of cohesin-SA1 and cohesin-SA2 in 3D chromosome organization. Nat. Struct. Mol. Biol. 2018, 25, 496–504. [Google Scholar] [CrossRef]
- Viny, A.D.; Bowman, R.L.; Liu, Y.; Lavallee, V.P.; Eisman, S.E.; Xiao, W.; Durham, B.H.; Navitski, A.; Park, J.; Braunstein, S.; et al. Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation. Cell Stem Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Losada, A. Specialized functions of cohesins STAG1 and STAG2 in 3D genome architecture. Curr. Opin. Genet. Dev. 2020, 61, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Cuartero, S.; Weiss, F.D.; Dharmalingam, G.; Guo, Y.; Ing-Simmons, E.; Masella, S.; Robles-Rebollo, I.; Xiao, X.; Wang, Y.-F.; Barozzi, I.; et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat. Immunol. 2018, 19, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Ing-Simmons, E.; Vaid, R.; Bing, X.Y.; Levine, M.; Mannervik, M.; Vaquerizas, J.M. Independence of chromatin conformation and gene regulation during Drosophila dorsoventral patterning. Nat. Genet. 2021, 53, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Gimenez, G.; Taylor, T.; Khatoon, U.; Day, R.; Morison, I.M.; Horsfield, J.A. BET inhibition prevents aberrant RUNX1 and ERG transcription in STAG2 mutant leukaemia cells. J. Mol. Cell Biol. 2020, 12, 397–399. [Google Scholar] [CrossRef]
- Antony, J.; Dasgupta, T.; Rhodes, J.M.; McEwan, M.V.; Print, C.G.; O’Sullivan, J.M.; Horsfield, J.A. Cohesin modulates transcription of estrogen-responsive genes. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2015, 1849, 257–269. [Google Scholar] [CrossRef]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef]
- Ryu, M.J.; Kim, B.J.; Lee, J.W.; Lee, M.W.; Choi, H.K.; Kim, S.T. Direct interaction between cohesin complex and DNA replication machinery. Biochem. Biophys. Res. Commun. 2006, 341, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Tittel-Elmer, M.; Lengronne, A.; Davidson, M.B.; Bacal, J.; François, P.; Hohl, M.; Petrini, J.H.; Pasero, P.; Cobb, J.A. Cohesin Association to Replication Sites Depends on Rad50 and Promotes Fork Restart. Mol. Cell 2012, 48, 98–108. [Google Scholar] [CrossRef]
- Benedict, B.; van Schie, J.J.M.; Oostra, A.B.; Balk, J.A.; Wolthuis, R.M.F.; Riele, H.t.; de Lange, J. WAPL-Dependent Repair of Damaged DNA Replication Forks Underlies Oncogene-Induced Loss of Sister Chromatid Cohesion. Dev. Cell 2020, 52, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Maldonado, D.; Byrum, A.K.; Jackson, J.; Wessel, S.; Lemaçon, D.; Guitton-Sert, L.; Quinet, A.; Tirman, S.; Graziano, S.; Masson, J.-Y.; et al. Perturbing cohesin dynamics drives MRE11 nuclease-dependent replication fork slowing. Nucleic Acids Res. 2019, 47, 1294–1310. [Google Scholar] [CrossRef] [PubMed]
- Frattini, C.; Villa-Hernández, S.; Pellicanò, G.; Jossen, R.; Katou, Y.; Shirahige, K.; Bermejo, R. Cohesin Ubiquitylation and Mobilization Facilitate Stalled Replication Fork Dynamics. Mol. Cell 2017, 68, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.; Ruiz-Torres, M.; Rodríguez-Acebes, S.; Lafarga, V.; Rodríguez-Corsino, M.; Megías, D.; Cisneros, D.A.; Peters, J.-M.; Méndez, J.; Losada, A. PDS5 proteins are required for proper cohesin dynamics and participate in replication fork protection. J. Biol. Chem. 2020, 295, 146–157. [Google Scholar] [CrossRef]
- Caron, P.; Aymard, F.; Iacovoni, J.S.; Briois, S.; Canitrot, Y.; Bugler, B.; Massip, L.; Losada, A.; Legube, G. Cohesin Protects Genes against γH2AX Induced by DNA Double-Strand Breaks. PLoS Genet. 2012, 8, e1002460. [Google Scholar] [CrossRef]
- Ström, L.; Lindroos, H.B.; Shirahige, K.; Sjögren, C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell 2004, 16, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Ström, L.; Karlsson, C.; Lindroos, H.B.; Wedahl, S.; Katou, Y.; Shirahige, K.; Sjögren, C. Postreplicative Formation of Cohesion Is Required for Repair and Induced by a Single DNA Break. Science 2007, 317, 242–245. [Google Scholar] [CrossRef]
- Kong, X.; Ball, A.R., Jr.; Pham, H.X.; Zeng, W.; Chen, H.Y.; Schmiesing, J.A.; Kim, J.S.; Berns, M.; Yokomori, K. Distinct functions of human cohesin-SA1 and cohesin-SA2 in double-strand break repair. Mol. Cell Biol. 2014, 34, 685–698. [Google Scholar] [CrossRef]
- Hellmuth, S.; Gutiérrez-Caballero, C.; Llano, E.; Pendás, A.M.; Stemmann, O. Local activation of mammalian separase in interphase promotes double-strand break repair and prevents oncogenic transformation. EMBO J. 2018, 37, e99184. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-J.; Li, Y.; Zhang, J.; Xi, Y.; Li, Y.; Yang, T.; Jung, S.Y.; Pan, X.; Chen, R.; Li, W.; et al. Genome-wide Reinforcement of Cohesin Binding at Pre-existing Cohesin Sites in Response to Ionizing Radiation in Human Cells. J. Biol. Chem. 2010, 285, 22784–22792. [Google Scholar] [CrossRef] [PubMed]
- So, E.Y.; Ausman, M.; Saeki, T.; Ouchi, T. Phosphorylation of SMC1 by ATR is required for desferrioxamine (DFO)-induced apoptosis. Cell Death Dis. 2011, 2, e128. [Google Scholar] [CrossRef] [PubMed]
- Watrin, E.; Peters, J.-M. The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 2009, 28, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Meisenberg, C.; Pinder, S.I.; Hopkins, S.R.; Wooller, S.K.; Benstead-Hume, G.; Pearl, F.M.G.; Jeggo, P.A.; Downs, J.A. Repression of Transcription at DNA Breaks Requires Cohesin throughout Interphase and Prevents Genome Instability. Mol. Cell 2019, 73, 212–223. [Google Scholar] [CrossRef]
- Bose, T.; Lee, K.K.; Lu, S.; Xu, B.; Harris, B.; Slaughter, B.; Unruh, J.; Garrett, A.; McDowell, W.; Box, A.; et al. Cohesin proteins promote ribosomal RNA production and protein translation in yeast and human cells. PLoS Genet. 2012, 8, e1002749. [Google Scholar] [CrossRef]
- Harris, B.; Bose, T.; Lee, K.K.; Wang, F.; Lu, S.; Ross, R.T.; Zhang, Y.; French, S.L.; Beyer, A.L.; Slaughter, B.D.; et al. Cohesion promotes nucleolar structure and function. Mol. Biol. Cell 2014, 25, 337–346. [Google Scholar] [CrossRef]
- Lu, S.; Lee, K.K.; Harris, B.; Xiong, B.; Bose, T.; Saraf, A.; Hattem, G.; Florens, L.; Seidel, C.; Gerton, J.L. The cohesin acetyltransferase Eco1 coordinates rDNA replication and transcription. EMBO Rep. 2014, 15, 609–617. [Google Scholar] [CrossRef]
- Chin, C.V.; Antony, J.; Ketharnathan, S.; Labudina, A.; Gimenez, G.; Parsons, K.M.; He, J.; George, A.J.; Pallotta, M.M.; Musio, A.; et al. Cohesin mutations are synthetic lethal with stimulation of WNT signaling. eLife 2020, 9, e61405. [Google Scholar] [CrossRef] [PubMed]
- Viny, A.D.; Ott, C.J.; Spitzer, B.; Rivas, M.; Meydan, C.; Papalexi, E.; Yelin, D.; Shank, K.; Reyes, J.; Chiu, A.; et al. Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J. Exp. Med. 2015, 212, 1819–1832. [Google Scholar] [CrossRef]
- Costantino, L.; Hsieh, T.-H.S.; Lamothe, R.; Darzacq, X.; Koshland, D. Cohesin residency determines chromatin loop patterns. eLife 2020, 9, e59889. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brito, I.L.; Villén, J.; Gygi, S.P.; Amon, A.; Moazed, D. Inhibition of homologous recombination by a cohesin-associated clamp complex recruited to the rDNA recombination enhancer. Genes Dev. 2006, 20, 2887–2901. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ganley, A.R.D. Recombination Regulation by Transcription-Induced Cohesin Dissociation in rDNA Repeats. Science 2005, 309, 1581–1584. [Google Scholar] [CrossRef] [PubMed]
- Piché, J.; Van Vliet, P.P.; Pucéat, M.; Andelfinger, G. The expanding phenotypes of cohesinopathies: One ring to rule them all! Cell Cycle 2019, 18, 2828–2848. [Google Scholar] [CrossRef]
- Krantz, I.D.; McCallum, J.; DeScipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004, 36, 631–635. [Google Scholar] [CrossRef]
- Tonkin, E.T.; Wang, T.J.; Lisgo, S.; Bamshad, M.J.; Strachan, T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004, 36, 636–641. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Klein, S.D.; Signer, R.H.; Vilain, E.; Martinez-Agosto, J.A. Mutations in STAG2 cause an X-linked cohesinopathy associated with undergrowth, developmental delay, and dysmorphia: Expanding the phenotype in males. Mol. Genet. Genom. Med. 2019, 7, e00501. [Google Scholar] [CrossRef]
- Soardi, F.C.; Machado-Silva, A.; Linhares, N.D.; Zheng, G.; Qu, Q.; Pena, H.B.; Martins, T.M.M.; Vieira, H.G.S.; Pereira, N.B.; Melo-Minardi, R.C.; et al. Familial STAG2 germline mutation defines a new human cohesinopathy. NPJ Genom. Med. 2017, 2, 7. [Google Scholar] [CrossRef]
- Vega, H.; Waisfisz, Q.; Gordillo, M.; Sakai, N.; Yanagihara, I.; Yamada, M.; van Gosliga, D.; Kayserili, H.; Xu, C.; Ozono, K.; et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat. Genet. 2005, 37, 468–470. [Google Scholar] [CrossRef]
- Tomkins, D.J.; Sisken, J.E. Abnormalities in the cell-division cycle in Roberts syndrome fibroblasts: A cellular basis for the phenotypic characteristics? Am. J. Hum. Genet. 1984, 36, 1332–1340. [Google Scholar] [PubMed]
- Kruszka, P.; Berger, S.I.; Casa, V.; Dekker, M.R.; Gaesser, J.; Weiss, K.; Martinez, A.F.; Murdock, D.R.; Louie, R.J.; Prijoles, E.J.; et al. Cohesin complex-associated holoprosencephaly. Brain 2019, 142, 2631–2643. [Google Scholar] [CrossRef] [PubMed]
- Chetaille, P.; Preuss, C.; Burkhard, S.; Côté, J.M.; Houde, C.; Castilloux, J.; Piché, J.; Gosset, N.; Leclerc, S.; Wünnemann, F.; et al. Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm. Nat. Genet. 2014, 46, 1245–1249. [Google Scholar] [CrossRef]
- Kumar, R.; Corbett, M.A.; Van Bon, B.W.M.; Gardner, A.; Woenig, J.A.; Jolly, L.A.; Douglas, E.; Friend, K.; Tan, C.; Van Esch, H.; et al. Increased STAG2 dosage defines a novel cohesinopathy with intellectual disability and behavioral problems. Hum. Mol. Genet. 2015, 24, 7171–7181. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Leiserson, M.D.M.; Vandin, F.; Wu, H.-T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Vasquez, J.C.; Kundishora, A.J.; Elsamadicy, A.A.; Beckta, J.M.; Sule, A.; Marks, A.M.; Leelatian, N.; Huttner, A.; Bindra, R.S.; et al. Persistent Stag2 Mutation Despite Multimodal Therapy in Recurrent Pediatric Glioblastoma. NPJ Genom. Med. 2020, 5, 23. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The Genomic Landscape of Pediatric Ewing Sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [PubMed]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic Landscape of Ewing Sarcoma Defines an Aggressive Subtype with Co-Association of STAG2 and TP53 Mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Balbas-Martinez, C.; Sagrera, A.; Carrillo-de-Santa-Pau, E.; Earl, J.; Marquez, M.; Vazquez, M.; Lapi, E.; Castro-Giner, F.; Beltran, S.; Bayes, M.; et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat. Genet. 2013, 45, 1464. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef]
- Solomon, D.A.; Kim, J.-S.; Bondaruk, J.; Shariat, S.F.; Wang, Z.-F.; Elkahloun, A.G.; Ozawa, T.; Gerard, J.; Zhuang, D.; Zhang, S.; et al. Frequent truncating mutations of STAG2 in bladder cancer. Nat. Genet. 2013, 45, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.F.; Platt, F.M.; Hurst, C.D.; Thygesen, H.H.; Knowles, M.A. Frequent inactivating mutations of STAG2 in bladder cancer are associated with low tumour grade and stage and inversely related to chromosomal copy number changes. Hum. Mol. Genet. 2014, 23, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Kon, A.; Shih, L.Y.; Minamino, M.; Sanada, M.; Shiraishi, Y.; Nagata, Y.; Yoshida, K.; Okuno, Y.; Bando, M.; Nakato, R.; et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013, 45, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Bollin, R.; Gehlhaar, M.; Walter, C.; Dugas, M.; Suchanek, K.J.; Kirchner, A.; Huang, L.; Chaturvedi, A.; Wichmann, M.; et al. Mutations in the cohesin complex in acute myeloid leukemia: Clinical and prognostic implications. Blood 2014, 123, 914–920. [Google Scholar] [CrossRef]
- Thota, S.; Viny, A.D.; Makishima, H.; Spitzer, B.; Radivoyevitch, T.; Przychodzen, B.; Sekeres, M.A.; Levine, R.L.; Maciejewski, J.P. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014, 124, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Toki, T.; Okuno, Y.; Kanezaki, R.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; Park, M.J.; Terui, K.; Suzuki, H.; et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. 2013. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Hill, V.K.; Kim, J.-S.; Waldman, T. Cohesin mutations in human cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 2016, 1866, 1–11. [Google Scholar] [CrossRef]
- Katainen, R.; Dave, K.; Pitkänen, E.; Palin, K.; Kivioja, T.; Välimäki, N.; Gylfe, A.E.; Ristolainen, H.; Hänninen, U.A.; Cajuso, T.; et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 2015, 47, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-H.; Hou, H.-A.; Tang, J.-L.; Kuo, Y.-Y.; Chiu, Y.-C.; Lin, C.-C.; Liu, C.-Y.; Tseng, M.-H.; Lin, T.-Y.; Liu, M.-C.; et al. Prognostic impacts and dynamic changes of cohesin complex gene mutations in de novo acute myeloid leukemia. Blood Cancer J. 2017, 7, 663. [Google Scholar] [CrossRef] [PubMed]
- Heimbruch, K.E.; Meyer, A.E.; Agrawal, P.; Viny, A.D.; Rao, S. A Cohesive Look at Leukemogenesis: The Cohesin Complex and Other Driving Mutations in Aml. Neoplasia 2021, 23, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Leylek, T.R.; Jeusset, L.M.; Lichtensztejn, Z.; McManus, K.J. Reduced Expression of Genes Regulating Cohesion Induces Chromosome Instability that May Promote Cancer and Impact Patient Outcomes. Sci. Rep. 2020, 10, 592. [Google Scholar] [CrossRef]
- Kim, J.-S.; He, X.; Orr, B.; Wutz, G.; Hill, V.; Peters, J.-M.; Compton, D.A.; Waldman, T. Intact Cohesion, Anaphase, and Chromosome Segregation in Human Cells Harboring Tumor-Derived Mutations in STAG2. PLoS Genet. 2016, 12, e1005865. [Google Scholar] [CrossRef]
- Mullenders, J.; Aranda-Orgilles, B.; Lhoumaud, P.; Keller, M.; Pae, J.; Wang, K.; Kayembe, C.; Rocha, P.P.; Raviram, R.; Gong, Y.; et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 2015, 212, 1833–1850. [Google Scholar] [CrossRef]
- Rittenhouse, N.L.; Carico, Z.M.; Liu, Y.F.; Stefan, H.C.; Arruda, N.L.; Zhou, J.; Dowen, J.M. Functional impact of cancer-associated cohesin variants on gene expression and cellular identity. Genetics 2021, 217. [Google Scholar] [CrossRef]
- Li, X.; Zhang, T.W.; Tang, J.L.; Fa, P.P.; Lu, J.X.; Qi, F.M.; Cai, Z.M.; Liu, C.X.; Sun, X.J. Loss of STAG2 causes aneuploidy in normal human bladder cells. Genet. Mol. Res. 2015, 14, 2638–2646. [Google Scholar] [CrossRef]
- Solomon, D.A.; Kim, T.; Diaz-Martinez, L.A.; Fair, J.; Elkahloun, A.G.; Harris, B.T.; Toretsky, J.A.; Rosenberg, S.A.; Shukla, N.; Ladanyi, M.; et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 2011, 333, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Horsfield, J.A.; Anagnostou, S.H.; Hu, J.K.; Cho, K.H.; Geisler, R.; Lieschke, G.; Crosier, K.E.; Crosier, P.S. Cohesin-dependent regulation of Runx genes. Development 2007, 134, 2639–2649. [Google Scholar] [CrossRef]
- Leeke, B.; Marsman, J.; O’Sullivan, J.M.; Horsfield, J.A. Cohesin mutations in myeloid malignancies: Underlying mechanisms. Exp. Hematol. Oncol. 2014, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Marsman, J.; O’Neill, A.C.; Kao, B.R.; Rhodes, J.M.; Meier, M.; Antony, J.; Monnich, M.; Horsfield, J.A. Cohesin and CTCF differentially regulate spatiotemporal runx1 expression during zebrafish development. Biochim. Biophys. Acta 2014, 1839, 50–61. [Google Scholar] [CrossRef] [PubMed]
- McEwan, M.V.; Eccles, M.R.; Horsfield, J.A. Cohesin Is Required for Activation of MYC by Estradiol. PLoS ONE 2012, 7, e49160. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Song, S.-H.; Kang, J.-Y.; Park, J.; Kim, H.-P.; Han, S.-W.; Kim, T.-Y. Reduced cohesin destabilizes high-level gene amplification by disrupting pre-replication complex bindings in human cancers with chromosomal instability. Nucleic Acids Res. 2016, 44, 558–572. [Google Scholar] [CrossRef]
- Yun, J.; Song, S.H.; Kim, H.P.; Han, S.W.; Yi, E.C.; Kim, T.Y. Dynamic cohesin-mediated chromatin architecture controls epithelial-mesenchymal plasticity in cancer. EMBO Rep. 2016, 17, 1343–1359. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, C.; Shen, Y.; Xavy, S.; Zhao, F.; Reinisch, A.; Li, R.; Corces, M.R.; Flynn, R.A.; Buenrostro, J.D.; Chan, S.M.; et al. Leukemia-Associated Cohesin Mutants Dominantly Enforce Stem Cell Programs and Impair Human Hematopoietic Progenitor Differentiation. Cell Stem Cell 2015, 17, 675–688. [Google Scholar] [CrossRef]
- Rhodes, J.M.; Bentley, F.K.; Print, C.G.; Dorsett, D.; Misulovin, Z.; Dickinson, E.J.; Crosier, K.E.; Crosier, P.S.; Horsfield, J.A. Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev. Biol. 2010, 344, 637–649. [Google Scholar] [CrossRef]
- Galeev, R.; Baudet, A.; Kumar, P.; Rundberg, N.A.; Nilsson, B.; Soneji, S.; Törngren, T.; Borg, Å.; Kvist, A.; Larsson, J. Genome-wide RNAi Screen Identifies Cohesin Genes as Modifiers of Renewal and Differentiation in Human HSCs. Cell Rep. 2016, 14, 2988–3000. [Google Scholar] [CrossRef]
- Fisher, J.B.; Peterson, J.; Reimer, M.; Stelloh, C.; Pulakanti, K.; Gerbec, Z.J.; Abel, A.M.; Strouse, J.M.; Strouse, C.; McNulty, M.; et al. The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of HoxA7 and HoxA9. Leukemia 2017, 31, 712–719. [Google Scholar] [CrossRef]
- Sasca, D.; Yun, H.; Giotopoulos, G.; Szybinski, J.; Evan, T.; Wilson, N.K.; Gerstung, M.; Gallipoli, P.; Green, A.R.; Hills, R.; et al. Cohesin-dependent regulation of gene expression during differentiation is lost in cohesin-mutated myeloid malignancies. Blood 2019, 134, 2195–2208. [Google Scholar] [CrossRef]
- Chen, Z.; Amro, E.M.; Becker, F.; Hölzer, M.; Rasa, S.M.M.; Njeru, S.N.; Han, B.; Di Sanzo, S.; Chen, Y.; Tang, D.; et al. Cohesin-mediated NF-κB signaling limits hematopoietic stem cell self-renewal in aging and inflammation. J. Exp. Med. 2019, 216, 152. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Nitzsche, A.; Paszkowski-Rogacz, M.; Matarese, F.; Janssen-Megens, E.M.; Hubner, N.C.; Schulz, H.; de Vries, I.; Ding, L.; Huebner, N.; Mann, M.; et al. RAD21 Cooperates with Pluripotency Transcription Factors in the Maintenance of Embryonic Stem Cell Identity. PLoS ONE 2011, 6, e19470. [Google Scholar] [CrossRef] [PubMed]
- Noutsou, M.; Li, J.; Ling, J.; Jones, J.; Wang, Y.; Chen, Y.; Sen, G.L. The Cohesin Complex Is Necessary for Epidermal Progenitor Cell Function through Maintenance of Self-Renewal Genes. Cell Rep. 2017, 20, 3005–3013. [Google Scholar] [CrossRef][Green Version]
- Khaminets, A.; Ronnen-Oron, T.; Baldauf, M.; Meier, E.; Jasper, H. Cohesin controls intestinal stem cell identity by maintaining association of Escargot with target promoters. eLife 2020, 9, e48160. [Google Scholar] [CrossRef]
- Surdez, D.; Zaidi, S.; Grossetête, S.; Laud-Duval, K.; Ferre, A.S.; Mous, L.; Vourc’h, T.; Tirode, F.; Pierron, G.; Raynal, V.; et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Xu, H.; Yan, M.; Patra, J.; Natrajan, R.; Yan, Y.; Swagemakers, S.; Tomaszewski, J.M.; Verschoor, S.; Millar, E.K.; van der Spek, P.; et al. Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 2011, 13, R9. [Google Scholar] [CrossRef]
- De Campos Silva, T.; Deb, S.; Xu, H.; Yan, Y.; Fox, S.B.; Mortensen, N.; McKay, M. Effect of RAD21 overexpression on prognosis in KRAS-mutant colorectal carcinomas. J. Clin. Oncol. 2014, 32, 453. [Google Scholar] [CrossRef]
- Xu, W.; Ying, Y.; Shan, L.; Feng, J.; Zhang, S.; Gao, Y.; Xu, X.; Yao, Y.; Zhu, C.; Mao, W. Enhanced expression of cohesin loading factor NIPBL confers poor prognosis and chemotherapy resistance in non-small cell lung cancer. J. Transl. Med. 2015, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Xu, H.; Tuynman, J.; George, J.; Yan, Y.; Li, J.; Ward, R.L.; Mortensen, N.; Hawkins, N.J.; McKay, M.J.; et al. RAD21 cohesin overexpression is a prognostic and predictive marker exacerbating poor prognosis in KRAS mutant colorectal carcinomas. Br. J. Cancer 2014, 110, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yan, Y.; Deb, S.; Rangasamy, D.; Germann, M.; Malaterre, J.; Eder, N.C.; Ward, R.L.; Hawkins, N.J.; Tothill, R.W.; et al. Cohesin Rad21 Mediates Loss of Heterozygosity and Is Upregulated via Wnt Promoting Transcriptional Dysregulation in Gastrointestinal Tumors. Cell Rep. 2014, 9, 1781–1797. [Google Scholar] [CrossRef]
- Su, X.A.; Ma, D.; Parsons, J.V.; Replogle, J.M.; Amatruda, J.F.; Whittaker, C.A.; Stegmaier, K.; Amon, A. RAD21 is a driver of chromosome 8 gain in Ewing sarcoma to mitigate replication stress. Genes Dev. 2021, 35, 556–572. [Google Scholar] [CrossRef]
- Benedetti, L.; Cereda, M.; Monteverde, L.; Desai, N.; Ciccarelli, F.D. Synthetic lethal interaction between the tumour suppressor STAG2 and its paralog STAG1. Oncotarget 2017, 8, 37619–37632. [Google Scholar] [CrossRef]
- Dasgupta, T.; Antony, J.; Braithwaite, A.W.; Horsfield, J.A. HDAC8 Inhibition Blocks SMC3 Deacetylation and Delays Cell Cycle Progression without Affecting Cohesin-dependent Transcription in MCF7 Cancer Cells. J. Biol. Chem. 2016, 291, 12761–12770. [Google Scholar] [CrossRef]
- van der Lelij, P.; Lieb, S.; Jude, J.; Wutz, G.; Santos, C.P.; Falkenberg, K.; Schlattl, A.; Ban, J.; Schwentner, R.; Hoffmann, T.; et al. Synthetic lethality between the cohesin subunits STAG1 and STAG2 in diverse cancer contexts. eLife 2017, 6. [Google Scholar] [CrossRef]
- van der Lelij, P.; Newman, J.A.; Lieb, S.; Jude, J.; Katis, V.; Hoffmann, T.; Hinterndorfer, M.; Bader, G.; Kraut, N.; Pearson, M.A.; et al. STAG1 vulnerabilities for exploiting cohesin synthetic lethality in STAG2-deficient cancers. Life Sci. Alliance 2020, 3, e202000725. [Google Scholar] [CrossRef]
- McLellan, J.L.; O’Neil, N.J.; Barrett, I.; Ferree, E.; van Pel, D.M.; Ushey, K.; Sipahimalani, P.; Bryan, J.; Rose, A.M.; Hieter, P. Synthetic lethality of cohesins with PARPs and replication fork mediators. PLoS Genet. 2012, 8, e1002574. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Valton, A.-L.; Gorelov, R.A.; Vallurupalli, M.; Krill-Burger, J.M.; Holmes, A.; Landers, C.C.; Haydu, J.E.; Malolepsza, E.; Hartigan, C.; et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight 2021, 6, e142149. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, H.; Van der Jeught, K.; Li, Y.; Liu, S.; Zhang, L.; Fang, Y.; Zhang, X.; Radovich, M.; Schneider, B.P.; et al. Somatic mutation of the cohesin complex subunit confers therapeutic vulnerabilities in cancer. J. Clin. Investig. 2018, 128, 2951–2965. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lieberman, P.M. Mechanism of Glycyrrhizic Acid Inhibition of Kaposi’s Sarcoma-Associated Herpesvirus: Disruption of CTCF-Cohesin-Mediated RNA Polymerase II Pausing and Sister Chromatid Cohesion. J. Virol. 2011, 85, 11159–11169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ge, G.; Meyer, R.; Sethi, S.; Basu, D.; Pradhan, S.; Zhao, Y.-J.; Li, X.-N.; Cai, W.-W.; El-Naggar, A.K.; et al. Overexpression of Separase induces aneuploidy and mammary tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 13033–13038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Scorsone, K.; Ge, G.; Kaffes, C.C.; Dobrolecki, L.E.; Mukherjee, M.; Lewis, M.T.; Berg, S.; Stephan, C.C.; Pati, D. Identification and Characterization of Separase Inhibitors (Sepins) for Cancer Therapy. J. Biomol. Screen. 2014, 19, 878–889. [Google Scholar] [CrossRef]
- Zhang, N.; Pati, D. Separase Inhibitor Sepin-1 Inhibits Foxm1 Expression and Breast Cancer Cell Growth. J. Cancer Sci. Ther. 2018, 10, 517. [Google Scholar] [CrossRef]
- Tothova, Z.; Krill-Burger, J.M.; Popova, K.D.; Landers, C.C.; Sievers, Q.L.; Yudovich, D.; Belizaire, R.; Aster, J.C.; Morgan, E.A.; Tsherniak, A.; et al. Multiplex CRISPR/Cas9-Based Genome Editing in Human Hematopoietic Stem Cells Models Clonal Hematopoiesis and Myeloid Neoplasia. Cell Stem Cell 2017, 21, 547–555. [Google Scholar] [CrossRef]
- Heimbruch, K.E.; Fisher, J.B.; Stelloh, C.T.; Phillips, E.; Reimer, M.H., Jr.; Wargolet, A.J.; Meyer, A.E.; Pulakanti, K.; Viny, A.D.; Loppnow, J.J.; et al. DOT1L inhibitors block abnormal self-renewal induced by cohesin loss. Sci. Rep. 2021, 11, 7288. [Google Scholar] [CrossRef]
- Grazioli, P.; Parodi, C.; Mariani, M.; Bottai, D.; Di Fede, E.; Zulueta, A.; Avagliano, L.; Cereda, A.; Tenconi, R.; Wierzba, J.; et al. Lithium as a possible therapeutic strategy for Cornelia de Lange syndrome. Cell Death Discov. 2021, 7, 34. [Google Scholar] [CrossRef]
- Mazzola, M.; Deflorian, G.; Pezzotta, A.; Ferrari, L.; Fazio, G.; Bresciani, E.; Saitta, C.; Ferrari, L.; Fumagalli, M.; Parma, M.; et al. NIPBL: A new player in myeloid cell differentiation. Haematologica 2019, 104, 1332–1341. [Google Scholar] [CrossRef]
- Smith, J.S.; Lappin, K.M.; Craig, S.G.; Liberante, F.G.; Crean, C.M.; McDade, S.S.; Thompson, A.; Mills, K.I.; Savage, K.I. Chronic loss of STAG2 leads to altered chromatin structure contributing to de-regulated transcription in AML. J. Transl. Med. 2020, 18, 339. [Google Scholar] [CrossRef]
- Zhang, W.; Jin, J.; Wang, Y.; Fang, L.; Min, L.; Wang, X.; Ding, L.; Weng, L.; Xiao, T.; Zhou, T.; et al. PD-L1 regulates genomic stability via interaction with cohesin-SA1 in the nucleus. Signal Transduct. Target. Ther. 2021, 6, 81. [Google Scholar] [CrossRef]
- Brough, R.; Bajrami, I.; Vatcheva, R.; Natrajan, R.; Reis-Filho, J.S.; Lord, C.J.; Ashworth, A. APRIN is a cell cycle specific BRCA2-interacting protein required for genome integrity and a predictor of outcome after chemotherapy in breast cancer. EMBO J. 2012, 31, 1160–1176. [Google Scholar] [CrossRef]
- Bailey, M.L.; O’Neil, N.J.; van Pel, D.M.; Solomon, D.A.; Waldman, T.; Hieter, P. Glioblastoma Cells Containing Mutations in the Cohesin Component STAG2 Are Sensitive to PARP Inhibition. Am. Assoc. Cancer Res. 2014, 13, 724–732. [Google Scholar] [CrossRef]
- Chang, J.; Zhang, B.; Heath, H.; Galjart, N.; Wang, X.; Milbrandt, J. Nicotinamide adenine dinucleotide (NAD)–regulated DNA methylation alters CCCTC-binding factor (CTCF)/cohesin binding and transcription at the BDNF locus. Proc. Natl. Acad. Sci. USA 2010, 107, 21836–21841. [Google Scholar] [CrossRef] [PubMed]
- Pherson, M.; Misulovin, Z.; Gause, M.; Dorsett, D. Cohesin occupancy and composition at enhancers and promoters are linked to DNA replication origin proximity in Drosophila. Genome Res. 2019, 29, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Canudas, S.; Smith, S. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J. Cell Biol. 2009, 187, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Chu, P.-Y.; Liao, W.-T.; Wu, M.-Y.; Tsui, K.-H.; Lin, L.-T.; Huang, C.-H.; Chen, L.-L.; Li, C.-J. Glycyrrhizic acid induces human MDA-MB-231 breast cancer cell death and autophagy via the ROS-mitochondrial pathway. Oncol. Rep. 2018, 39, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Qi, J.; Singh, S.; Hua, W.K.; Cai, Q.; Chao, S.W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A.; et al. HDAC8 Inhibition Specifically Targets Inv(16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation. Cell Stem Cell 2015, 17, 597–610. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef]
- Zhang, N.; Sarkar, A.K.; Pati, D. Toxicity study of separase inhibitor–Sepin-1 in Sprague-Dawley rats. Pathol. Res. Pract. 2020, 216, 152730. [Google Scholar] [CrossRef]
- Nagao, K.; Adachi, Y.; Yanagida, M. Separase-mediated cleavage of cohesin at interphase is required for DNA repair. Nature 2004, 430, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Mintzas, K.; Heuser, M. Emerging strategies to target the dysfunctional cohesin complex in cancer. Expert Opin. Ther. Targets 2019, 23, 525–537. [Google Scholar] [CrossRef]
- Kang, J.Y.; Song, S.H.; Yun, J.; Jeon, M.S.; Kim, H.P.; Han, S.W.; Kim, T.Y. Disruption of CTCF/cohesin-mediated high-order chromatin structures by DNA methylation downregulates PTGS2 expression. Oncogene 2015, 34, 5677–5684. [Google Scholar] [CrossRef]
- Nanavaty, V.; Abrash, E.W.; Hong, C.; Park, S.; Fink, E.E.; Li, Z.; Sweet, T.J.; Bhasin, J.M.; Singuri, S.; Lee, B.H.; et al. DNA Methylation Regulates Alternative Polyadenylation via CTCF and the Cohesin Complex. Mol. Cell 2020, 78, 752–764. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, A.S.; Roe, J.S.; Mok, B.Y.L.; Hohmann, A.F.; Shi, J.; Vakoc, C.R. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016, 15, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Olley, G.; Ansari, M.; Bengani, H.; Grimes, G.R.; Rhodes, J.; von Kriegsheim, A.; Blatnik, A.; Stewart, F.J.; Wakeling, E.; Carroll, N.; et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange–like syndrome. Nat. Genet. 2018, 50, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Roe, J.-S.; Mercan, F.; Rivera, K.; Pappin, D.J.; Vakoc, C.R. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol. Cell 2015, 58, 1028–1039. [Google Scholar] [CrossRef]
- Chen, H.-S.; De Leo, A.; Wang, Z.; Kerekovic, A.; Hills, R.; Lieberman, P.M. BET-Inhibitors Disrupt Rad21-Dependent Conformational Control of KSHV Latency. PLOS Pathog. 2017, 13, e1006100. [Google Scholar] [CrossRef] [PubMed]
- Mill, C.P.; Fiskus, W.; DiNardo, C.D.; Qian, Y.; Raina, K.; Rajapakshe, K.; Perera, D.; Coarfa, C.; Kadia, T.M.; Khoury, J.D.; et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood 2019, 134, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Schuster, K.; Leeke, B.; Meier, M.; Wang, Y.; Newman, T.; Burgess, S.; Horsfield, J.A. A neural crest origin for cohesinopathy heart defects. Hum. Mol. Genet. 2015, 24, 7005–7016. [Google Scholar] [CrossRef] [PubMed]
- Oginuma, M.; Harima, Y.; Tarazona, O.A.; Diaz-Cuadros, M.; Michaut, A.; Ishitani, T.; Xiong, F.; Pourquié, O. Intracellular pH controls WNT downstream of glycolysis in amniote embryos. Nature 2020, 584, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Cukrov, D.; Newman, T.A.C.; Leask, M.; Leeke, B.; Sarogni, P.; Patimo, A.; Kline, A.D.; Krantz, I.D.; Horsfield, J.A.; Musio, A. Antioxidant treatment ameliorates phenotypic features of SMC1A-mutated Cornelia de Lange syndrome in vitro and in vivo. Hum. Mol. Genet. 2018, 27, 3002–3011. [Google Scholar] [CrossRef]
- Xu, B.; Lee, K.K.; Zhang, L.; Gerton, J.L. Stimulation of mTORC1 with L-leucine Rescues Defects Associated with Roberts Syndrome. PLoS Genet. 2013, 9, e1003857. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H.; Kim, S.H.; Trousil, S.; Frederick, D.T.; Piris, A.; Yuan, P.; Cai, L.; Gu, L.; Li, M.; Lee, J.H.; et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat. Med. 2016, 22, 1056–1061. [Google Scholar] [CrossRef]
- Carey, A.; Edwards, D.K.t.; Eide, C.A.; Newell, L.; Traer, E.; Medeiros, B.C.; Pollyea, D.A.; Deininger, M.W.; Collins, R.H.; Tyner, J.W.; et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017, 18, 3204–3218. [Google Scholar] [CrossRef]
- Ding, S.; Diep, J.; Feng, N.; Ren, L.; Li, B.; Ooi, Y.S.; Wang, X.; Brulois, K.F.; Yasukawa, L.L.; Li, X.; et al. STAG2 deficiency induces interferon responses via cGAS-STING pathway and restricts virus infection. Nat. Commun. 2018, 9, 1485. [Google Scholar] [CrossRef] [PubMed]
- Siwek, W.; Tehrani, S.S.H.; Mata, J.F.; Jansen, L.E.T. Activation of Clustered IFNγ Target Genes Drives Cohesin-Controlled Transcriptional Memory. Mol. Cell 2020, 80, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Balakrishnan, K.; Malaterre, J.; Beasley, M.; Yan, Y.; Essers, J.; Appeldoorn, E.; Thomaszewski, J.M.; Vazquez, M.; Verschoor, S.; et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 2010, 5, e12112. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Sehrawat, A.; Eroglu, Z.; Somlo, G.; Hickey, R.; Yadav, S.; Liu, X.; Awasthi, Y.C.; Awasthi, S. Role of SMC1 in Overcoming Drug Resistance in Triple Negative Breast Cancer. PLoS ONE 2013, 8, e64338. [Google Scholar] [CrossRef]
- Hurtado-Bagès, S.; Knobloch, G.; Ladurner, A.G.; Buschbeck, M. The taming of PARP1 and its impact on NAD+ metabolism. Mol. Metab. 2020, 38, 100950. [Google Scholar] [CrossRef]
- Bajrami, I.; Kigozi, A.; Van Weverwijk, A.; Brough, R.; Frankum, J.; Lord, C.J.; Ashworth, A. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol. Med. 2012, 4, 1087–1096. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Huang, S.N.; Wutz, G.; Tang, W.; Zagnoli-Vieira, G.; Callen, E.; Wong, N.; Day, A.; Peters, J.M.; et al. Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Mol. Cell 2019, 75, 252–266. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome Organization Drives Chromosome Fragility. Cell 2017, 170, 507–521. [Google Scholar] [CrossRef]
- Gothe, H.J.; Bouwman, B.A.M.; Gusmao, E.G.; Piccinno, R.; Petrosino, G.; Sayols, S.; Drechsel, O.; Minneker, V.; Josipovic, N.; Mizi, A.; et al. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Mol. Cell 2019, 75, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Uusküla-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Agent | Mode of Action | Impact on Cohesin Mutant Cells |
|---|---|---|
| Inactivation of STAG1 | Synthetic lethal | Specific to STAG2 mutant cells [145,147,148,151]. |
| Glycyrrhizic acid | Blocks SMC3 acetylation and interaction with RAD21 [152] | Not tested. |
| PCI-30451 | Inhibits HDAC8 [25,146] | Not tested. |
| Sepin-1 | Inhibits separase | Inhibits growth. Sensitises separase-overexpressing breast cancers [153,154,155]. |
| MK-8745 ZM 44743 | Inhibitors of Aurora kinase B | Differentially inhibits MCF10A cells with deletion mutations in RAD21, SMC3 and STAG2 [80]. |
| P276-00 | Inhibits cyclin-dependent kinase | Differentially inhibits MCF10A cells with deletion mutations in RAD21, SMC3 and STAG2 [80]. |
| Decitabine Azacytidine | Hypomethylating agents | Effective in myeloid dysplasia patients with STAG2 or RAD21 mutations [109]. Differentially inhibits CD34+ cells heterozygous for SMC3 mutation [156]. |
| JQ1 | Bromodomain and extra-terminal (BET) protein inhibitor | Decreases aberrant RUNX1 and ERG transcription in STAG2 mutant K562 leukaemia cells [59]. |
| I-BET-762 RVX-208 | Bromodomain and extra-terminal (BET) protein inhibitor | Differentially inhibits MCF10A cells with deletion mutations in RAD21, SMC3 and STAG2 [80]. |
| EPZ-4777 EPZ-5676 | DOTL1 inhibitors | Blocks abnormal self-renewal of mouse haematopoietic stem cells heterozygous for Rad21 or Smc3 mutation. Reduces aberrant HoxA7/9 expression in cohesin mutant cells [157]. |
| LY209031 | GSK3 inhibitor | Differentially inhibits MCF10A cells with deletion mutations in RAD21, SMC3 and STAG2 [80]. Differentially inhibits CMK STAG2 mutant leukaemia cells [80]. Causes enhanced β-catenin stabilization in cohesin mutant cells [80]. |
| Lithium | GSK3 inhibitor | Rescued cell proliferation defects in Drosophila CdLS model and CdLS lymphoblastoid cells [158]. |
| Indomethacin | Non-steroidal anti-inflammatory Wnt signalling inhibitor | Reverses the proliferation of myeloid progenitors in Nipbl mutant zebrafish [159]. |
| WAY-600 AZD2014 | mTOR inhibitor | Differentially inhibits MCF10A cells with deletion mutations in RAD21, SMC3 and STAG2 [80]. |
| Ipatasertib | BRAF inhibitor | Differentially inhibits MCF10A cells with deletion mutations in RAD21, SMC3 and STAG2 [80]. |
| SAR131675 | VEGFR-3-tyrosine kinase | Differentially inhibits MCF10A cells with deletion mutations in RAD21, SMC3 and STAG2 [80]. |
| VX-702 | P38-MAPK/MEK inhibitor | Differentially inhibits MCF10A cells with deletion mutations in RAD21, SMC3 and STAG2 [80]. |
| Selumetinib Trametinib | P38-MAPK/MEK inhibitors | Differentially inhibits STAG2 mutant OCI-AML3 cells [160]. |
| Interferon | Exogenous addition of interferon | Rescues LPS-induced inflammatory response in Rad21-depleted macrophages [57]. |
| Anti-PDL1 | PDL1 inhibiton | Inhibits growth of triple-negative breast cancer cells with low Sororin and high PDL1 expression [31]. Inhibits cohesin–STAG1 function in HeLa cells [161]. |
| Benzamide Olaparib Veliparib Rucaparib ABT-888 Talazoparib | PARP inhibitors | Differential inhibition in: Cohesin-depleted colon neoplastic cells [149]. PDS5B-depleted breast cancer cells [162]. STAG2 mutant glioblastoma, Ewing sarcoma, hTERT-positive retinal pigmented epithelial cells and leukaemia cells (U937) [15,150,163]. |
| FK866 | Nampt inhibitor. Causes hypermethylation and reduces cohesin binding in neurons [164] | Not tested. |
| Cyclophosphamide 5-fluorouracil | DNA alkylating agents | Differential inhibition in: RAD21-depleted MDA-MB-231 breast cancer cells [139]. |
| Cyclophosphamide Gemcitabine Temozolomide Cisplatin | DNA alkylating agents | STAG2 mutant glioblastoma, Ewing sarcoma, hTERT-positive retinal pigmented epithelial cells [15]. |
| VX-970 AZD6738 | ATR kinase inhibitors | Differentially inhibits STAG2 mutant glioblastoma, Ewing sarcoma, hTERT-positive retinal pigmented epithelial cells [15]. |
| Doxorubicin Etoposide Topotecan | Topoisomerase targeting agents | Differentially inhibits STAG2 mutant glioblastoma, Ewing sarcoma and hTERT-positive retinal pigmented epithelial cells [15]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antony, J.; Chin, C.V.; Horsfield, J.A. Cohesin Mutations in Cancer: Emerging Therapeutic Targets. Int. J. Mol. Sci. 2021, 22, 6788. https://doi.org/10.3390/ijms22136788
Antony J, Chin CV, Horsfield JA. Cohesin Mutations in Cancer: Emerging Therapeutic Targets. International Journal of Molecular Sciences. 2021; 22(13):6788. https://doi.org/10.3390/ijms22136788
Chicago/Turabian StyleAntony, Jisha, Chue Vin Chin, and Julia A. Horsfield. 2021. "Cohesin Mutations in Cancer: Emerging Therapeutic Targets" International Journal of Molecular Sciences 22, no. 13: 6788. https://doi.org/10.3390/ijms22136788
APA StyleAntony, J., Chin, C. V., & Horsfield, J. A. (2021). Cohesin Mutations in Cancer: Emerging Therapeutic Targets. International Journal of Molecular Sciences, 22(13), 6788. https://doi.org/10.3390/ijms22136788