
Can Developments in Tissue Optical Clearing Aid Super-Resolution Microscopy Imaging?

 , ,
, ,
Abstract
:1. Introduction
2. Overview of the Existing Methods
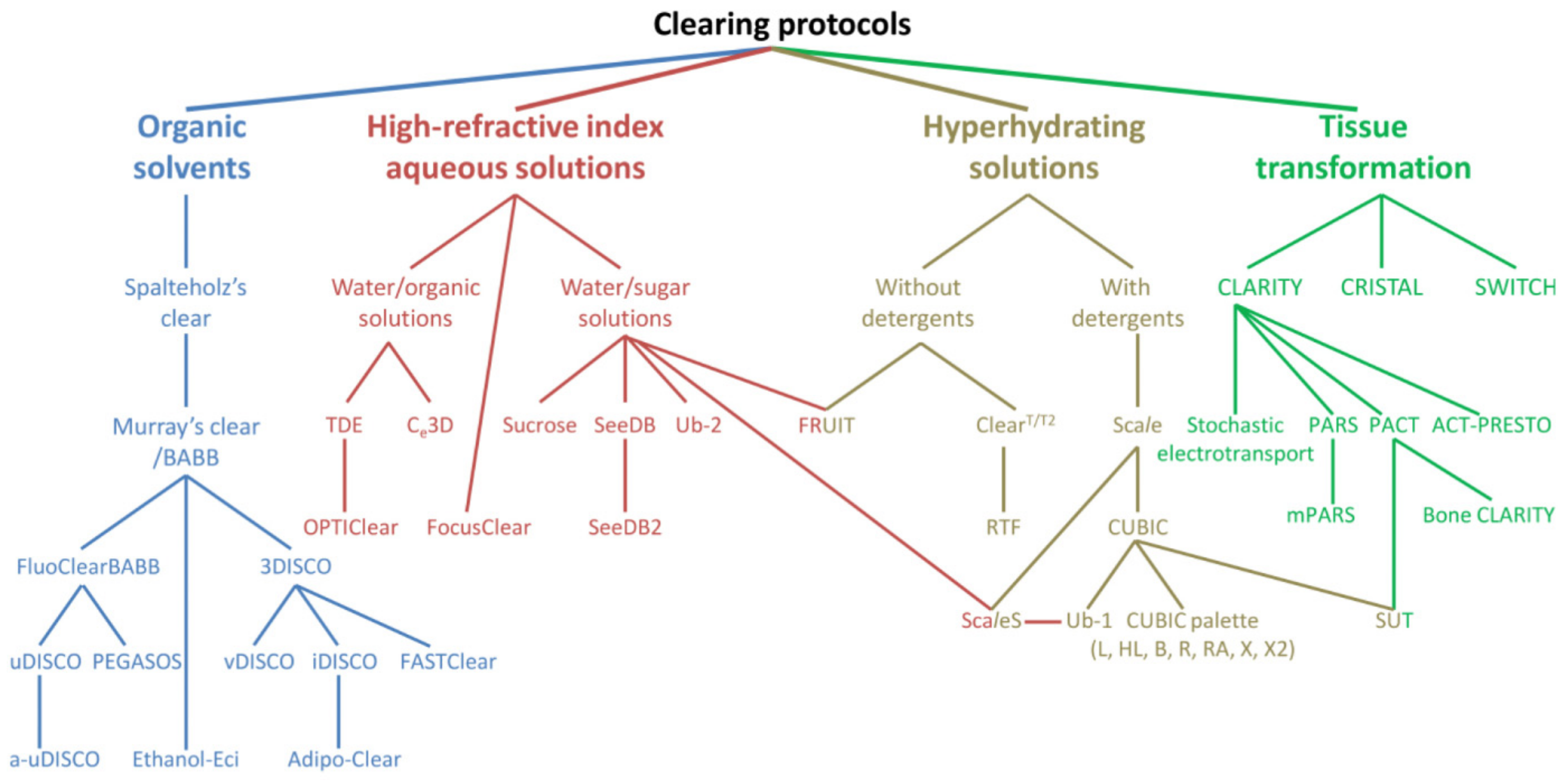
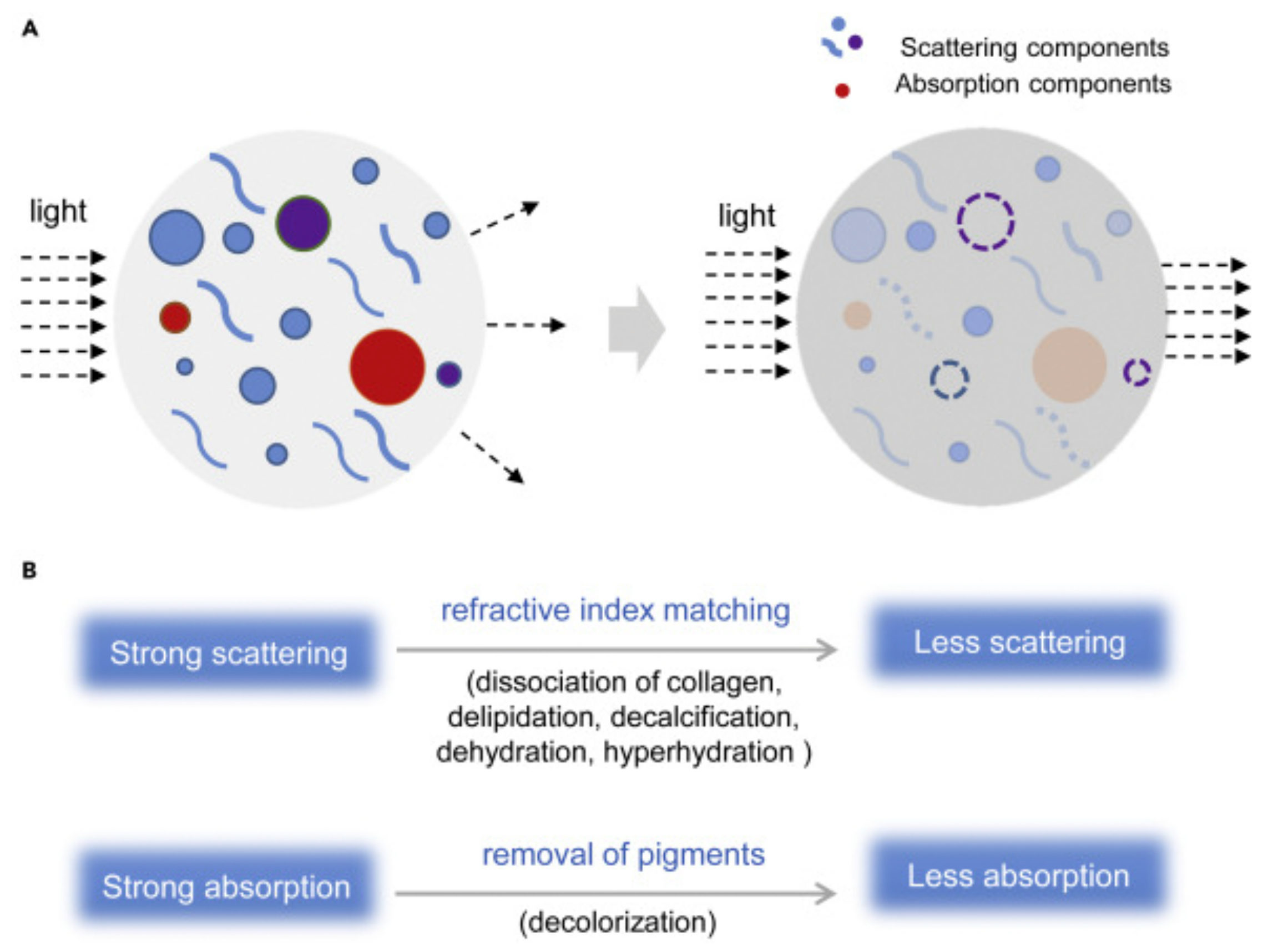
2.1. Tissue Optical Clearing (TOC) Techniques
2.2. SRM Techniques
3. Approaches to Address Technical Challenges in SRM of Thick Samples with Tissue Clearing
3.1. Spherical Aberration
3.2. Suboptimal Intensity of Fluorescence
3.3. Insufficient and Heterogeneous Molecular Probe Labelling
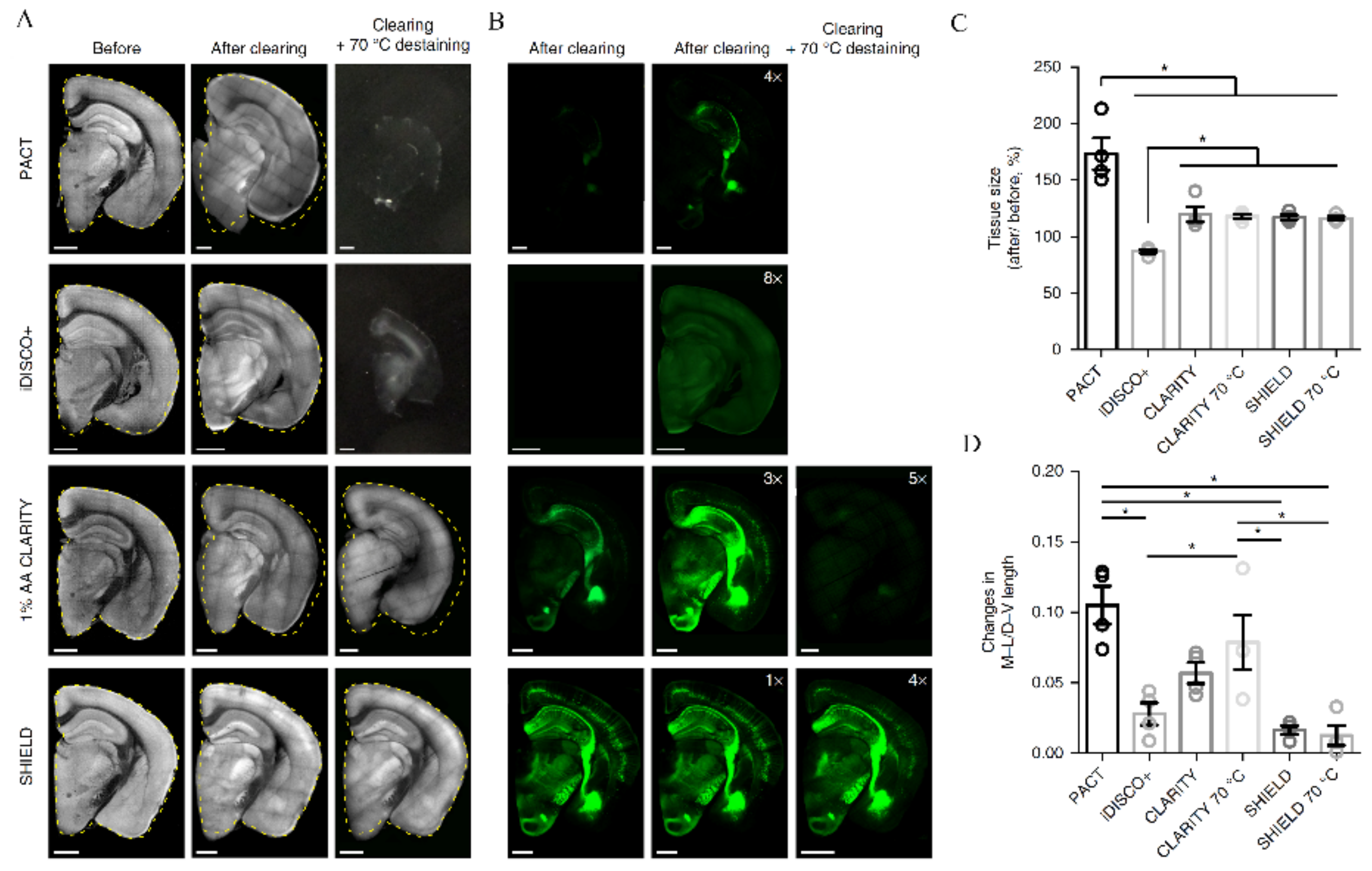
4. Limitations of TOC in the Context of SRM
5. Selected Applications of TOC in SRM
6. Summary and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, D.J. Progress and Perspectives for Atomic-Resolution Electron Microscopy. Ultramicroscopy 2008, 108, 159–166. [Google Scholar] [CrossRef]
- Smith, D.J. Ultimate Resolution in the Electron Microscope? Mater. Today 2008, 11, 30–38. [Google Scholar] [CrossRef]
- Susaki, E.A.; Tainaka, K.; Perrin, D.; Yukinaga, H.; Kuno, A.; Ueda, H.R. Advanced CUBIC Protocols for Whole-Brain and Whole-Body Clearing and Imaging. Nat. Protoc. 2015, 10, 1709–1727. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.-H.; Tang, W.-C.; Liu, Y.-T.; Chang, S.-W.; Wu, F.C.M.; Chen, C.-Y.; Tsai, Y.-C.; Yang, S.-M.; Kuo, C.-W.; Okada, Y.; et al. Lightsheet Localization Microscopy Enables Fast, Large-Scale, and Three-Dimensional Super-Resolution Imaging. Commun. Biol. 2019, 2, 1–10. [Google Scholar] [CrossRef]
- Susaki, E.A.; Ueda, H.R. Whole-Body and Whole-Organ Clearing and Imaging Techniques with Single-Cell Resolution: Toward Organism-Level Systems Biology in Mammals. Cell Chem. Biol. 2016, 23, 137–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möckl, L.; Moerner, W.E. Super-Resolution Microscopy with Single Molecules in Biology and Beyond–Essentials, Current Trends, and Future Challenges. J. Am. Chem. Soc. 2020, 142, 17828–17844. [Google Scholar] [CrossRef] [PubMed]
- Sydor, A.M.; Czymmek, K.J.; Puchner, E.M.; Mennella, V. Super-Resolution Microscopy: From Single Molecules to Supramolecular Assemblies. Trends Cell Biol. 2015, 25, 730–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangindertael, J.; Camacho, R.; Sempels, W.; Mizuno, H.; Dedecker, P.; Janssen, K.P.F. An Introduction to Optical Super-Resolution Microscopy for the Adventurous Biologist. Methods Appl. Fluoresc. 2018, 6, 022003. [Google Scholar] [CrossRef]
- Igarashi, M.; Nozumi, M.; Wu, L.-G.; Cella Zanacchi, F.; Katona, I.; Barna, L.; Xu, P.; Zhang, M.; Xue, F.; Boyden, E. New Observations in Neuroscience Using Superresolution Microscopy. J. Neurosci. 2018, 38, 9459–9467. [Google Scholar] [CrossRef]
- Okada, Y. Current Limitations in Super-Resolution Fluorescence Microscopy for Biological Specimens: How Deep Can We Go from the Cover Glass? In Proceedings of the Biomedical Imaging and Sensing Conference; International Society for Optics and Photonics, Yokohama, Japan, 18 April 2017. [Google Scholar]
- Gao, R.; Asano, S.M.; Upadhyayula, S.; Pisarev, I.; Milkie, D.E.; Liu, T.-L.; Singh, V.; Graves, A.; Huynh, G.H.; Zhao, Y.; et al. Cortical Column and Whole-Brain Imaging with Molecular Contrast and Nanoscale Resolution. Science 2019, 363. [Google Scholar] [CrossRef]
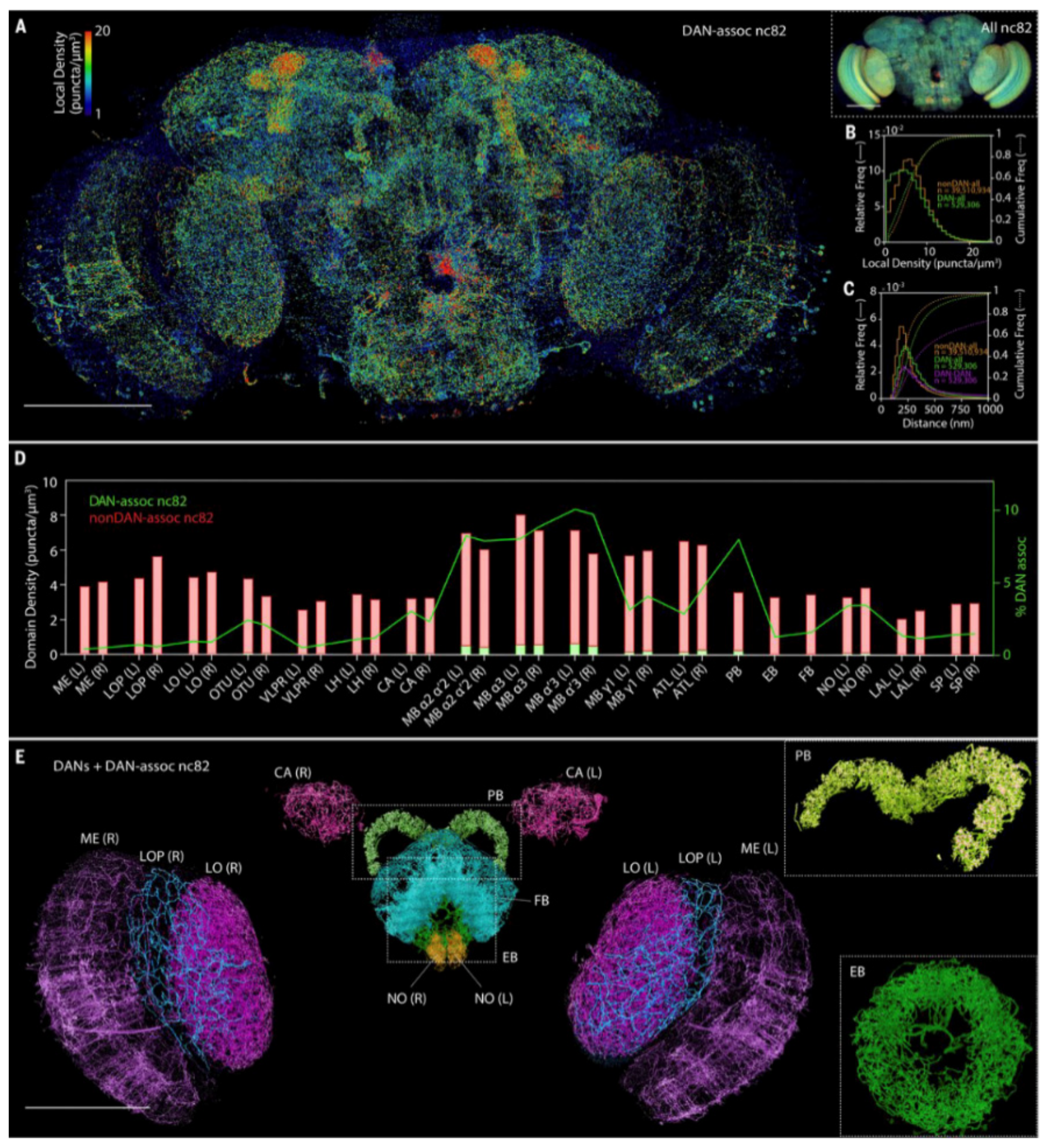
- Lin, H.-Y.; Chu, L.-A.; Yang, H.; Hsu, K.-J.; Lin, Y.-Y.; Lin, K.-H.; Chu, S.-W.; Chiang, A.-S. Imaging through the Whole Brain of Drosophila at λ/20 Super-Resolution. iScience 2019, 14, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Costantini, I.; Cicchi, R.; Silvestri, L.; Vanzi, F.; Pavone, F.S. In-Vivo and Ex-Vivo Optical Clearing Methods for Biological Tissues: Review. Biomed. Opt. Express 2019, 10, 5251–5267. [Google Scholar] [CrossRef]
- Sdobnov, A.Y.; Darvin, M.E.; Genina, E.A.; Bashkatov, A.N.; Lademann, J.; Tuchin, V.V. Recent Progress in Tissue Optical Clearing for Spectroscopic Application. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2018, 197, 216–229. [Google Scholar] [CrossRef]
- Dodt, H.-U.; Leischner, U.; Schierloh, A.; Jährling, N.; Mauch, C.P.; Deininger, K.; Deussing, J.M.; Eder, M.; Zieglgänsberger, W.; Becker, K. Ultramicroscopy: Three-Dimensional Visualization of Neuronal Networks in the Whole Mouse Brain. Nat. Methods 2007, 4, 331–336. [Google Scholar] [CrossRef]
- Tsai, P.S.; Kaufhold, J.P.; Blinder, P.; Friedman, B.; Drew, P.J.; Karten, H.J.; Lyden, P.D.; Kleinfeld, D. Correlations of Neuronal and Microvascular Densities in Murine Cortex Revealed by Direct Counting and Colocalization of Nuclei and Vessels. J. Neurosci. 2009, 29, 14553–14570. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.; Wallace, J.; Kim, S.-Y.; Kalyanasundaram, S.; Andalman, A.S.; Davidson, T.J.; Mirzabekov, J.J.; Zalocusky, K.A.; Mattis, J.; Denisin, A.K.; et al. Structural and Molecular Interrogation of Intact Biological Systems. Nature 2013, 497, 332–337. [Google Scholar] [CrossRef]
- Susaki, E.A.; Tainaka, K.; Perrin, D.; Kishino, F.; Tawara, T.; Watanabe, T.M.; Yokoyama, C.; Onoe, H.; Eguchi, M.; Yamaguchi, S.; et al. Whole-Brain Imaging with Single-Cell Resolution Using Chemical Cocktails and Computational Analysis. Cell 2014, 157, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Matryba, P.; Kaczmarek, L.; Gołąb, J. Advances in Ex Situ Tissue Optical Clearing. Laser Photonics Rev. 2019, 13, 1800292. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ma, Y.; Yu, T.; Zhu, D. Quantitative Assessment of Optical Clearing Methods in Various Intact Mouse Organs. J. Biophotonics 2019, 12, e201800134. [Google Scholar] [CrossRef]
- Yi, Y.; Men, Y.; Jing, D.; Luo, W.; Zhang, S.; Feng, J.Q.; Liu, J.; Ge, W.-P.; Wang, J.; Zhao, H. 3-Dimensional Visualization of Implant-Tissue Interface with the Polyethylene Glycol Associated Solvent System Tissue Clearing Method. Cell Prolif. 2019, 52, e12578. [Google Scholar] [CrossRef] [Green Version]
- DePas, W.H.; Starwalt-Lee, R.; Van Sambeek, L.; Ravindra Kumar, S.; Gradinaru, V.; Newman, D.K. Exposing the Three-Dimensional Biogeography and Metabolic States of Pathogens in Cystic Fibrosis Sputum via Hydrogel Embedding, Clearing, and RRNA Labeling. mBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Höök, P.; Brito-Robinson, T.; Kim, O.; Narciso, C.; Goodson, H.V.; Weisel, J.W.; Alber, M.S.; Zartman, J.J. Whole Blood Clot Optical Clearing for Nondestructive 3D Imaging and Quantitative Analysis. Biomed. Opt. Express 2017, 8, 3671–3686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.S.; Lichtman, J.W. Clarifying Tissue Clearing. Cell 2015, 162, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Zhu, J.; Li, D.; Zhu, D. Physical and Chemical Mechanisms of Tissue Optical Clearing. iScience 2021, 24, 102178. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gaviro, M.V.; Sanderson, D.; Ripoll, J.; Desco, M. Biomedical Applications of Tissue Clearing and Three-Dimensional Imaging in Health and Disease. iScience 2020, 23, 101432. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Costantini, I.; Sacconi, L.; Pavone, F.S. Clearing of Fixed Tissue: A Review from a Microscopist’s Perspective. J. Biomed. Opt. 2016, 21, 081205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, D.; Zhang, S.; Luo, W.; Gao, X.; Men, Y.; Ma, C.; Liu, X.; Yi, Y.; Bugde, A.; Zhou, B.O.; et al. Tissue Clearing of Both Hard and Soft Tissue Organs with the PEGASOS Method. Cell Res. 2018, 28, 803–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKey, J.; Cameron, L.A.; Lewis, D.; Batchvarov, I.S.; Capel, B. Combined IDISCO and CUBIC Tissue Clearing and Lightsheet Microscopy for in Toto Analysis of the Adult Mouse Ovary†. Biol. Reprod. 2020, 102, 1080–1089. [Google Scholar] [CrossRef]
- Matryba, P.; Wolny, A.; Pawłowska, M.; Sosnowska, A.; Rydzyńska, Z.; Jasiński, M.; Stefaniuk, M.; Gołąb, J. Tissue Clearing-Based Method for Unobstructed Three-Dimensional Imaging of Mouse Penis with Subcellular Resolution. J. Biophotonics 2020, 13, e202000072. [Google Scholar] [CrossRef]
- Orlich, M.; Kiefer, F. A Qualitative Comparison of Ten Tissue Clearing Techniques. Histol. Histopathol. 2018, 33, 181–199. [Google Scholar] [CrossRef]
- Wan, P.; Zhu, J.; Xu, J.; Li, Y.; Yu, T.; Zhu, D. Evaluation of Seven Optical Clearing Methods in Mouse Brain. Neurophotonics 2018, 5, 035007. [Google Scholar] [CrossRef] [Green Version]
- Matryba, P.; Sosnowska, A.; Wolny, A.; Bozycki, L.; Greig, A.; Grzybowski, J.; Stefaniuk, M.; Nowis, D.; Gołąb, J. Systematic Evaluation of Chemically Distinct Tissue Optical Clearing Techniques in Murine Lymph Nodes. J. Immunol. 2020, 204, 1395–1407. [Google Scholar] [CrossRef]
- Pan, C.; Cai, R.; Quacquarelli, F.P.; Ghasemigharagoz, A.; Lourbopoulos, A.; Matryba, P.; Plesnila, N.; Dichgans, M.; Hellal, F.; Ertürk, A. Shrinkage-Mediated Imaging of Entire Organs and Organisms Using UDISCO. Nat. Methods 2016, 13, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Alon, S.; Huynh, G.H.; Boyden, E.S. Expansion Microscopy: Enabling Single Cell Analysis in Intact Biological Systems. FEBS J. 2019, 286, 1482–1494. [Google Scholar] [CrossRef]
- Tainaka, K.; Murakami, T.C.; Susaki, E.A.; Shimizu, C.; Saito, R.; Takahashi, K.; Hayashi-Takagi, A.; Sekiya, H.; Arima, Y.; Nojima, S.; et al. Chemical Landscape for Tissue Clearing Based on Hydrophilic Reagents. Cell Rep. 2018, 24, 2196–2210.e9. [Google Scholar] [CrossRef] [Green Version]
- Schermelleh, L.; Ferrand, A.; Huser, T.; Eggeling, C.; Sauer, M.; Biehlmaier, O.; Drummen, G.P.C. Super-Resolution Microscopy Demystified. Nat. Cell Biol. 2019, 21, 72–84. [Google Scholar] [CrossRef]
- Sigal, Y.M.; Zhou, R.; Zhuang, X. Visualizing and Discovering Cellular Structures with Super-Resolution Microscopy. Science 2018, 361, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Bates, M.; Zhuang, X. Super Resolution Fluorescence Microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef] [Green Version]
- Egner, A.; Hell, S.W. Fluorescence Microscopy with Super-Resolved Optical Sections. Trends Cell Biol. 2005, 15, 207–215. [Google Scholar] [CrossRef]
- Bewersdorf, J.; Schmidt, R.; Hell, S.W. Comparison of I5M and 4Pi-Microscopy. J. Microsc. 2006, 222, 105–117. [Google Scholar] [CrossRef]
- Szczurek, A.; Contu, F.; Hoang, A.; Dobrucki, J.; Mai, S. Aqueous Mounting Media Increasing Tissue Translucence Improve Image Quality in Structured Illumination Microscopy of Thick Biological Specimen. Sci. Rep. 2018, 8, 13971. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Kawakami, R.; Shigemoto, R.; Nemoto, T. Super-resolution Structural Analysis of Dendritic Spines Using Three-dimensional Structured Illumination Microscopy in Cleared Mouse Brain Slices. Eur. J. Neurosci. 2018, 47, 1033–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halpern, A.R.; Alas, G.C.M.; Chozinski, T.J.; Paredez, A.R.; Vaughan, J.C. Hybrid Structured Illumination Expansion Microscopy Reveals Microbial Cytoskeleton Organization. ACS Nano 2017, 11, 12677–12686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.-C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A.; Liu, Z.; et al. Lattice Light-Sheet Microscopy: Imaging Molecules to Embryos at High Spatiotemporal Resolution. Science 2014, 346. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, E.J. A Surface Contact Microscope for the Study of Cell Movements. Nature 1956, 178, 1194. [Google Scholar] [CrossRef]
- Hell, S.; Stelzer, E.H.K. Properties of a 4Pi Confocal Fluorescence Microscope. J. Opt. Soc. Am. A JOSAA 1992, 9, 2159–2166. [Google Scholar] [CrossRef]
- Gustafsson, M.G. Surpassing the Lateral Resolution Limit by a Factor of Two Using Structured Illumination Microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Dertinger, T.; Colyer, R.; Iyer, G.; Weiss, S.; Enderlein, J. Fast, Background-Free, 3D Super-Resolution Optical Fluctuation Imaging (SOFI). Proc. Natl. Acad. Sci. USA 2009, 106, 22287–22292. [Google Scholar] [CrossRef] [Green Version]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence Microscopy with Diffraction Resolution Barrier Broken by Stimulated Emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Tillberg, P.W.; Boyden, E.S. Optical Imaging. Expansion Microscopy. Science 2015, 347, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Hell, S.W.; Willig, K.; Dyba, M.; Jakobs, S.; Kastrup, L.; Westphal, V. Nanoscale resolution with focused light: STED and other RESOLFT microscopy concepts. In Handbook of Biological Confocal Microscopy; Pawley, J.B., Ed.; Springer Science + Business Media: New York, NY, USA, 2006; pp. 571–579. [Google Scholar]
- Angibaud, J.; Mascalchi, P.; Poujol, C.; Nägerl, U.V. A Simple Tissue Clearing Method for Increasing the Depth Penetration of STED Microscopy of Fixed Brain Slices. J. Phys. D Appl. Phys. 2020, 53, 184001. [Google Scholar] [CrossRef]
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence Nanoscopy in Cell Biology. Nat. Rev. Mol. Cell Biol. 2017, 18, 685–701. [Google Scholar] [CrossRef]
- Zwettler, F.U.; Reinhard, S.; Gambarotto, D.; Bell, T.D.M.; Hamel, V.; Guichard, P.; Sauer, M. Molecular Resolution Imaging by Post-Labeling Expansion Single-Molecule Localization Microscopy (Ex-SMLM). Nat. Commun. 2020, 11, 3388. [Google Scholar] [CrossRef]
- Ke, M.-T.; Nakai, Y.; Fujimoto, S.; Takayama, R.; Yoshida, S.; Kitajima, T.S.; Sato, M.; Imai, T. Super-Resolution Mapping of Neuronal Circuitry With an Index-Optimized Clearing Agent. Cell Rep. 2016, 14, 2718–2732. [Google Scholar] [CrossRef] [Green Version]
- Bekkouche, B.M.B.; Fritz, H.K.M.; Rigosi, E.; O’Carroll, D.C. Comparison of Transparency and Shrinkage During Clearing of Insect Brains Using Media With Tunable Refractive Index. Front. Neuroanat. 2020, 14. [Google Scholar] [CrossRef]
- Glaser, A.K.; Reder, N.P.; Chen, Y.; Yin, C.; Wei, L.; Kang, S.; Barner, L.A.; Xie, W.; McCarty, E.F.; Mao, C.; et al. Multi-Immersion Open-Top Light-Sheet Microscope for High-Throughput Imaging of Cleared Tissues. Nat. Commun. 2019, 10, 2781. [Google Scholar] [CrossRef] [Green Version]
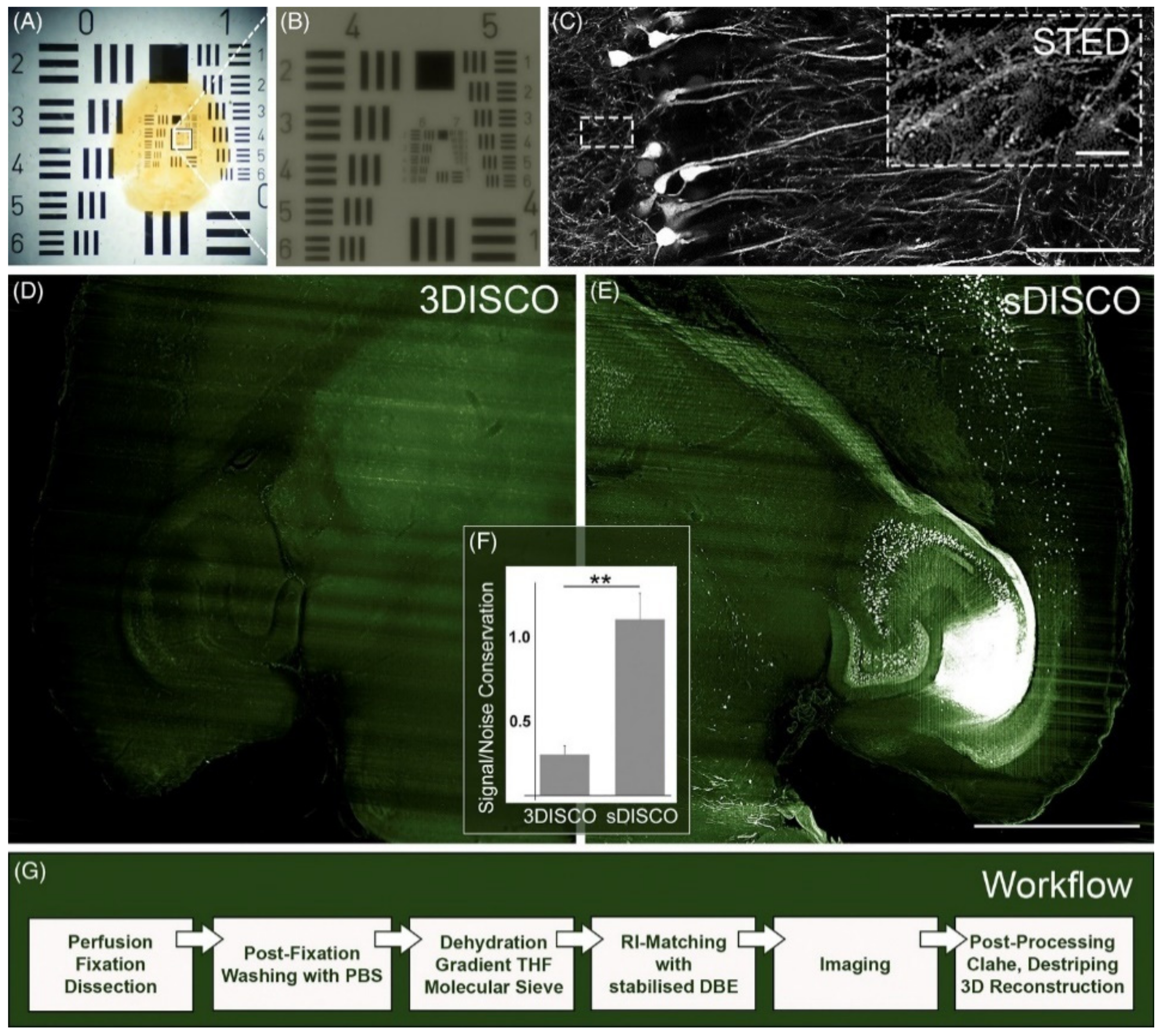
- Hahn, C.; Becker, K.; Saghafi, S.; Pende, M.; Avdibašić, A.; Foroughipour, M.; Heinz, D.E.; Wotjak, C.T.; Dodt, H.-U. High-Resolution Imaging of Fluorescent Whole Mouse Brains Using Stabilised Organic Media (SDISCO). J. Biophotonics 2019, 12, e201800368. [Google Scholar] [CrossRef] [Green Version]
- Sauerbeck, A.D.; Gangolli, M.; Reitz, S.J.; Salyards, M.H.; Kim, S.H.; Hemingway, C.; Gratuze, M.; Makkapati, T.; Kerschensteiner, M.; Holtzman, D.M.; et al. SEQUIN Multiscale Imaging of Mammalian Central Synapses Reveals Loss of Synaptic Connectivity Resulting from Diffuse Traumatic Brain Injury. Neuron 2020, 107, 257–273.e5. [Google Scholar] [CrossRef]
- Narasimhan, A.; Venkataraju, K.U.; Mizrachi, J.; Albeanu, D.F.; Osten, P. Oblique Light-Sheet Tomography: Fast and High Resolution Volumetric Imaging of Mouse Brains. bioRxiv 2017, 132423. [Google Scholar] [CrossRef] [Green Version]
- Mizrachi, J.; Narasimhan, A.; Qi, X.; Drewes, R.; Palaniswamy, R.; Wu, Z.; Osten, P. Super-Resolution Light-Sheet Fluorescence Microscopy by SOFI. bioRxiv 2020. [Google Scholar] [CrossRef]
- Van der Velde, J.H.M.; Oelerich, J.; Huang, J.; Smit, J.H.; Aminian Jazi, A.; Galiani, S.; Kolmakov, K.; Gouridis, G.; Eggeling, C.; Herrmann, A.; et al. A Simple and Versatile Design Concept for Fluorophore Derivatives with Intramolecular Photostabilization. Nat. Commun. 2016, 7, 10144. [Google Scholar] [CrossRef] [PubMed]
- Donnert, G.; Keller, J.; Medda, R.; Andrei, M.A.; Rizzoli, S.O.; Lührmann, R.; Jahn, R.; Eggeling, C.; Hell, S.W. Macromolecular-Scale Resolution in Biological Fluorescence Microscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 11440–11445. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.; Zahn, J.; Maglione, M.; Sigrist, S.J.; Marquard, J.; Chojnacki, J.; Kräusslich, H.-G.; Sahl, S.J.; Engelhardt, J.; Hell, S.W. Ultrafast, Temporally Stochastic STED Nanoscopy of Millisecond Dynamics. Nat. Methods 2015, 12, 827–830. [Google Scholar] [CrossRef]
- Beater, S.; Holzmeister, P.; Pibiri, E.; Lalkens, B.; Tinnefeld, P. Choosing Dyes for Cw-STED Nanoscopy Using Self-Assembled Nanorulers. Phys. Chem. Chem. Phys. 2014, 16, 6990–6996. [Google Scholar] [CrossRef] [Green Version]
- Ertürk, A.; Becker, K.; Jährling, N.; Mauch, C.P.; Hojer, C.D.; Egen, J.G.; Hellal, F.; Bradke, F.; Sheng, M.; Dodt, H.-U. Three-Dimensional Imaging of Solvent-Cleared Organs Using 3DISCO. Nat. Protoc. 2012, 7, 1983–1995. [Google Scholar] [CrossRef]
- Kirschnick, N.; Drees, D.; Redder, E.; Erapaneedi, R.; Pereira da Graca, A.; Schäfers, M.; Jiang, X.; Kiefer, F. Rapid Methods for the Evaluation of Fluorescent Reporters in Tissue Clearing and the Segmentation of Large Vascular Structures. iScience 2021, 24, 102650. [Google Scholar] [CrossRef]
- Schwarz, M.K.; Scherbarth, A.; Sprengel, R.; Engelhardt, J.; Theer, P.; Giese, G. Fluorescent-Protein Stabilization and High-Resolution Imaging of Cleared, Intact Mouse Brains. PLoS ONE 2015, 10, e0124650. [Google Scholar] [CrossRef] [Green Version]
- Dennison, C.; Lovrien, R. Three Phase Partitioning: Concentration and Purification of Proteins. Protein Expr. Purif. 1997, 11, 149–161. [Google Scholar] [CrossRef]
- Li, Y.; Xu, J.; Wan, P.; Yu, T.; Zhu, D. Optimization of GFP Fluorescence Preservation by a Modified UDISCO Clearing Protocol. Front. Neuroanat. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Masselink, W.; Reumann, D.; Murawala, P.; Pasierbek, P.; Taniguchi, Y.; Bonnay, F.; Meixner, K.; Knoblich, J.A.; Tanaka, E.M. Broad Applicability of a Streamlined Ethyl Cinnamate-Based Clearing Procedure. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Klingberg, A.; Hasenberg, A.; Ludwig-Portugall, I.; Medyukhina, A.; Männ, L.; Brenzel, A.; Engel, D.R.; Figge, M.T.; Kurts, C.; Gunzer, M. Fully Automated Evaluation of Total Glomerular Number and Capillary Tuft Size in Nephritic Kidneys Using Lightsheet Microscopy. J. Am. Soc. Nephrol. 2017, 28, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Yu, T.; Xu, J.; Wan, P.; Ma, Y.; Zhu, J.; Li, Y.; Gong, H.; Luo, Q.; Zhu, D. FDISCO: Advanced Solvent-Based Clearing Method for Imaging Whole Organs. Sci. Adv. 2019, 5, 8355. [Google Scholar] [CrossRef] [Green Version]
- Epp, J.R.; Niibori, Y.; Liz Hsiang, H.-L.; Mercaldo, V.; Deisseroth, K.; Josselyn, S.A.; Frankland, P.W. Optimization of CLARITY for Clearing Whole-Brain and Other Intact Organs(1,2,3). eNeuro 2015, 2. [Google Scholar] [CrossRef] [Green Version]
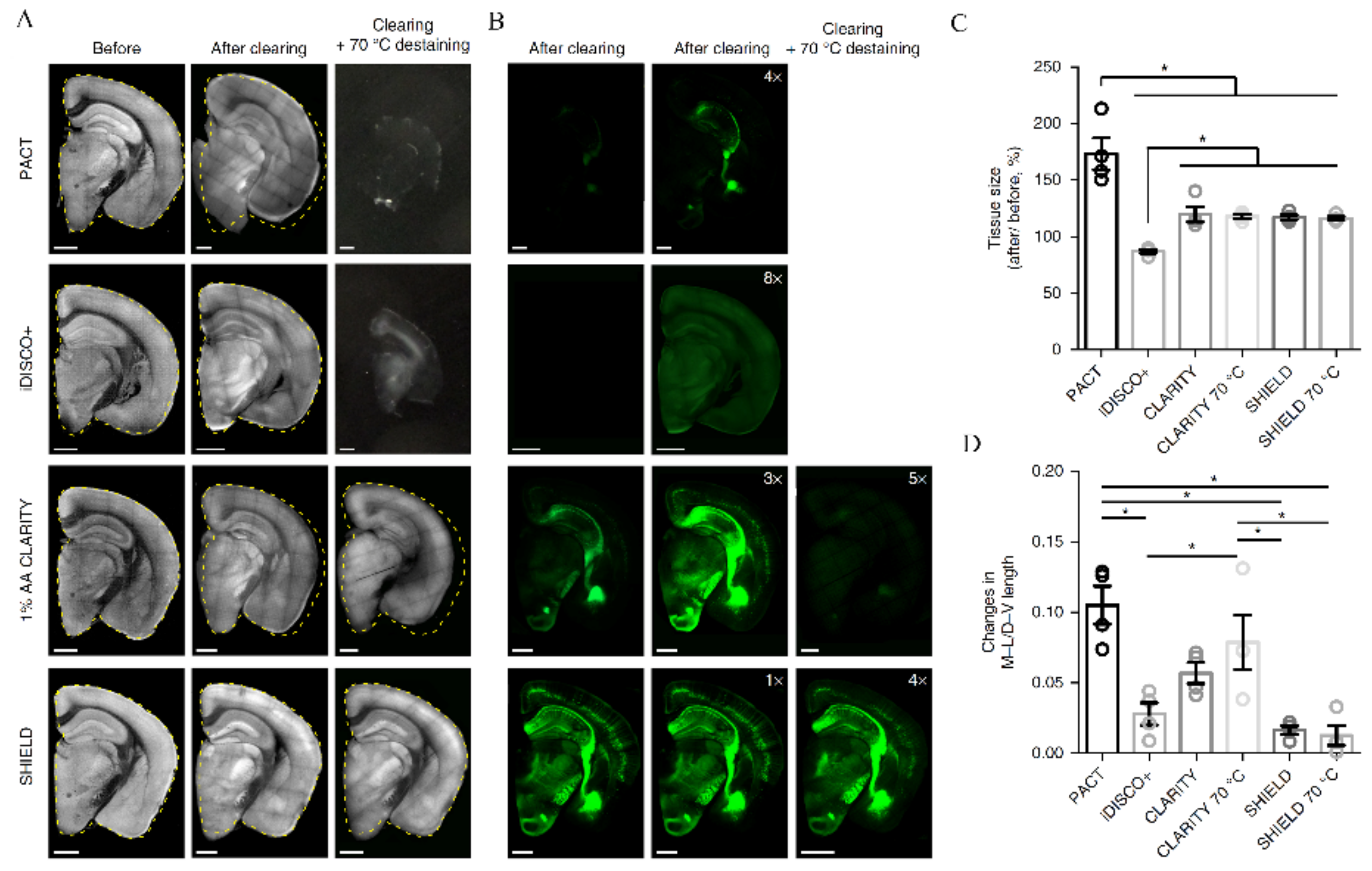
- Yu, T.; Qi, Y.; Zhu, J.; Xu, J.; Gong, H.; Luo, Q.; Zhu, D. Elevated-Temperature-Induced Acceleration of PACT Clearing Process of Mouse Brain Tissue. Sci. Rep. 2017, 7, 38848. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.-G.; Sohn, C.H.; Chen, R.; McCue, M.; Yun, D.H.; Drummond, G.T.; Ku, T.; Evans, N.B.; Oak, H.C.; Trieu, W.; et al. Protection of Tissue Physicochemical Properties Using Polyfunctional Crosslinkers. Nat. Biotechnol. 2019, 37, 73–83. [Google Scholar] [CrossRef]
- Sakaguchi, R.; Leiwe, M.N.; Imai, T. Bright Multicolor Labeling of Neuronal Circuits with Fluorescent Proteins and Chemical Tags. Elife 2018, 7. [Google Scholar] [CrossRef]
- Kingston, B.R.; Syed, A.M.; Ngai, J.; Sindhwani, S.; Chan, W.C.W. Assessing Micrometastases as a Target for Nanoparticles Using 3D Microscopy and Machine Learning. Proc. Natl. Acad. Sci. USA 2019, 116, 14937–14946. [Google Scholar] [CrossRef] [Green Version]
- García-Pinel, B.; Porras-Alcalá, C.; Ortega-Rodríguez, A.; Sarabia, F.; Prados, J.; Melguizo, C.; López-Romero, J.M. Lipid-Based Nanoparticles: Application and Recent Advances in Cancer Treatment. Nanomaterials 2019, 9, 638. [Google Scholar] [CrossRef] [Green Version]
- Syed, A.M.; MacMillan, P.; Ngai, J.; Wilhelm, S.; Sindhwani, S.; Kingston, B.R.; Wu, J.L.Y.; Llano-Suárez, P.; Lin, Z.P.; Ouyang, B.; et al. Liposome Imaging in Optically Cleared Tissues. Nano Lett. 2020, 20, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.; Nabel, E.M.; Murdock, M.H.; Lao-Peregrin, C.; Tsoulfas, P.; Blackmore, M.G.; Lee, F.S.; Liston, C.; Morishita, H.; Petsko, G.A. MGreenLantern: A Bright Monomeric Fluorescent Protein with Rapid Expression and Cell Filling Properties for Neuronal Imaging. Proc. Natl. Acad. Sci. USA 2020, 117, 30710–30721. [Google Scholar] [CrossRef]
- Scott, D.J.; Gunn, N.J.; Yong, K.J.; Wimmer, V.C.; Veldhuis, N.A.; Challis, L.M.; Haidar, M.; Petrou, S.; Bathgate, R.A.D.; Griffin, M.D.W. A Novel Ultra-Stable, Monomeric Green Fluorescent Protein For Direct Volumetric Imaging of Whole Organs Using CLARITY. Sci. Rep. 2018, 8, 667. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brenna, C.; Daniele, C.; Rudolf, R.; Heuveline, V.; Gretz, N. A Cationic near Infrared Fluorescent Agent and Ethyl-Cinnamate Tissue Clearing Protocol for Vascular Staining and Imaging. Sci. Rep. 2019, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Legant, W.R.; Shao, L.; Grimm, J.B.; Brown, T.A.; Milkie, D.E.; Avants, B.B.; Lavis, L.D.; Betzig, E. High-Density Three-Dimensional Localization Microscopy across Large Volumes. Nat. Methods 2016, 13, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
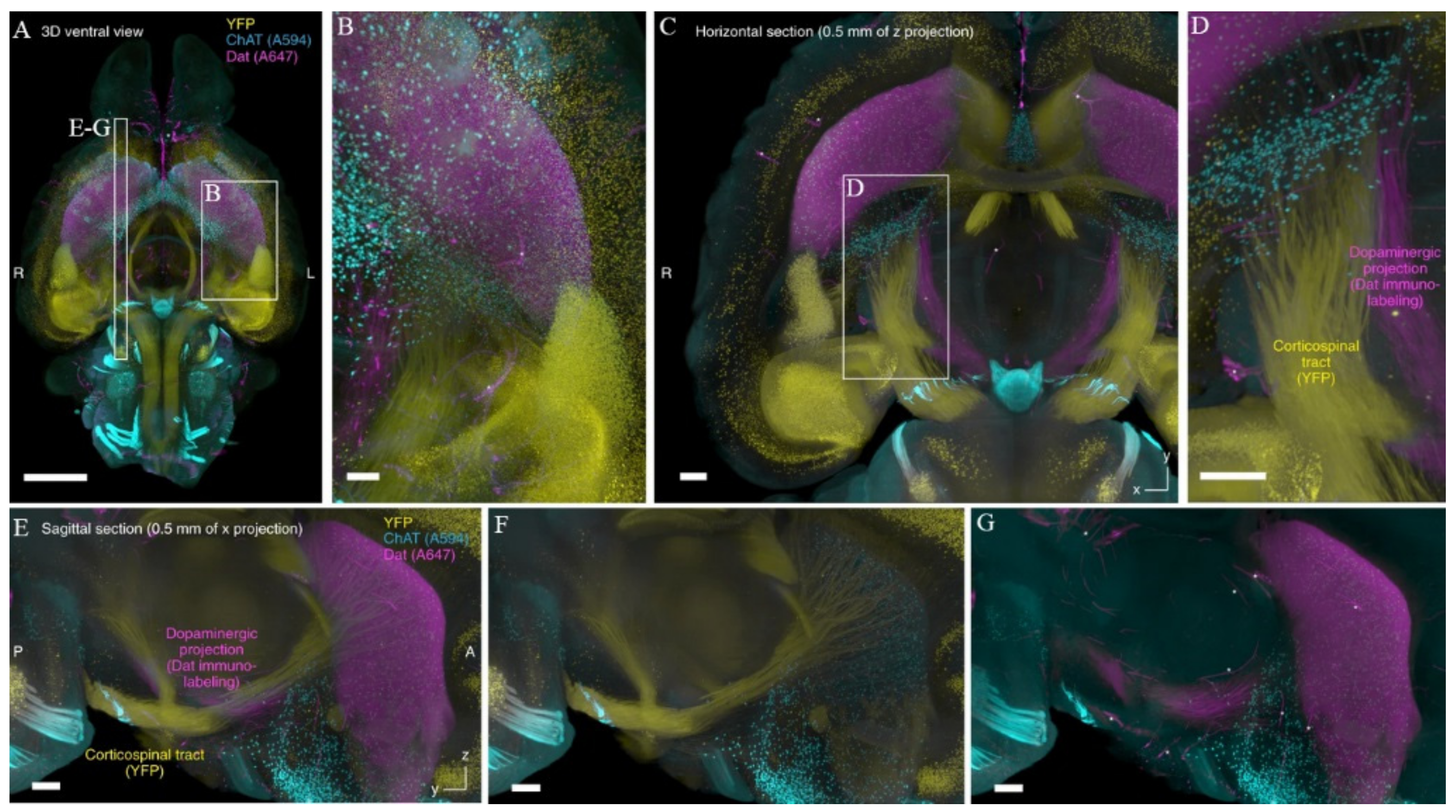
- Cai, R.; Pan, C.; Ghasemigharagoz, A.; Todorov, M.I.; Förstera, B.; Zhao, S.; Bhatia, H.S.; Parra-Damas, A.; Mrowka, L.; Theodorou, D.; et al. Panoptic Imaging of Transparent Mice Reveals Whole-Body Neuronal Projections and Skull-Meninges Connections. Nat. Neurosci. 2019, 22, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Treweek, J.B.; Gradinaru, V. Extracting Structural and Functional Features of Widely Distributed Biological Circuits with Single Cell Resolution via Tissue Clearing and Delivery Vectors. Curr. Opin. Biotechnol. 2016, 40, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renier, N.; Wu, Z.; Simon, D.J.; Yang, J.; Ariel, P.; Tessier-Lavigne, M. IDISCO: A Simple, Rapid Method to Immunolabel Large Tissue Samples for Volume Imaging. Cell 2014, 159, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Merz, S.F.; Korste, S.; Bornemann, L.; Michel, L.; Stock, P.; Squire, A.; Soun, C.; Engel, D.R.; Detzer, J.; Lörchner, H.; et al. Contemporaneous 3D Characterization of Acute and Chronic Myocardial I/R Injury and Response. Nat. Commun. 2019, 10, 2312. [Google Scholar] [CrossRef]
- Pavlova, I.P.; Shipley, S.C.; Lanio, M.; Hen, R.; Denny, C.A. Optimization of Immunolabeling and Clearing Techniques for Indelibly-Labeled Memory Traces. Hippocampus 2018, 28, 523–535. [Google Scholar] [CrossRef]
- Yang, B.; Treweek, J.B.; Kulkarni, R.P.; Deverman, B.E.; Chen, C.-K.; Lubeck, E.; Shah, S.; Cai, L.; Gradinaru, V. Single-Cell Phenotyping within Transparent Intact Tissue through Whole-Body Clearing. Cell 2014, 158, 945–958. [Google Scholar] [CrossRef] [Green Version]
- Biswas, L.; Chen, J.; de Angelis, J.; Chatzis, A.; Nanchahal, J.; Dustin, M.L.; Ramasamy, S.K.; Kusumbe, A.P. SUMIC: A Simple Ultrafast Multicolor Immunolabelling and Clearing Approach for Whole-Organ and Large Tissue 3D Imaging. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hofmann, J.; Gadjalova, I.; Mishra, R.; Ruland, J.; Keppler, S.J. Efficient Tissue Clearing and Multi-Organ Volumetric Imaging Enable Quantitative Visualization of Sparse Immune Cell Populations During Inflammation. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Lee, E.; Choi, J.; Jo, Y.; Kim, J.Y.; Jang, Y.J.; Lee, H.M.; Kim, S.Y.; Lee, H.-J.; Cho, K.; Jung, N.; et al. ACT-PRESTO: Rapid and Consistent Tissue Clearing and Labeling Method for 3-Dimensional (3D) Imaging. Sci. Rep. 2016, 6, 18631. [Google Scholar] [CrossRef] [Green Version]
- Fiorelli, R.; Sidhu, G.S.; Cebrián-Silla, A.; Melendez, E.L.; Mehta, S.; Garcia-Verdugo, J.M.; Sanai, N. Enhanced Tissue Penetration of Antibodies through Pressurized Immunohistochemistry. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fiorelli, R.; Sidhu, G.S. Systems, Devices, and Methods for Improved Tissue Treatment. U.S. Patent No 10,697,869, 30 June 2020. [Google Scholar]
- Kim, S.-Y.; Cho, J.H.; Murray, E.; Bakh, N.; Choi, H.; Ohn, K.; Ruelas, L.; Hubbert, A.; McCue, M.; Vassallo, S.L.; et al. Stochastic Electrotransport Selectively Enhances the Transport of Highly Electromobile Molecules. Proc. Natl. Acad. Sci. USA 2015, 112, E6274–E6283. [Google Scholar] [CrossRef] [Green Version]
- Na, M.; Kim, K.; Lim, H.R.; Ha, C.M.; Chang, S. Rapid Immunostaining Method for Three-Dimensional Volume Imaging of Biological Tissues by Magnetic Force-Induced Focusing of the Electric Field. Brain Struct. Funct. 2021, 226, 297–309. [Google Scholar] [CrossRef]
- Murray, E.; Cho, J.H.; Goodwin, D.; Ku, T.; Swaney, J.; Kim, S.-Y.; Choi, H.; Park, Y.-G.; Park, J.-Y.; Hubbert, A.; et al. Simple, Scalable Proteomic Imaging for High-Dimensional Profiling of Intact Systems. Cell 2015, 163, 1500–1514. [Google Scholar] [CrossRef] [Green Version]
- Yun, D.H.; Park, Y.-G.; Cho, J.H.; Kamentsky, L.; Evans, N.B.; Albanese, A.; Xie, K.; Swaney, J.; Sohn, C.H.; Tian, Y.; et al. Ultrafast Immunostaining of Organ-Scale Tissues for Scalable Proteomic Phenotyping. bioRxiv 2019, 660373. [Google Scholar] [CrossRef] [Green Version]
- Susaki, E.A.; Shimizu, C.; Kuno, A.; Tainaka, K.; Li, X.; Nishi, K.; Morishima, K.; Ono, H.; Ode, K.L.; Saeki, Y.; et al. Versatile Whole-Organ/Body Staining and Imaging Based on Electrolyte-Gel Properties of Biological Tissues. Nat. Commun. 2020, 11, 1982. [Google Scholar] [CrossRef]
- Li, J.; Czajkowsky, D.M.; Li, X.; Shao, Z. Fast Immuno-Labeling by Electrophoretically Driven Infiltration for Intact Tissue Imaging. Sci. Rep. 2015, 5, 10640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossolani, G.D.P.; Pintelon, I.; Detrez, J.D.; Buckinx, R.; Thys, S.; Zanoni, J.N.; Vos, W.H.D.; Timmermans, J.-P. Comparative Analysis Reveals Ce3D as Optimal Clearing Method for in Toto Imaging of the Mouse Intestine. Neurogastroenterol. Motil. 2019, 31, e13560. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.C.; Mano, T.; Saikawa, S.; Horiguchi, S.A.; Shigeta, D.; Baba, K.; Sekiya, H.; Shimizu, Y.; Tanaka, K.F.; Kiyonari, H.; et al. A Three-Dimensional Single-Cell-Resolution Whole-Brain Atlas Using CUBIC-X Expansion Microscopy and Tissue Clearing. Nat. Neurosci. 2018, 21, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Huang, L.; Zheng, Y.; Song, Y.; Xu, Q.; Wang, J.; Si, K.; Duan, S.; Gong, W. Ultrafast Optical Clearing Method for Three-Dimensional Imaging with Cellular Resolution. Proc. Natl. Acad. Sci. USA 2019, 116, 11480–11489. [Google Scholar] [CrossRef] [Green Version]
- Hou, B.; Zhang, D.; Zhao, S.; Wei, M.; Yang, Z.; Wang, S.; Wang, J.; Zhang, X.; Liu, B.; Fan, L.; et al. Scalable and DiI-Compatible Optical Clearance of the Mammalian Brain. Front. Neuroanat. 2015, 9, 19. [Google Scholar] [CrossRef]
- Chen, L.; Li, G.; Li, Y.; Li, Y.; Zhu, H.; Tang, L.; French, P.; McGinty, J.; Ruan, S. UbasM: An Effective Balanced Optical Clearing Method for Intact Biomedical Imaging. Sci. Rep. 2017, 7, 12218. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Shen, Y.; Ding, L.; Yang, C.-Y.; Tan, H.; Wang, H.; Zhu, Q.; Xu, R.; Wu, F.; Xu, C.; et al. High-Throughput Whole-Brain Mapping of Rhesus Monkey at Micron Resolution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Unnersjö-Jess, D.; Scott, L.; Blom, H.; Brismar, H. Super-Resolution Stimulated Emission Depletion Imaging of Slit Diaphragm Proteins in Optically Cleared Kidney Tissue. Kidney Int. 2016, 89, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unnersjö-Jess, D.; Scott, L.; Sevilla, S.Z.; Patrakka, J.; Blom, H.; Brismar, H. Confocal Super-Resolution Imaging of the Glomerular Filtration Barrier Enabled by Tissue Expansion. Kidney Int. 2018, 93, 1008–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Øie, C.I.; Mönkemöller, V.; Hübner, W.; Schüttpelz, M.; Mao, H.; Ahluwalia, B.S.; Huser, T.R.; McCourt, P. New Ways of Looking at Very Small Holes—Using Optical Nanoscopy to Visualize Liver Sinusoidal Endothelial Cell Fenestrations. Nanophotonics 2018, 7, 575–596. [Google Scholar] [CrossRef]
- Pende, M.; Vadiwala, K.; Schmidbaur, H.; Stockinger, A.W.; Murawala, P.; Saghafi, S.; Dekens, M.P.S.; Becker, K.; Revilla-i-Domingo, R.; Papadopoulos, S.-C.; et al. A Versatile Depigmentation, Clearing, and Labeling Method for Exploring Nervous System Diversity. Sci. Adv. 2020, 6, 0365. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Zhang, D.; Wang, C.; Feng, R.; Li, X.; Wen, Y.; Xu, H.; Zhang, X.S.; Yang, X.; et al. A Versatile Tiling Light Sheet Microscope for Imaging of Cleared Tissues. Cell Rep. 2020, 33, 108349. [Google Scholar] [CrossRef]
- Chu, L.-A.; Lu, C.-H.; Yang, S.-M.; Liu, Y.-T.; Feng, K.-L.; Tsai, Y.-C.; Chang, W.-K.; Wang, W.-C.; Chang, S.-W.; Chen, P.; et al. Rapid Single-Wavelength Lightsheet Localization Microscopy for Clarified Tissue. Nat. Commun. 2019, 10, 4762. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q.; Liu, L.; Zhou, Z.; Ruan, Z.; Kong, L.; Li, Y.; Wang, Y.; Zhong, N.; Chai, R.; et al. TeraVR Empowers Precise Reconstruction of Complete 3-D Neuronal Morphology in the Whole Brain. Nat. Commun. 2019, 10, 3474. [Google Scholar] [CrossRef] [Green Version]

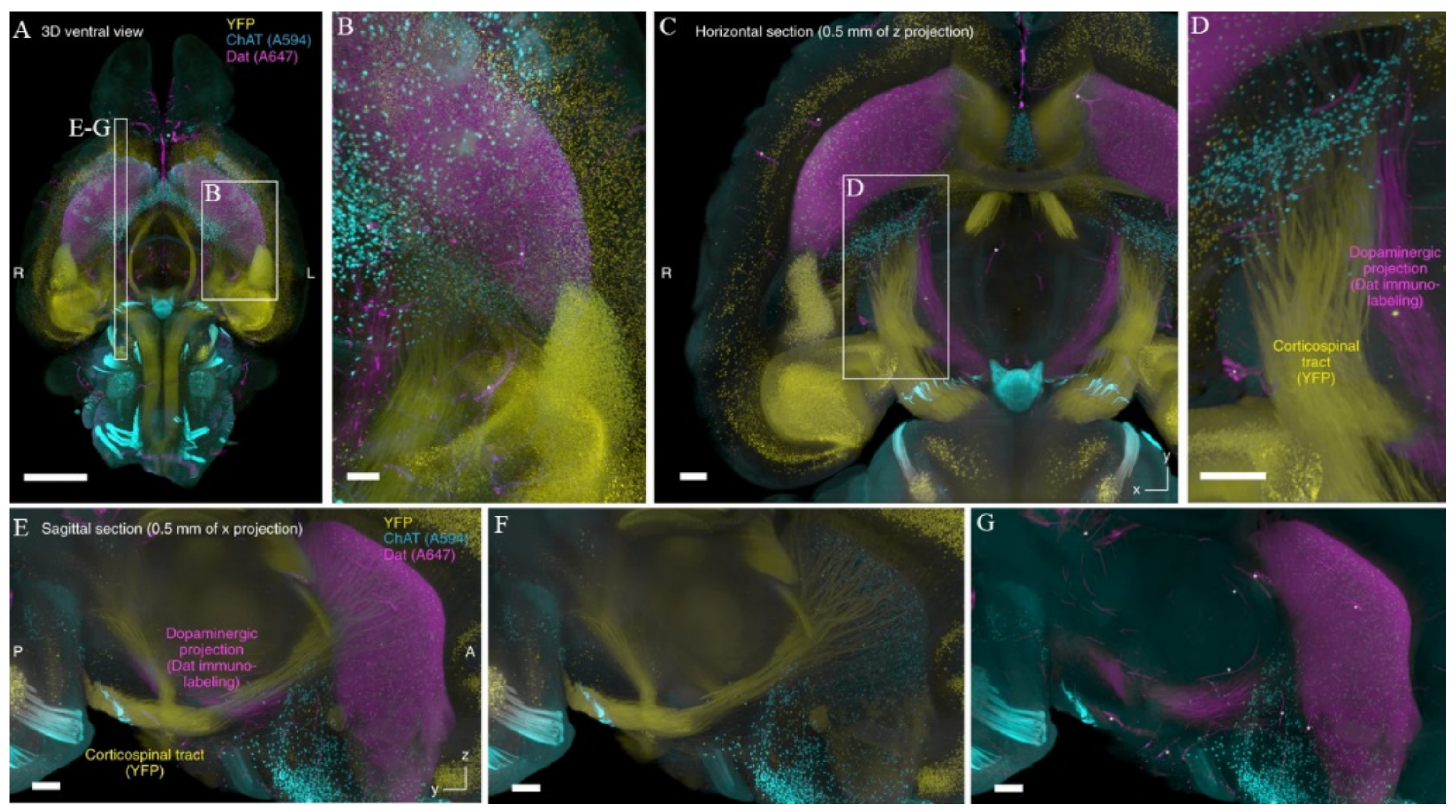
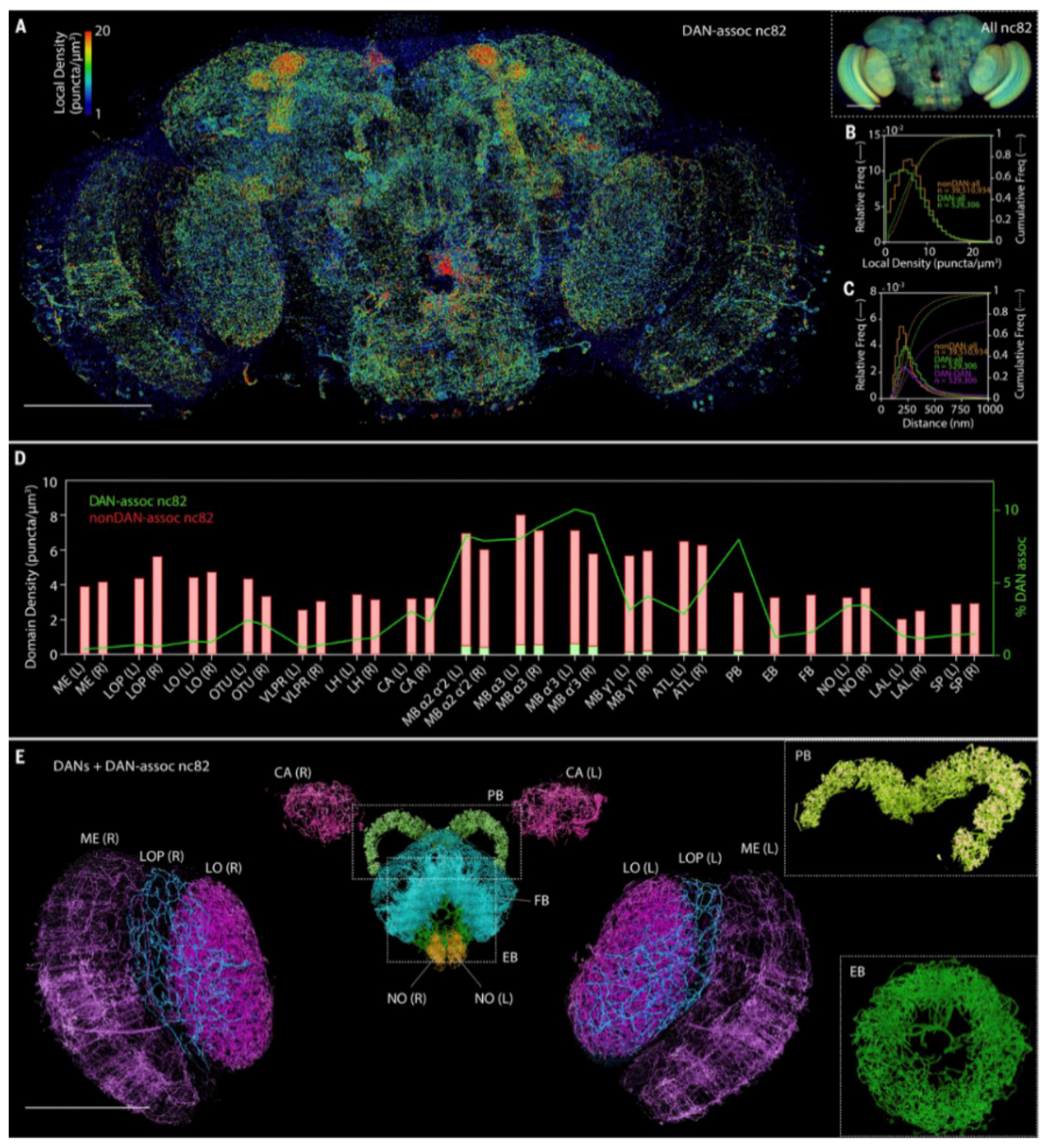

{kind=link}
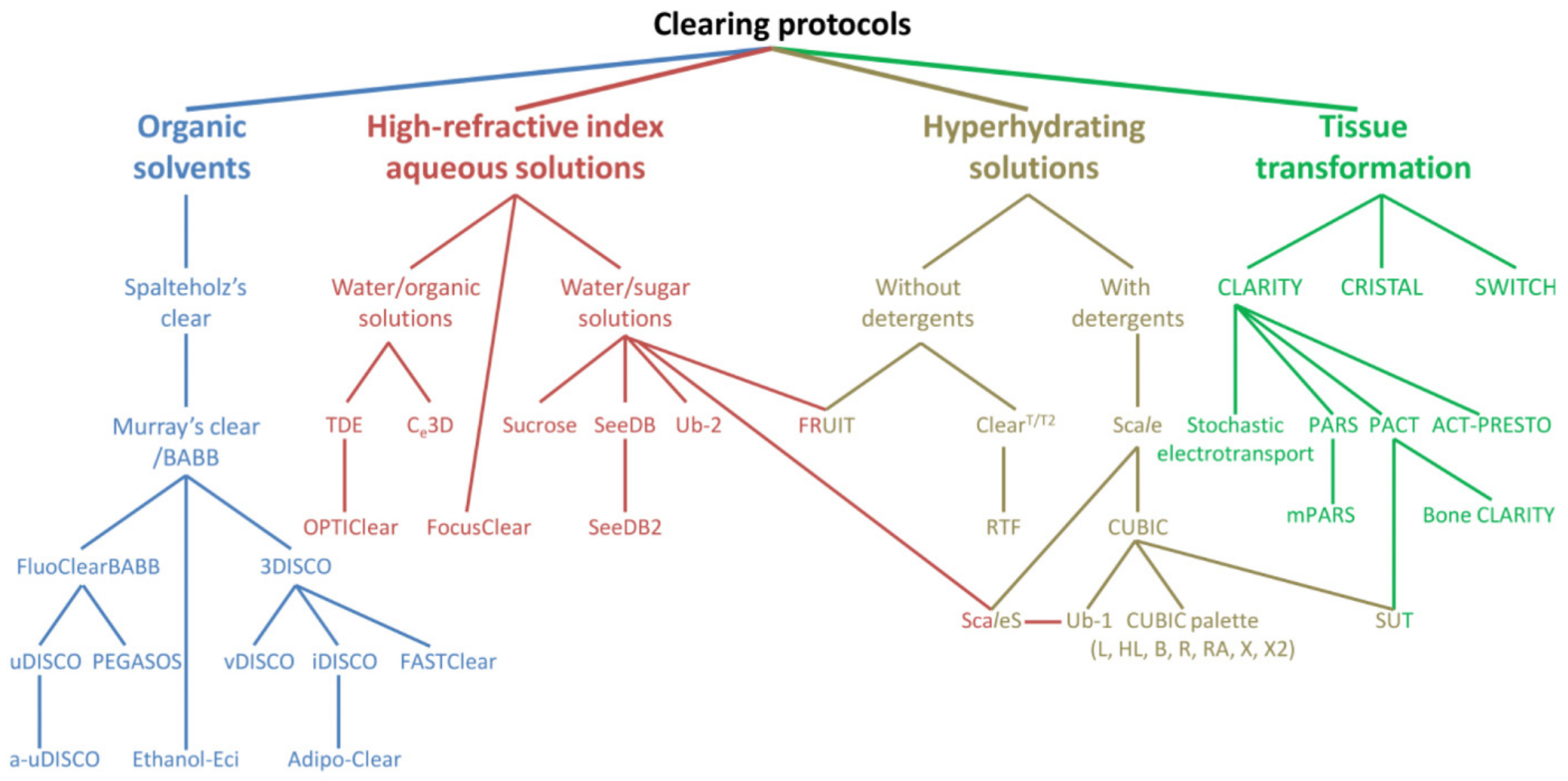
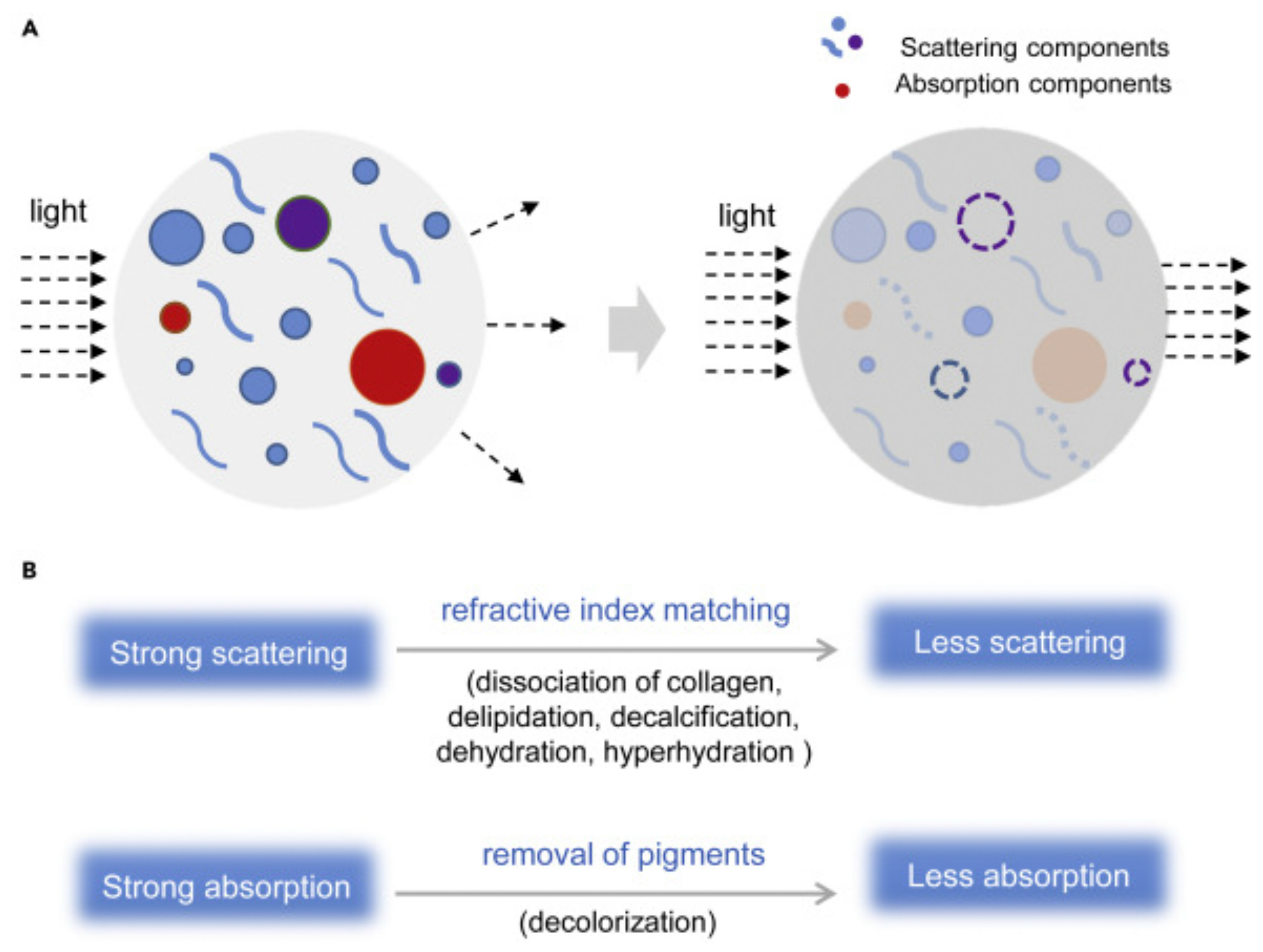{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SRM Technique | Acronym | Principle | Major Advantages | Major Disadvantages | Lateral Resolution (nm) | Axial Resolution (nm) |
|---|---|---|---|---|---|---|
| total internal reflection microscopy [46] | TIRF | evanescent wave selectively illuminates and excites fluorophores in a restricted region of the specimen | inherent Z- sectioning | only the outer layer of the specimen can be imaged | 200–300 | ~100 |
| 4Pi microscope [47] | 4Pi | increase of the effective numerical aperture by using two opposing lenses | high resolution in 3D representations | complex setup | ~110 | 100-150 |
| structured illumination microscopy [48] | SIM | illumination of the sample with a structured pattern generated from a coherent light source | fast, does not require special sample preparation | requires refractive index homogeneity along the optical path | 100–130 | 300-400 |
| super- resolution optical fluctuation imaging [49] | SOFI | correlations between adjacent pixels are calculated to separate signal coming from different fluorophores | no setup modifications, easy to combine with other modalities | requires special fluorophores | 100–130 | 300-400 |
| stimulated emission depletion microscopy [50] | STED | selective deactivation of fluorophores by stimulated emission in donut-shaped region spatially restricts fluorophores spontaneous emission | high lateral resolution | sample is prone to photodamage | ~50 | ~150 |
| single molecule localization microscopy [51,52] | SMLM | controlled switching on/off the fluorophores | high localization precision, relatively easy to upgrade using existing hardware | lower time resolution compared to other SRM techniques, complex sample preparation, challenging in vivo imaging | ~20 | ~50 |
| expansion microscopy [53] | ExM | isotropic swelling of a sample using polymers | compatible with standard imaging and staining techniques | nonuniform expansion of some biological structures, expansion affects fluorophore’s structure | 25–70 | ~200 |
| TOC | Microscopy | Objective | Specimen | Observable Depth and Achievable Resolution | Time of Clearing |
|---|---|---|---|---|---|
| CFM3 (RI 1.518) [55] | STED | 100×, NA 1.40, oil | 40-μm-thick mouse brain sections | sufficient resolution for detection of dendritic spine necks (which are known to be thinner than the diffraction limit) at 40-μm depth | 5 min |
| SeeDB2S (RI 1.518) [58] | STED | 63×, NA 1.40, WD 0.19, oil | thin mouse brain slices | sub-diffraction images up to ~120 μm in depth (limit set by the WD of an objective lens) | a few hours (for relatively thin samples) to 2 days (adult half brain samples) |
| SR-SIM | 100×, NA 1.46, WD 0.11, oil | HEK293T cells (~10-μm-thick) | HEK293T cells labeled with membrane EGFP, MitoTracker, and DAPI could be fully resolved | a few hours (for relatively thin samples) to 2 days (adult half brain samples) | |
| SeeDB2G (RI 1.46) [58] | confocal | 63×, NA 1.30, WD 0.30, glycerol | entire fly brain (~300-μm-thick) | comprehensive maps of bsh-positive neurons in a whole brain | a few hours (for relatively thin samples) to 2 days (adult half brain samples) |
| LUCID#2 (RI 1.496) [43] | SIM | 100×, NA 1.49, oil | 150-μm-thick mouse brain sections | at a depth of 10 μm, lateral and axial FWHMs were 163 ± 1 and 583 ± 4 nm; maintained at depths from 10 to 60 μm | 6 h |
| Rapiclear 1.49 (RI 1.49) [59] | confocal | 63×, NA 1.30, oil | fly brains (freshly dissected dimensions ~3000 by 1500 by 400 μm) | sufficient to capture very fine neurites (diameter of between 136 and 271 nm) up to ~100-μm depth | 5 h of ethanol pretreatment + overnight incubation in Rapiclear |
| H71VE (RI ~1.50) [42] | SIM | 63×, 1.40 NA, oil | 10-µm-thick paraffin-embedded mouse spleen tissue sections | average modulation contrast-to-noise ratio = 10.1, that remained constant across the entire imaging depth | ~1 h |
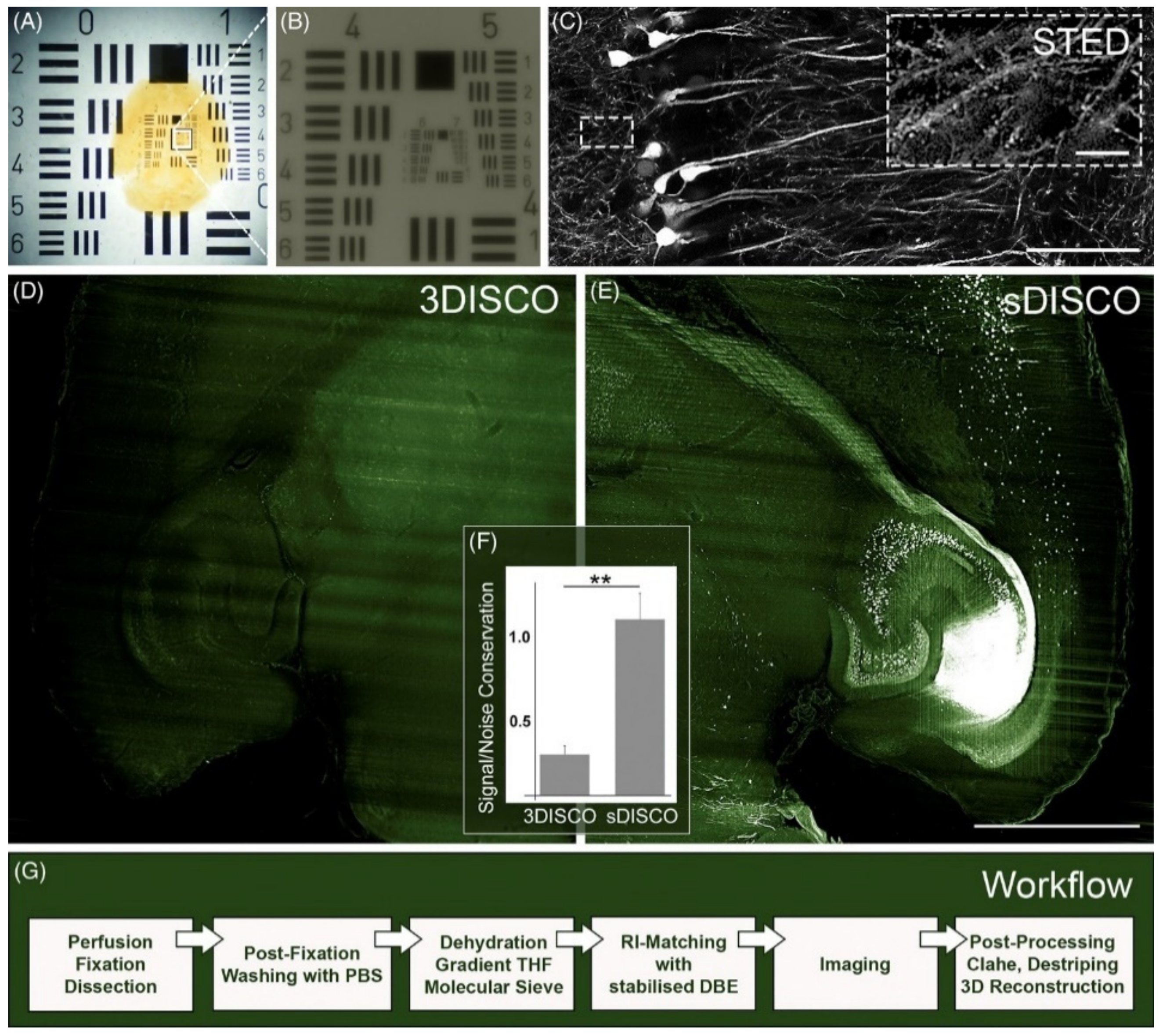
| sDISCO (RI 1.56) [61] | STED | 100×, NA 1.40, oil | 600-μm-thick mouse brain sections | sufficient to visualize single dendritic spines; depth was not studied | days |
| Mowiol 4-88 (RI 1.46) [62] | image scanning microscopy implemented with the Airyscan microscope | 63×, NA 1.40; or 100×, NA 1.46; oil | ~50-μm-thick mouse brain sections | 140 × 140 × 350 nm (XYZ) at ~50-μm depth | Not described |
| protein -retention ExM [11] | lattice light-sheet microscopy | 25×, NA 1.10, WD 2.00 | D. melanogaster brain | ~60 × 60 × 90 nm for 4× expansion in the entire fly brain | Few days |
| FocusClear (RI 1.45) [12] | DMI6000 microscope equipped with CSU spinning disk confocal scan head | 63×, NA 1.3, glycerol | D. melanogaster brain | intact fly brain with 20-nm lateral resolution at ~200-μm depth | 1–2 days |
| Modified CUBIC (mCUBIC described in [63]) [64] | light-sheet fluorescence microscopy and super-resolution optical fluctuation imaging | 16×, NA 0.80, WD 3.00 | 300-μm-thick mouse brain sections | 50 × 50 nm lateral pixel size | 1 day |
| TOC Method Name/Acronym | Key Chemical | Anti-Bleaching Step | pH | Compatible Fluorophores |
|---|---|---|---|---|
| FluoClearBABB [71] | tert-butanol | not included | 9.5 | eGFP and mRFP (imaging 271 days after TOC) |
| uDISCO [34] | tert-butanol, diphenyl ether | tocopherol | not adjusted | GFP (imaging 35 days after TOC), RFP, Texas Red, AF 568 and 647 |
| a-uDISCO [73] | tert-butanol, diphenyl ether | tocopherol | 9.0–9.5 | GFP (imaging 42 days after TOC) |
| 2Eci [74] | 1-propanol, ethyl cinnamate | not included | 9.0 | GFP, mCherry, AF 488, 568 and 647, Brainbow (GFP, RFP, CFP and YFP) |
| Eci [75] | ethanol, ethyl cinnamate | not included | 9.0 | eGFP, eYFP, AF 647 |
| PEGASOS [28] | tert-butanol, Quadrol, poly(ethylene glycol) methacrylate, benzyl benzoate | not included | 9.0 | eGFP, tdTomato, FITC, AF 488, 568 |
| sDISCO [61] | tetrahydrofuran, dibenzyl ether | propyl gallate, cleared and stored at 4 °C | dehydration in THF in PBS (pH = 8.3) | eGFP, YFP, tdTomato |
| FDISCO [76] | tetrahydrofuran, dibenzyl ether | cleared and stored at 4 °C | THF and dibenzyl ether of pH = 9.0 | GFP, YFP, LEL-Dylight649, Cy5, tdTomato |
| Main Mechanism | Acronym | Key Features | Sample | Antibodies/Markers Tested | Time of Procedure |
|---|---|---|---|---|---|
| pressure-assisted | PARS [93] | continuous perfusion of clearing agents and antibodies at 1 mL/min rate | mouse body and rat brain | GFAP nanobody (rat brain), anti-mouse IgG antibody (mouse brain), antitubulin (mouse kidney) | 3 weeks |
| pressure-assisted | vDISCO [88] | continuous perfusion of clearing agents and antibodies under increased pressure (160–230 mmHg) | mouse body | updated list of anti/nanobodies available at http://www.discotechnologies.org/vDISCO/ | 3 weeks |
| pressure-assisted | PRESTO [96] | centrifugal, c-PRESTO and syringe-based, s-PRESTO, variants available | mouse kidney, lung, liver, testis | anti-collagen type IV, acetylated tubulin, and laminin Abs | 1–2 days |
| pressure-assisted | pIHC [97] | IHC with use of N2 at 225 KPa | up to 1-mm-thick mouse and human brain samples | numerous, e.g., Olig2, Ki67, Iba1, NF, MAP2, NeuN, Lectin, GFAP | hours to 3 days, depending on sample thickness |
| digestion of extracellular matrix | EMOVI [95] | saponin-based fixation and hyaluronidase-based matrix digestion | various mouse organs (entire, halves and thick slices) | numerous Abs (related to immunology) were tested: anti-CD11c, CD3, CD21, MHCII, LYVE-1, CD31. | 6–9 days |
| digestion of extracellular matrix | SUMIC [94] | collagenase A-based matrix digestion | thick slices of mouse organs and human endocrine gland tissue samples | over 35 Abs tested and verified as compatible | 2–3 days |
| Electro- phoresis | eTANGO [99] | continuous rotation of sample that is placed between two parallel electrodes | entire mouse organs (brain, intestine, heart) | Dylight 594-conjugated tomato lectin | 1 day + clearing |
| Electro- phoresis | EFIC [100] | magnetic force focuses the electric field by bending it onto the sample | CLARITY pre-cleared 1-mm-thick and not cleared, 150-μm-thick, rodent brain samples | numerous Abs, e.g., anti-NeuN, Iba1, GFAP, Neurofilament 200, Myelin Basic protein, Parvalbumin | hours + clearing |
| modification of probe affinity | SWITCH [101] | SWITCH-OFF that inhibits Abs binding (but allows diffusion) and SWITCH-ON that increases Abs binding | 100-μm- to 1-mm-thick mouse and human brain samples | numerous Abs validated, possibility to perform 22 rounds of immunostaining and stripping | 1–2 days |
| modification of probe affinity | eFLASH [102] | eTANGO combined with sodium deoxycholate to control the labeling affinity for various antibodies in a concentration- and pH-dependent manner | mouse and marmoset brains, organoids | numerous Abs, e.g., anti-NeuN, Iba1, GFAP, Parvalbumin, ChAT, c-Fos, NPY | 1 day + clearing |
| modification of probe affinity | CUBIC-Histo VIsion [103] | modulation of interaction between Abs and tissue with Triton X-100 or Quadrol, 1-step staining approach, staining in over RT, digestion with hyaluronidase or collagenase | mouse, marmoset and human brain samples | numerous Abs validated | 1–8 weeks + clearing |
| delipidation | iDISCO [90] | Severe, methanol-based permeabilization and delipidation, some Abs incompatible with methanol pretreatment | adult mouse organs, mouse embryos | numerous, list available at https://idisco.info/validated-antibodies/ | ~week mouse embryo ~month adult mouse brain |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matryba, P.; Łukasiewicz, K.; Pawłowska, M.; Tomczuk, J.; Gołąb, J. Can Developments in Tissue Optical Clearing Aid Super-Resolution Microscopy Imaging? Int. J. Mol. Sci. 2021, 22, 6730. https://doi.org/10.3390/ijms22136730
Matryba P, Łukasiewicz K, Pawłowska M, Tomczuk J, Gołąb J. Can Developments in Tissue Optical Clearing Aid Super-Resolution Microscopy Imaging? International Journal of Molecular Sciences. 2021; 22(13):6730. https://doi.org/10.3390/ijms22136730
Chicago/Turabian StyleMatryba, Paweł, Kacper Łukasiewicz, Monika Pawłowska, Jacek Tomczuk, and Jakub Gołąb. 2021. "Can Developments in Tissue Optical Clearing Aid Super-Resolution Microscopy Imaging?" International Journal of Molecular Sciences 22, no. 13: 6730. https://doi.org/10.3390/ijms22136730
APA StyleMatryba, P., Łukasiewicz, K., Pawłowska, M., Tomczuk, J., & Gołąb, J. (2021). Can Developments in Tissue Optical Clearing Aid Super-Resolution Microscopy Imaging? International Journal of Molecular Sciences, 22(13), 6730. https://doi.org/10.3390/ijms22136730