1. Introduction
In the wake of the awareness and acceptance that autism spectrum disorder (ASD) is complicatedly heterogeneous and constantly rising [
1], very little is known of its etiology and pathophysiology. According to the DSM-5 [
2], current classifications of individuals with ASD house them under two core symptoms, which are: (1) deficits in social communication and social interaction and (2) restricted, repetitive patterns of behavior, interests, or activities. To date, subjective and clinical diagnosis has been the standard method of identifying children with the disorder, which has been helpful but is still far from ideal. This method risks late/missed diagnosis and, on occasion, ineffective therapeutic interventions [
3]. Hence, finding alternative methods to objectively and systematically identify children with ASD, and assimilate a more compressive understanding of where an individual lies on the spectrum, is an important research area, paving the way to better therapeutic strategies and personalized medicine.
Biomarker discovery has increasingly been documented as an impactful avenue in disease diagnosis and is essential for drug discovery and restricting the advancement of diverse groups of diseases. The constant emergence of new and innovative technologies has made it even more possible to identify and validate biomarkers, making them a viable way to provide patients with personalized medicine. Furthermore, evidence has shown that the proper development and machinery of the central nervous system relies on the complex and finely orchestrated spatiotemporal expression of non-coding RNA (ncRNA), and their capricious regulation contributes to irregularities in gene regulator networks, brain development, and their correlating neurodevelopmental disorder [
4]. Since ncRNAs impact biological processes, they could be utilized as attractive predictive and diagnostic biomarkers of ASD. Furthermore, due to the complexities and behavioral phenotypic heterogeneity of ASD, one must further understand the biological players that contribute to the disorder’s outcome and provide objective and reliable data to faction between these distinct groups and symptom severities. Ultimately, the goal is to cultivate tremendous leaps in diagnostic methods and manage personalized treatment strategies for ASD. This paper highlights the impact of utilizing cir-ncRNA biomarkers in ASD diagnosis and underlines an appreciation of their use as an emblem of ASD severity.
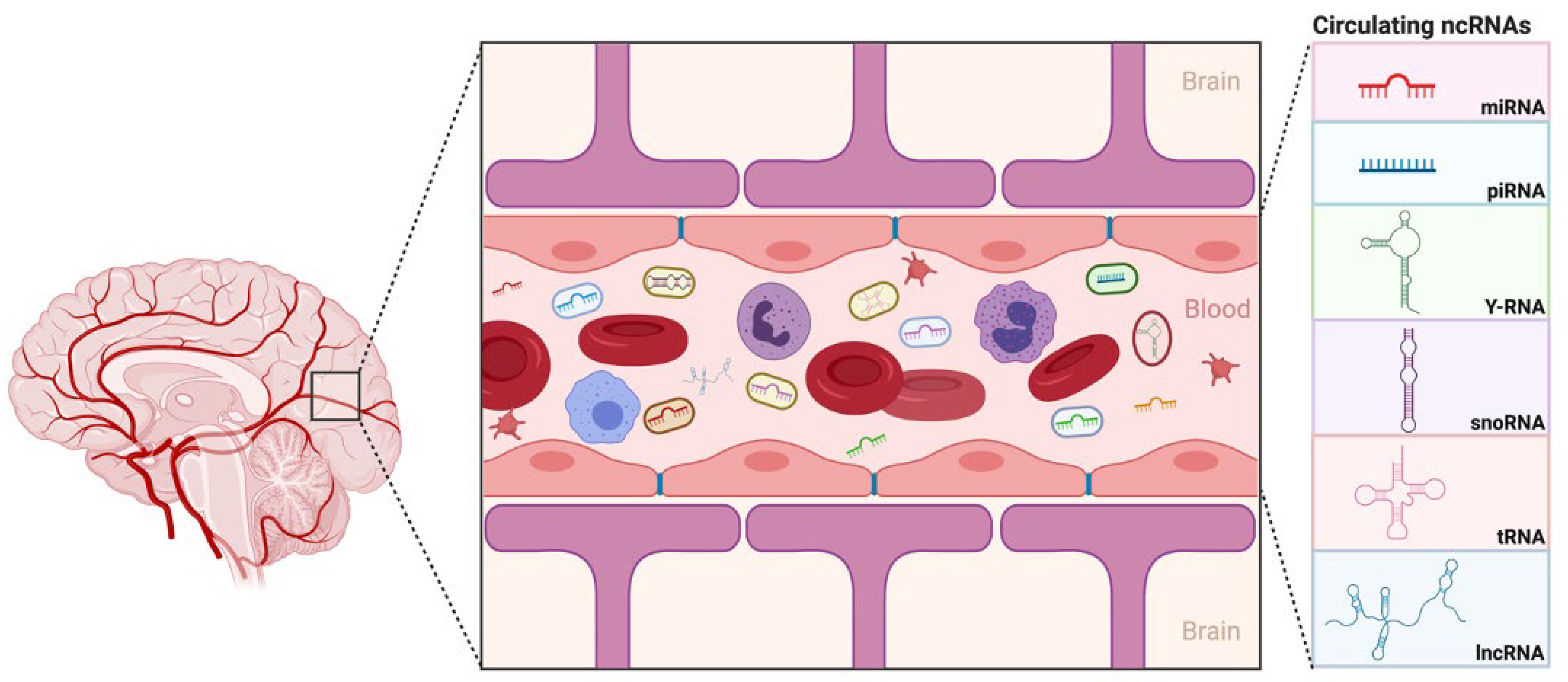
Mammalian cells have been recognized as investing energy in the production of endless diversities of small and large RNA transcripts that do not code for proteins, known as non-coding RNAs (ncRNAs). Traditionally, only ribosomal RNA (rRNA) and transfer RNA (tRNA) were considered ‘functional’ RNA, whereas all other non-coding transcripts were deemed ‘desert’ or ‘junk’ DNA. However, transcriptome data have expanded and shed light on the prominence and the seemingly infinite diversity and complexity of RNA biology, and their influence on numerous areas of cellular biology and molecular mechanisms that were once overlooked. Their ability to seemingly be secreted and circulated within exosomes, microvesicles, or RNA-binding proteins such as Argonaute 2, make them easily detectable and a less invasive biomarker of pathophysiological status [
5]. Some examples of ncRNAs and their influential roles in gene regulation or interference include small nuclear RNAs (snoRNAs; RNA splicing), small nucleolar RNAs (RNA modification), miRNAs (RNA degradation and/or translation inhibition), PIWI-interacting RNAs (piRNAs; gene silencing), Y-RNAs (DNA replication), transfer RNAs (tRNAs), and long non-coding RNAs (gene regulation) [
6]. Furthermore, ncRNAs were previously shown to be differentially regulated in neurodegenerative diseases [
7], while others have suggested that its rich repertoire within the human brain makes their involvement and dysregulation potentially crucial in the steps that lead to abnormal brain development and the etiology of neurodevelopmental disorders [
1,
8].
miRNAs are small and single-stranded RNAs of approximately 22 nucleotides that have been identified as crucial post-transcriptional inhibitors of gene expression. miRNAs act as regulators in “fine-tuning” the translational output of target messenger RNAs (mRNAs) by promoting their degradation and inhibiting translation while regulating a plethora of physiological pathways and metabolic processes. The expression pattern of miRNAs is dynamically regulated during neurogenesis, neuronal differentiation, maintenance and function and overall brain development, making their involvement in neurodevelopmental processes of interest [
1,
9]. Studies have shown the critical role of alterations in miRNA regulation and signaling in many neurodevelopmental disorders, such as Fragile X syndrome, Rett Syndrome, Down Syndrome, and Prader-Willi Angelman Syndrome [
8]. Since ASD has been associated with several of these conditions, it is interesting to investigate how miRNA dysregulation and/or expression patterns contribute.
Moreover, previous studies have proven that circulating miRNAs (cir-miRNAs) are emerging as significant biomarker for diagnosis and prognosis in human disorders and disease. It is understood that the miRNA repertoire that manifests itself in varying cell types is highly defined. In other words, the characteristic expression pattern of miRNA in different tissues, or cells, contributes to the shaping of specific features and functions. Some have even been identified as being solely expressed in particular tissues or cell types. Hence, this sheds light on the ability to identify specific miRNA expression patterns in varying diseases and disorders. Consequently, it has previously been shown that miRNA can be used as a diagnostic marker for certain types of cancers or cardiovascular conditions, as they contain specific profile characterizations [
10].
Subsequently, this highlights the importance and potential of cir-miRNAs as viable and reliable biomarkers for ASD. Assessing the expression patterns of miRNAs and their influential capacity towards neurodevelopmental disorders is an exciting area of exploration. Previous investigations have underlined their effects on the spatial localization or compartmentalization of protein translation in dendrites, axons, and synapses [
9]. Additionally, understanding the importance of their unique expression patterns can lead to new treatment strategies, in which miRNAs may be down-regulated, inhibited, up-regulated, or even replaced. In previous studies, multiple miRNAs have been identified as down- or up-regulated in blood, serum, and saliva, with the promise of their having relevance in ASD [
11,
12,
13]. A meta-analysis review has recently shown the possibility of using oxytocin and vasopressin as biomarkers; however, this is heterogeneous in psychiatric disorders and does not provide convincing findings that peripheral OT or ADH levels are altered in ASD compared to healthy controls [
14]. Moreover, a recent review yielded data on 67 environmental risk factors and 52 biomarkers. Only two associations, maternal overweight before or during pregnancy and SSRI use during pregnancy, might act as an independent risk factor for autism spectrum disorder [
15].
Furthermore, another compelling group of non-coding RNAs are the piRNAs, which are characterized as having 24–31 nucleotides and have a role in regulating gene expression and stability, and silencing transposons by driving PIWI proteins of the Argonaute family towards specific genomic loci [
8]. Once considered to be “dark matter”, piRNAs have emerged as potentially crucial regulators of disease outcomes, especially in cancer, and their aberrant expression is a distinct characteristic [
16]. Furthermore, their value for use as a molecular marker has increased due to their abundance in various body fluids, their capacity to not easily be degraded by ribonucleases, and the fact that they are short enough to pass through cell membranes [
7,
17].
Additionally, snoRNAs are another fascinating group of non-coding RNAs which are characterized as metabolically stable RNAs that consist of 60–300 nucleotides and accumulate within the nucleoli. snoRNAs are subdivided into two main classes, which possess distinctive and evolutionary conserved sequence elements: C (RUGAUGA)/D (CUGA) motifs and H (ANANNA)/ACA elements, which guide the 2’-O-methylation and pseudouridylation of ribosomal RNAs (rRNAs), respectively, regulating its modification, maturation, and stabilization. Emerging evidence has further expanded our understanding of their roles, including endonucleolytic processing, guiding small nuclear RNAs and mRNA, and regulating alternative splicing in pre-mRNAs [
18]. Previous investigations have shown that aberrant expression levels of snoRNAs, as well as mutations in its structure, influence physiological stress responses, including oxidative stress, tumorigenesis, metastasis, memory consolidation and learning, cholesterol, and another metabolic homeostasis, as well as being implicated in Prader–Willi syndrome, which has behavioral phenotypes that overlap with ASD [
18,
19]. Moreover, it is important to note that snoRNAs expression levels are stable and easily detectable in blood plasma, serum, and urine, making it a promising biomarker target.
To date, reliable biomarkers to diagnose ASD or define its severity do not exist. This study aims to comprehensively explore the potential of cir-ncRNAs as diagnostic biomarkers of ASD, as well as examine their expression profiles as a signature of ASD symptom severity. Our research plan used a sensitive, accurate, and cost-efficient methodology to profile ncRNAs. Consequently, we utilized next-generation sequencing (NGS) to identify novel non-coding species that could be functionally relevant as well as reliable biomarker targets. The dynamic expression pattern of cir-ncRNAs may be associated with the outcome of ASD and, further, distinguish between individuals that express either severe or mild symptoms of ASD.
3. Discussion
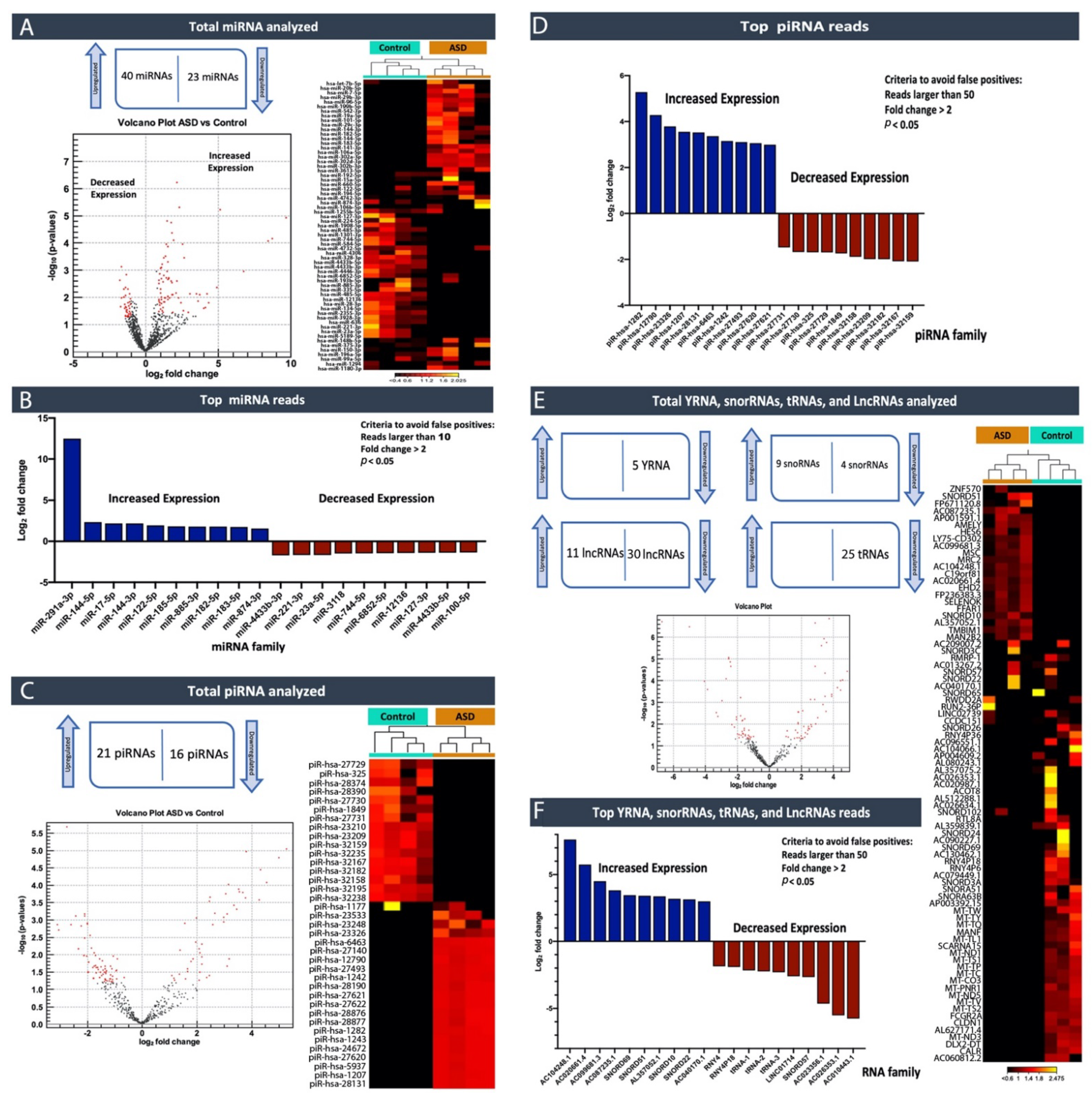
It is imperative to appreciate that the accurate and early diagnosis of neurodevelopmental disorders such as ASD, and timely intervention, can potentially alter the adverse developmental trajectories and characteristic severities. At present, a reliable biomarker or a combination of biomarkers that can be used for precise ASD diagnosis does not exist. Consequently, ASD is mainly identified through behavioral phenotypes and characteristics. This subjective analysis leaves room for misdiagnosis and potentially ineffective treatment strategies. Therefore, there is an essential need to uncover specific and reliable biomarkers that provide objective identification of ASD and better faction the spectrum to deliver more effective therapies. The recruitment of very young, healthy children and children with ASD to donate blood proved to be difficult. Consequently, the small number of controls recruited is a limitation of our study. However, our analysis shows significantly associated circulating non-coding RNAs (cir-ncRNAs) specific to individuals with ASD, and their roles were reinforced through the pathway analysis results, despite the small sample size of controls.
The non-coding genome is significantly larger than the genome’s protein-coding portion; more than 98% of the human genome does not encode proteins [
39]. Non-coding RNAs (ncRNAs) have a high potential to circulate in the blood and act as novel biomarkers for several conditions. Furthermore, cir-ncRNAs have recently been categorized as potential diagnostic markers for different conditions, including neurological disorders. Although there have been multiple studies associating circulating miRNAs with ASD, other non-coding RNAs have not been studied, and disorder causal genes have yet to be confirmed [
1]. Additionally, several studies have examined miRNAs in whole blood [
12], serum [
13], and saliva [
11], obtained from children with ASD compared to controls, but none of these studies isolated RNAs from plasma.
Recent findings have indicated the biological variable that impacted miRNA profiles in plasma and serum. These two biofluids may exhibit some differences in their miRNA contents, notably due to the coagulation process occurring during serum collection [
40]. The extent of platelet depletion in plasma samples is a critical variable, as well as the loss of cir-ncRNA through surface adsorption on the blood clot or through additional RNA degradation paired with the release of platelet-containing RNAs upon coagulation. In our study, we used platelet-depleted plasma, generated immediately after blood collection, which constitutes the preferred sample-collection process for cell-free circulating RNA biomarker discovery [
41]. To the best of our knowledge, our study is the first to use plasma obtained from children with ASD to delineate ncRNA as a diagnostic biomarker. We have utilized comprehensive, evidence-based data sources to introduce, for the first time, several potential circulating ncRNAs biomarkers, isolated from the plasma, with the aim of identifying ASD cases that tend to manifest severe symptoms of ASD vs. mild ones, and hence, this may even be implemented as a tool in early diagnosis and treatment approaches (
Figure 6).
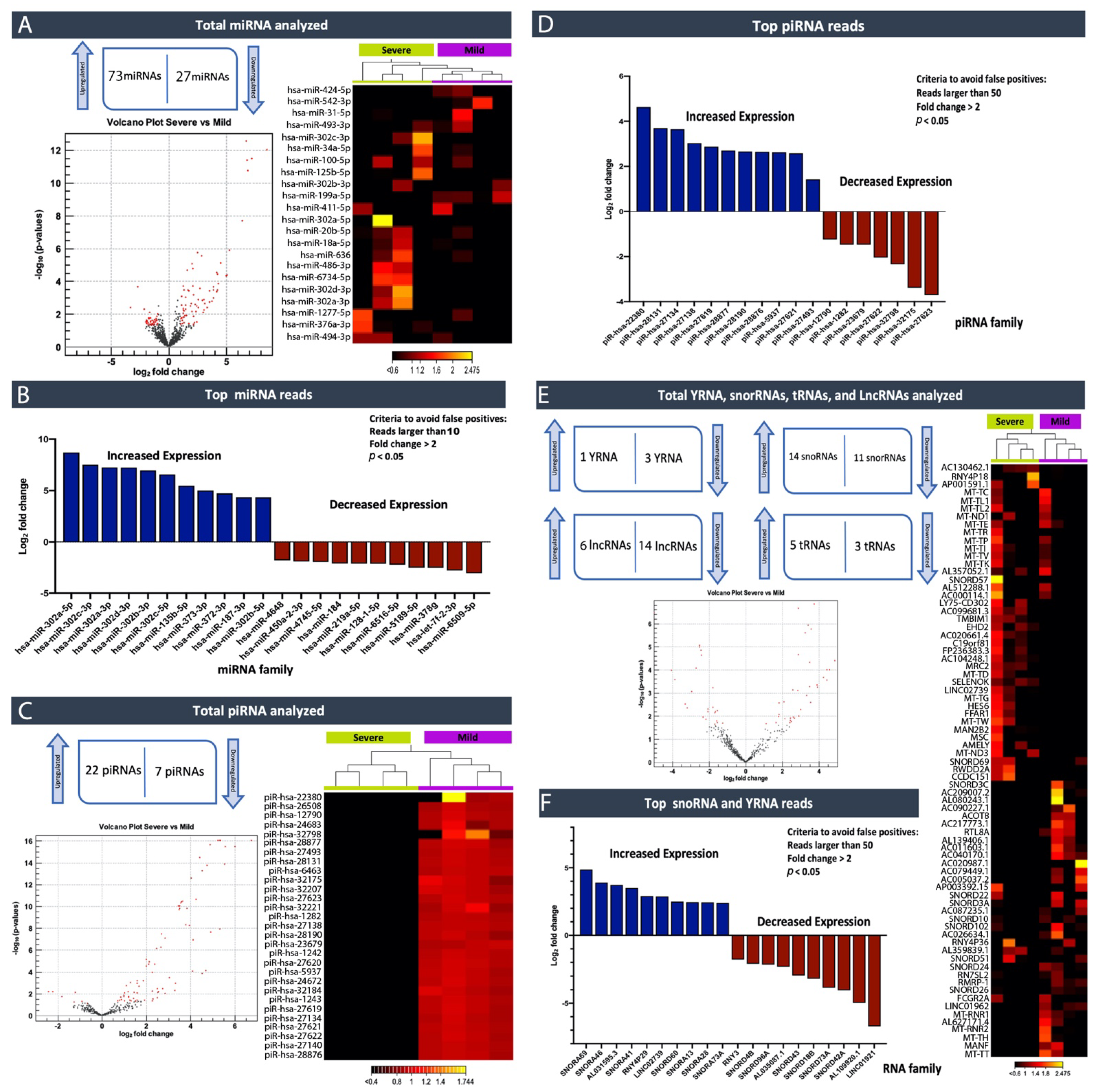
In this study, by profiling ncRNAs in the plasma of ASD individuals, we examined several key ncRNAs biotypes; miRNAs (the most abundant biotype), piRNAs, snoRNAs, Y-RNAs, tRNAs, and lncRNAs. Each group of subjects (ASD vs. Controls and ASD individuals that expressed severe symptoms vs. mild symptoms) appeared to have differences in terms of the circulating ncRNAs expression profiles. In particular, within the miRNA family miR-302, which displayed substantially high read counts, we observed that hsa-miR-302a-5p, hsa-miR-302c-3p, hsa-miR-302a-3p, hsa-miR-302d-3p, hsa-miR-302b-3p, hsa-miR-302c-5p and hsa-miR-302b-5p were expressed at significantly high levels in cases where individuals exhibited severe symptoms of ASD compared to those that expressed a few or milder forms of their defining characteristics. This finding, miR-302a in plasma, has confirmed Vasu et al.’s (2014) findings that miR-302a is differentially expressed in the serum of ASD. Moreover, miR-302a seems to be highly expressed in ASD cases with severe symptoms. The significant increase in the miR-302 family sparked our interest, as miR-302 was previously shown to negatively influence the neural development, neuronal receptors, and synaptic plasticity [
42]. We, therefore, aim to understand why children with higher expression levels of this family of miRNA show more severe symptoms. Consequently, miR-302a might be a biologically and analytically attractive biomarker for ASD with a severe symptomatic biomarker. Moreover, consistent findings have shown that the miR-302 family is critical in stem cell pluripotency and renewal, and somatic cell DNA demethylation [
20,
21,
22]. miR-135b-5p was another miRNA that was flagged in our study as being expressed at significantly higher levels in individuals who expressed severe symptoms versus mild ones. It was previously described that variable regulation of disrupted in schizophrenia 1 (DISC1) by miR-135b-5p in the brain may predispose one to neuropsychiatric phenotypes [
23]. Furthermore, a recent study has shown that miR-135 can serve as a biomarker of post-traumatic stress disorder (PTSD) and might be an important therapeutic target for dampening persistent and stress-enhanced memory [
43]. By analyzing these miRNA pathways further, and identifying proteins the associated with them, one can then develop clinical assays that can be used to diagnose objectively.
Furthermore, we believe that simultaneous measurements of RNA and proteins can increase detection specificity for ASD and revolutionize the current diagnosis paradigm. We aim to develop an assay which simultaneously interrogates large sets of RNAs and proteins, as a unique, sole biomarker for ASD is near impossible.
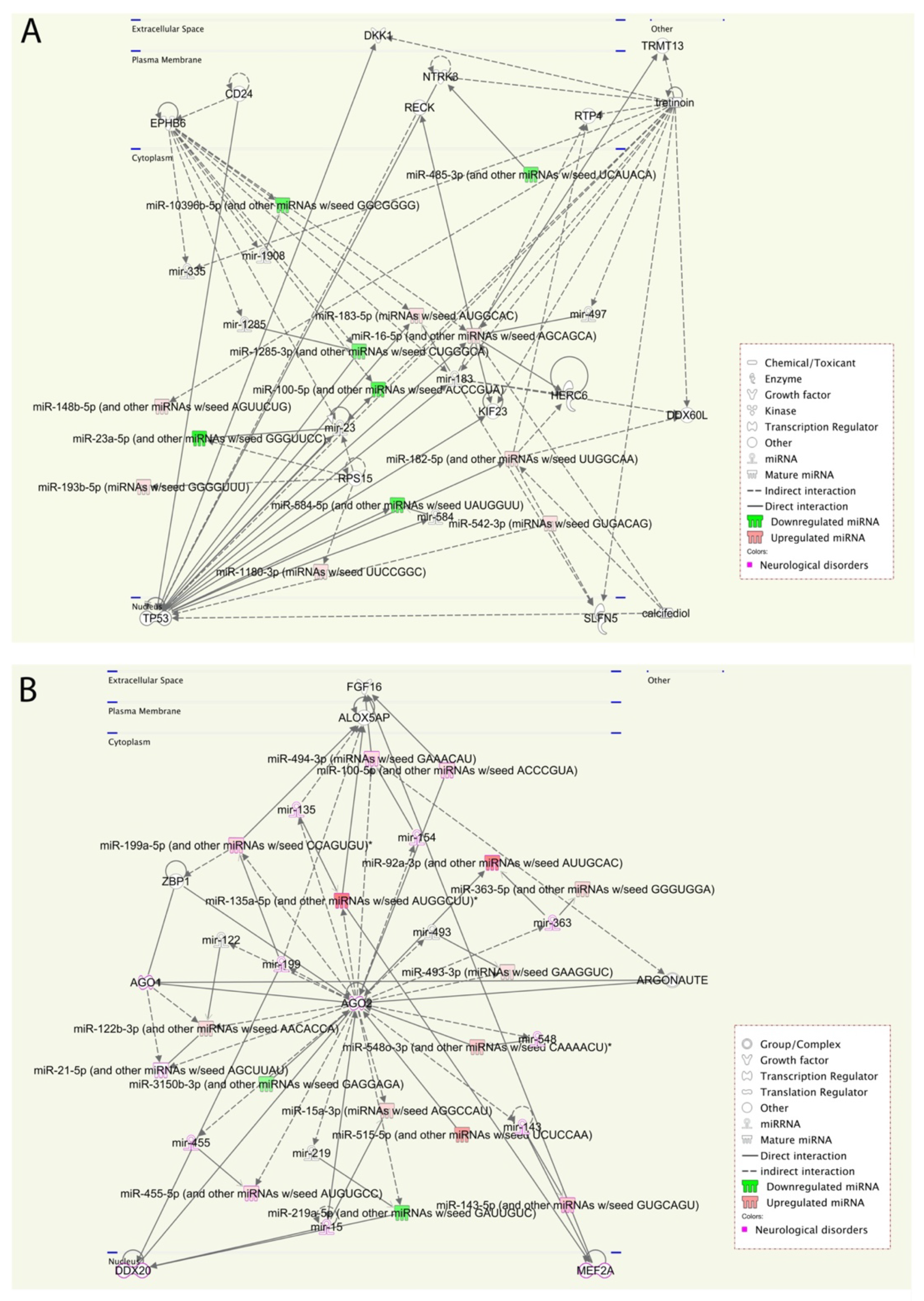
We performed a pathway enrichment analysis to better understand the miRNA’s biological implications in the context of the regulatory system. The pathway analysis revealed that a large number of pathways are neuro-related processes, and several enriched molecules/proteins are involved, such as DKK1, EPHB6, NTRK3, P53, AGO2, PTEN, and BRAF. DKK1 has been previously described as being involved in the brain [
25], whereas EPHB6, which plays important role in gut homeostasis, is generally considered as a candidate gene for ASD [
26]. Furthermore, a genetic association was found between the NTRK3 gene and both autism and Asperger syndrome [
27]. Additionally, altered P53 and p53-dependent pathways have been reported in autism [
24]. AGO2 mutations in some patients contribute to disturbances in terms of neurological development and the outcome of ASD [
28,
29]. In our cohort, AGO2 was identified in the pathway analysis of ASD vs. Control and was found to be more specific to individuals that expressed more severe symptoms of ASD. Consequently, we will explore the role of AGO2 further. These results are consistent with prior knowledge of ASD phenotypes, providing new insights into interpretations of the underlying molecular mechanisms in ASD.
The relevance of ncRNAs, especially miRNAs, to human diseases and disorders was initially extensively studied in cancer [
16,
17,
18,
44,
45]. Besides miRNAs, other ncRNAs, such as PIWI-interacting RNAs (piRNAs), act as key elements in cellular homeostasis and are crucial in transposon silencing during embryo development [
46]. While cir-miRNAs were proven to be highly stable in the blood, piRNAs have also emerged as being stably expressed in circulation [
40]. Remarkably, specific piRNAs have been useful in distinguishing between tumors and non-tumor tissues [
16,
17], and have been suggested to contribute to colorectal cancer development and risk, a concept of identification and exploration that may transcend into potentially aiding in a more objective classification of individuals within the ASD spectrum. Consequently, this suggests that it is an emerging, highly promising diagnostic and prognostic biomarker discovery and intervention area. Our study identified piRNAs that could, hypothetically, be used to help clinicians better faction between individuals, furthering more personalized treatment approaches.
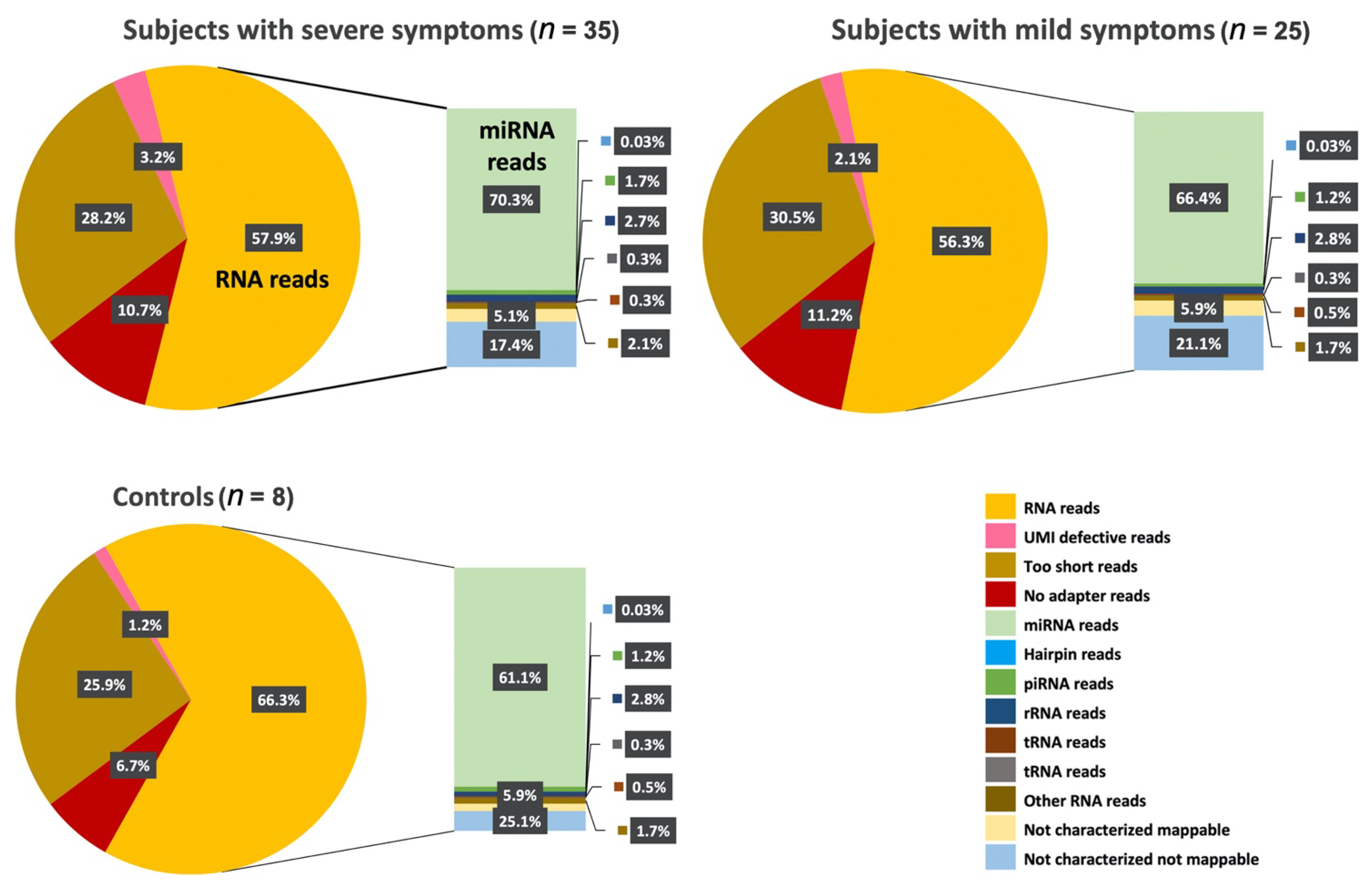
Excitingly, a growing number of isolation methods for profiling cir-ncRNAs are being developed, making this a captivating and promising area of research. One of the significant advantages of deep sequencing platforms is its ability to identify a considerable number of ncRNA transcripts. As we have shown in our study, high-throughput sequencing revealed the existence of other classes of ncRNAs, including snoRNAs, Y-RNAs, tRNAs, and lncRNAs, indicating a wide range of regulatory RNAs with a wide variety of processing mechanisms and functions.
Using small RNA high-throughput sequencing, we demonstrated that the ~110 nucleotides (nt) long Ro-associated Y-RNAs (also called RNYs or Y-RNAs) are present in plasma. Our investigations elucidated that Y-RNA, hY3, and pseudogene hY3P1 are differentially downregulated in individuals with more severe symptoms of ASD. RNY4 Pseudogene 28 and 29, were further identified to be differentially expressed in the severe symptom group. Y-RNAs have emerged as being important for the initiation of chromosomal DNA replication, RNA stability, and cellular responses to stress [
47]. As seen in the other types of non-coding RNA, past investigations into Y-RNA have mainly focused on cancer research, where an accumulation of investigations have shown that fragments of Y-RNAs displayed significant differential expression patterns, in both circulation and/or in tumor tissues as compared to controls [
48]. This concept is exciting, as this holds promise regarding the role of Y-RNA as a biomarker in ASD, providing a platform to differentiate between ASD’s characteristic and symptom states, as well as, potentially, other neurodevelopmental disorders that may share similar symptoms to ASD. However, Y-RNA research is relatively new, and more in-depth research is required to better understand its functioning.
On the other hand, snoRNAs also offer novel insights into, and promising perspectives for, ASD. According to our analysis, the SNORA69 (known as U69) is the most up-regulated small nucleolar RNA, whereas SNORD42A (U42) is the most down-regulated snoRNA in individuals that expressed more severe ASD symptoms. Interestingly, a microdeletion of a subtype of snoRNA (HBI-85), has been previously associated with Prader–Willi-Like Syndrome (PWLS) [
8]. Since PWLS has overlapping ASD characteristics (i.e., social difficulties), it is exciting to note the potential to utilize non-coding biomarkers as an objective identifier between similar neurodevelopmental conditions, further bringing us closer to better treatment strategies. While the exact roles of SNORA69 and SNORD42A were not specifically unveiled in the field of ASD, this suggests a promising area of investigation.
Collectively, our study supports and builds on other contributions in the field. To our knowledge, our study is the first to evaluate cir-ncRNAs in the context of two groups of symptom severities, in order to objectively identify differentiating flags between groups. Our study and consequent findings aim to improve on the currently existing platform of diagnosing children with ASD. Due to the complexities associated with ASD, one must develop a system to further appreciate the biological players that contribute to its molecular and phenotypic outcome. Furthermore, one must stress the importance of identifying objective and reliable data to faction ASD severities and characteristics within the spectrum and not solely rely on diagnostic comparisons of cases vs. controls. Opening our eyes to the possibility of assessing ASD within its spectrum is as essential as assessing ASD as compared to other neurodevelopmental conditions or control groups. This would potentially lead to even greater and exceptional management of personalized treatment strategies and outcomes.
Our capacity to truly understand ASD is limited by the subjectivity of analysis. While it is unlikely that one specific ncRNA is a factor between an individual without ASD and one with ASD, identifying cir-ncRNAs as antecedent biomarkers to identify the risk of developing ASD, as well as utilizing them as a tool to screen those already diagnosed with ASD and objectively assess ASD severity could revolutionize the current paradigm. In general, our pilot study identified ncRNAs as biomarkers indicative of altered neurodevelopment, and this impairment was measurable via alternations in their levels. Moreover, while we have identified specific ncRNAs that have significant expression levels between ASD and control, or within severe vs. mild symptomology groups, we believe that a panel of biomarkers, whether they are within an identified non-coding family, in combination with other ncRNA, or in association with other proteins, would provide a higher degree of specificity and sensitivity in terms of diagnosis, compared to a single biomarker.
Finally, while this study did not characterize the biological functions of cir-ncRNAs in vitro, it did substantiate a very sensitive methodology for profiling all circulating extracellular RNAs and explored their functional relationships using sophisticated, pathway-based analysis tools. The study suggests that one should prioritize the identified candidate ncRNAs for further validation in a larger cohort and perform rigorous functional studies.
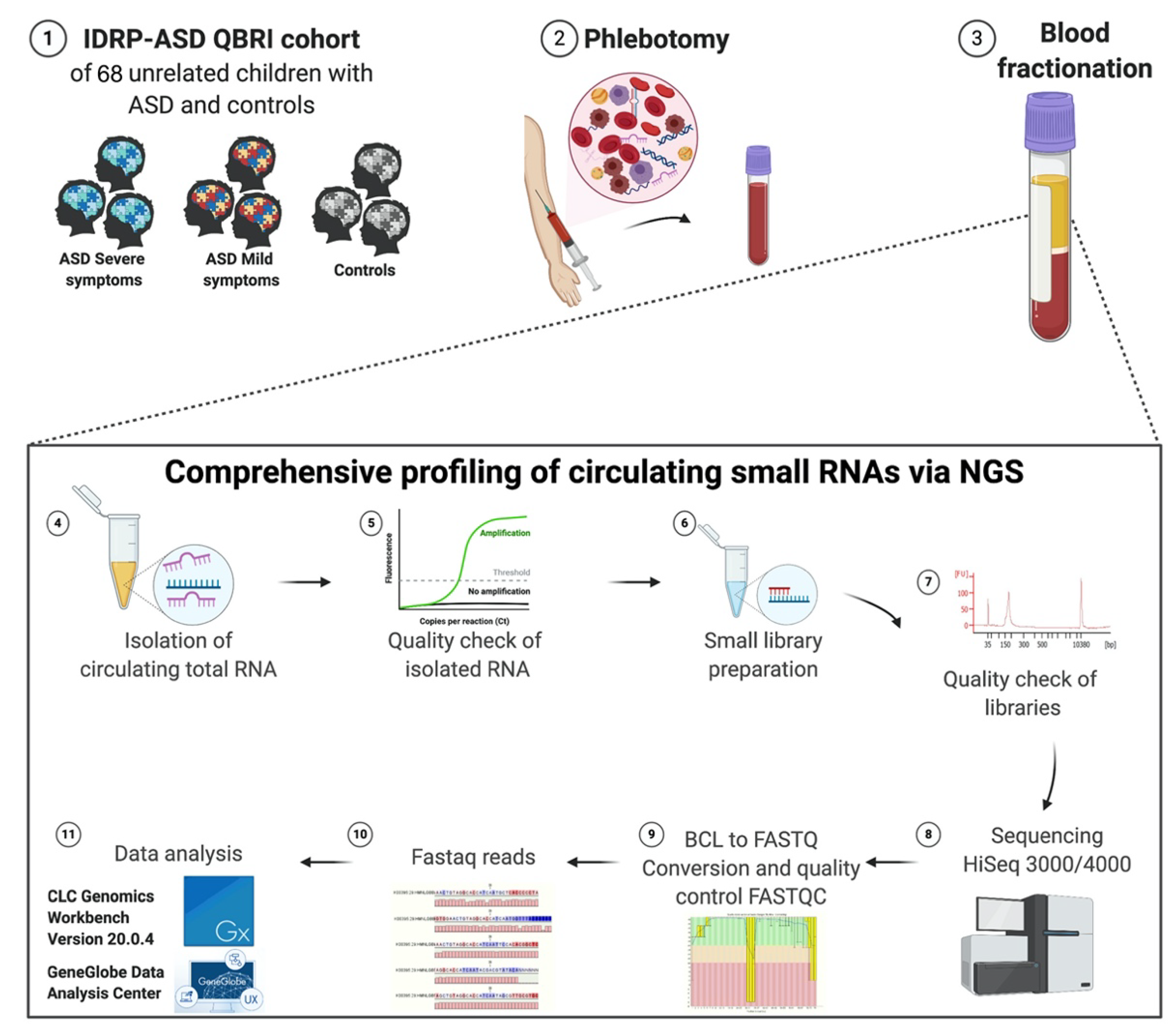
4. Materials and Methods
4.1. Subjects—The Interdisciplinary Research Program (IDRP) ASD Cohort
The samples utilized in this study were obtained from a depository belonging to Qatar Biomedical Research Institute (QBRI) Interdisciplinary Research Program (IDRP) entitled Identifying Potential Molecular Biomarkers for ASD. The umbrella study encompasses various disciplines and a blend of omic investigations to further our understanding of the fundamental underpinnings of ASD and establish diagnostic tools for its early detection. Children ranging in age from 3 to 15 years were recruited, alongside their parents. Recruitment was solely based on families that reside in Qatar. ASD cases were subdivided based on characteristic symptoms of ASD or were diagnosed as having ASD with an associated comorbidity (i.e., attention-deficit/hyperactivity disorder (ADHD), intellectual disability (ID), or epilepsy). This study’s strength lies in the varying attributes used to define the divisions within the cohort based on the absence of communication and social skills, symptomatology, and comorbidities. Age-matched control groups included siblings/healthy individuals from the general population and a neurodevelopmental disorder group of age-matched children that solely elicited ADHD, ID, or epilepsy.
For our current pilot study, we subdivided children into those that exhibited severe (
n = 35), mild symptoms (
n = 25), and controls (
n = 8). The clinical characteristics of the subjects are described in
Table 1.
4.2. ASD Assessment
Children were clinically assessed and diagnosed with ASD at the Rumailah Hospital and Shaffalah Center for Children with Special Needs, Doha, Qatar. All children were diagnosed through a specialized, multidisciplinary team (MDT) consisting of medical doctors, psychiatrists, clinical nurse specialists, community mental health nurses, psychologists, social workers, and occupational therapists. Furthermore, validated screening and diagnostic tests and tools, including the Diagnostic and Statistical Manual of Mental disorders (DSM-5), Autism Diagnostic Observation Schedule, Second Edition (ADOS), and Autism Diagnostic Interview-Revised (ADI-R), were used.
4.3. Severity Classification
To ensure that the analyzed samples were grouped accordingly, the following criteria was used: (1) ADOS-2 score to verify the initial clinical diagnosis, (2) DSM-5 severity levels, and (3) clinical assessment by experienced and qualified clinicians. Due to the complexity and heterogeneity of ASD, classifying an individual as having the disorder is a perplexing endeavor. Hence, to in order respect and be sensitive to the extensive and multifaced classification of ASD diagnosis, we have divided our findings into two groups, the first of which represents individuals that exhibit severe symptoms, displaying multiple, unambiguous characteristics of ASD, including severe behavioral phenotypes (i.e., significant alternations in social and language development), and those that show mild symptoms of ASD.
4.4. Collection of Human Blood/Plasma
The collection of blood samples complied with the national guidelines that oversee research investigations comprising vulnerable subjects such as children. Well-trained phlebotomists, with extensive experience working with children with special needs, were responsible for collecting venous blood samples. Furthermore, an EMLA cream was used with the local anesthesia to avoid and/or reduce pain sensitivity during blood withdrawal. Samples were collected into VACUETTE® tubes containing EDTA, centrifuged at 1800 rpm for 10 min, followed by plasma collection and re-centrifugation for 10 min at 3000 rpm. Finally, plasma samples were aliquoted into 200 μL aliquots and stored at −80 °C until further use.
4.5. RNA Isolation from Peripheral Blood Plasma
Frozen plasma samples were thawed in a 37 °C water bath. The thawed plasma samples were centrifuged at 400× g (~2000 rpm) for 2 min to remove cells and precipitated plasma proteins/lipids. According to the manufacturer’s instructions, cell-free (cf) plasma samples were transferred to new tubes for RNA isolation using the miRNeasy Serum/Plasma Advanced Kit (Cat. no. 217204, Qiagen, Germany). We optimized the recommended starting amount of plasma; due to the low quantity of cfRNA, we used 200 μL of plasma for total RNA extraction, with the addition of 52 Qiaseq miRNA Library QC Spike-ins (Cat. no.: 331541, Qiagen, Germany) as an internal control for miRNA expression profiling in plasma.
4.6. QIAseq miRNA Library Quality Check
The QIAseq miRNA Library QC qPCR Assay Kit (Cat. no. 331551, Qiagen, Germany) was used to evaluate RNA isolation quality before small RNA library preparation and to assess NGS performance post-sequencing. The kit provides 52 Spike-Ins controls with a qPCR panel that monitors the technical quality of the whole process from RNA isolation (by evaluating the reproducibility) to sequencing data analysis (by checking the reads). This method also enables the detection of enzymatic inhibitors or nucleases and hemolysis assessment (necessary for plasma miRNA identification). In brief, the procedure started during RNA isolation with the addition of 52 QIAseq miRNA Library QC Spike-Ins to the samples. The sample evaluation was determined using qRT-PCR. For the identification of RNA isolation efficiency, the calculation of delta CT for UniSp100 (CT: 31–34 range) and UniSp101 (CT: 25–28 range) was assessed and found to be within the recommended value of around 5–7. For the inhibitor detection, the UniSp6 was measured. The value was <2 CTs between any two samples. For hemolysis, delta CT (miR-23a–miR-451a) was less than 5, ensuring high-quality samples. A value of 5–7 was considered a borderline sample. Samples with a value > 7 were not used.
4.7. Small RNA Library Preparation
For the library construction and molecular indexing, the QIAseq miRNA Library Kit (96) (Cat. no. 331505, Qiagen, Germany) and QIAseq miRNA NGS 96 Index IL (Cat. no. 331565, Qiagen, Germany) were used. The gold standard approach for the normalization of circulating miRNAs utilizes equal amounts of biofluids and isolated total RNA, and the spike-ins are used as normalization controls. Thus, 5 μL of 15 μL total RNA column eluate was used for library preparation. RNA samples were subjected to 3′ and 5′ adaptor ligation targeting miRNAs by reverse transcription to generate the cDNA construct based on small RNA having 3′ and 5′ adaptor ligation. This reverse transcription step will help to enrich the RNA fragments with 3′ and 5′ adaptors on both ends. The reverse transcription (RT) primer contained an integrated Unique Molecular Indices (UMI). The RT primer binds to a region of the 3′ adapter and facilitates conversion of the 3′/5′ ligated miRNAs into cDNA while assigning a UMI to every miRNA molecule. During reverse transcription, a universal sequence is also added. The sample indexing primers recognize this during library amplification. cDNA constructs were purified using a streamlined magnetic bead-based method. Then, the unbiased amplification of libraries was accomplished using a dried universal forward primer from a plate paired with 1 of 96 dried reverse primers in the same plate (Cat. no. 331565, Qiagen, Germany).
Consequently, this assigned each sample a unique custom index. After the library amplification, a cleanup was performed, using the streamlined magnetic bead-based method. Validation of the libraries was performed using Agilent technologies 2100 Bioanalyzer with an Agilent High Sensitivity DNA assay (Cat. no. G2938-90020, Agilent Technologies, United States). A unique peak of around 141 bp was obtained (a purified library example is shown in
Figure 1).
4.8. Small RNA Deep Sequencing
cDNA libraries were measured based on the average size obtained from the bioanalyzer using the Qubit Fluorometer, Qubit HS dsDNA Assay Kit (Cat. no. Q32854, Thermo Fisher Scientific, United States). Libraries were diluted to 10 nM using a resuspension buffer and pooled with unique indexing for Illumina. The final dilution loaded was 3nM, with further clustering on cBot2, and sequencing on the Illumina platform was achieved using the HiSeq 3000/4000 SBS Kit (150 cycles). To discover novel miRNAs, we aimed to generate up to 20 million reads per sample. The adapters were trimmed. The raw data from the Illumina HiSeq 3000/4000 were converted from bcl2 to fastq format.
4.9. Sequencing Read Mapping and Small RNA Annotation
The raw sequence files from the Illumina HiSeq 3000/4000 in the
BCL format were converted to the
FASTQ format using the bcl2fastq v1.8.4 conversion tool software (Illumina, United States). Reads were filtered and adapters were trimmed. After adapter trimming, the read data were evaluated for quality using FASTQC to filter out reads with a quality score [
49].
4.10. UMI Analysis: The Geneglobe Data Analysis Center
The GeneGlobe data analysis center (accessed on 18 February 2020,
www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/) can align and report on the QIAseq miRNA spike-ins in addition to the aligned small/miRNA/piRNA from each sample. This QIAGEN analysis tool was used to assess the effectiveness of QIAseq’s UMIs. For the synthetic miRNA samples, the option ‘other’ was chosen for mapping, while ‘human’ was chosen for the human total RNA samples during the primary data analysis. The resulting count table included UMI and raw read counts for each miRNA in the samples. Before analyzing the correlation between UMI and raw read counts, the counts were rlog transformed.
Next-generation sequencing (NGS) allows not only the quantification of known miRNAs, but also the identification and quantification of novel miRNAs, isomiRs (miRNA variants), and other small RNA species that can be functionally relevant in diseases and is therefore used as potential disease biomarker (
Figure 2). miRNAs are identified by aligning the reads to miRBase (version 21), and the reads are tallied to generate total counts for each miRNA. Statistical significance (
p-Value) between two or more samples was calculated to generate differential expression profiles.
4.11. Differential Expression Analysis: CLC Genomics Workbench Version 20.0.4
Files were then exported to the CLC Genomics Workbench (version 20.0.4) for read mapping to the hg38 human genome version. This allowed for a single-mismatched base, down to 18 nucleotides. Analysis of the resulting data was performed using small RNA analysis tools in CLC Genomics Workbench. Spike-in reads were filtered out from the rest of the data. A “perfect match” setting was applied when mapping, filtering, and counting QIAaseq NGS Spike-in reads in a dataset. After counting the QIAseq NGS Spike-in reads, they should be normalized to the total number of reads per sample. After this normalization, correlation matrices should be plotted for all sample-to-sample comparisons. This is done to evaluate the sample-to-sample correlation in the sample set. The expected correlation should be R2 of 0.95–0.99. If samples deviate from these values, they could be technical outliers, and potentially excluded from downstream analysis.
Using the Biomedical Genomics Analysis plugin that supports the analysis of reads sequenced using the QIAseq miRNA Library Kit, the QIAGEN miRNA Quantification workflow quantified the expression in each sample miRNAs found in miRBase. Reads were first mapped to miRBase version 21 database (accessed on: 9 May 2020,
www.mirbase.org) and piRNABank database Human_piRNA_sequence_v1.0 (accessed on: 9 May 2020,
www.regulatoryrna.org/database/piRNA/) to assign reads to miRNAs and piRNAs, respectively, and to exclude them before mapping to the whole human genome. The unmapped reads from the QIAseq miRNA quantification workflow were collected and mapped using RNA-seq analysis to assign reads to other non-coding RNAs, such as Y-RNAs, snoRNAs, tRNAs, and lncRNAs.
The QIAseq miRNA Quantification tool allows for the grouping of miRNA into mature miRNA; the same mature miRNA may be produced from different precursor miRNAs, or on seed, as the same seed sequence may be found in different mature miRNAs. A custom database for piRNAs was also provided. This miRNA grouped on seed was used for further analysis through the Ingenuity Pathway Analysis (IPA) platform. The workflow calculates differential expressions for expression tables with associated metadata using multi-factorial statistics based on a negative binomial Generalized Linear Model (GLM). Both Grouped on Mature and Grouped on Seed expression tables can be used. Integrated Unique Molecular Indices enable the quantification of individual miRNA molecules, eliminating PCR and sequencing bias. For the differential expression analysis, miRNAs were deemed to be statistically differentially expressed if they had an expression of greater than 50 read counts at an absolute fold change > two and an adjusted p < 0.05.
4.12. Functional Enrichment Tests
We used the Ingenuity Pathway Analysis (IPA) system for pathway analysis and molecular networks to perform the candidate miRNAs’ functional enrichment tests. The IPA system provides a more comprehensive pathway resource based on manual collection. The rich information returned by IPA is also suitable for pathway crosstalk analysis, as it has almost all molecules, including their connections. Briefly, the IPA system implements Fisher’s exact test to determine the pathways that are enriched with miRNAs of interest. Furthermore, the IPA system’s network analysis searches for significant molecular networks in a commercial knowledge base, including integrative information from the literature, gene expression, and gene annotation.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}