
Isookanin Inhibits PGE2-Mediated Angiogenesis by Inducing Cell Arrest through Inhibiting the Phosphorylation of ERK1/2 and CREB in HMEC-1 Cells

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effects of Isookanin on PGE2-Induced Endothelial Cell Proliferation and Cytotoxicity
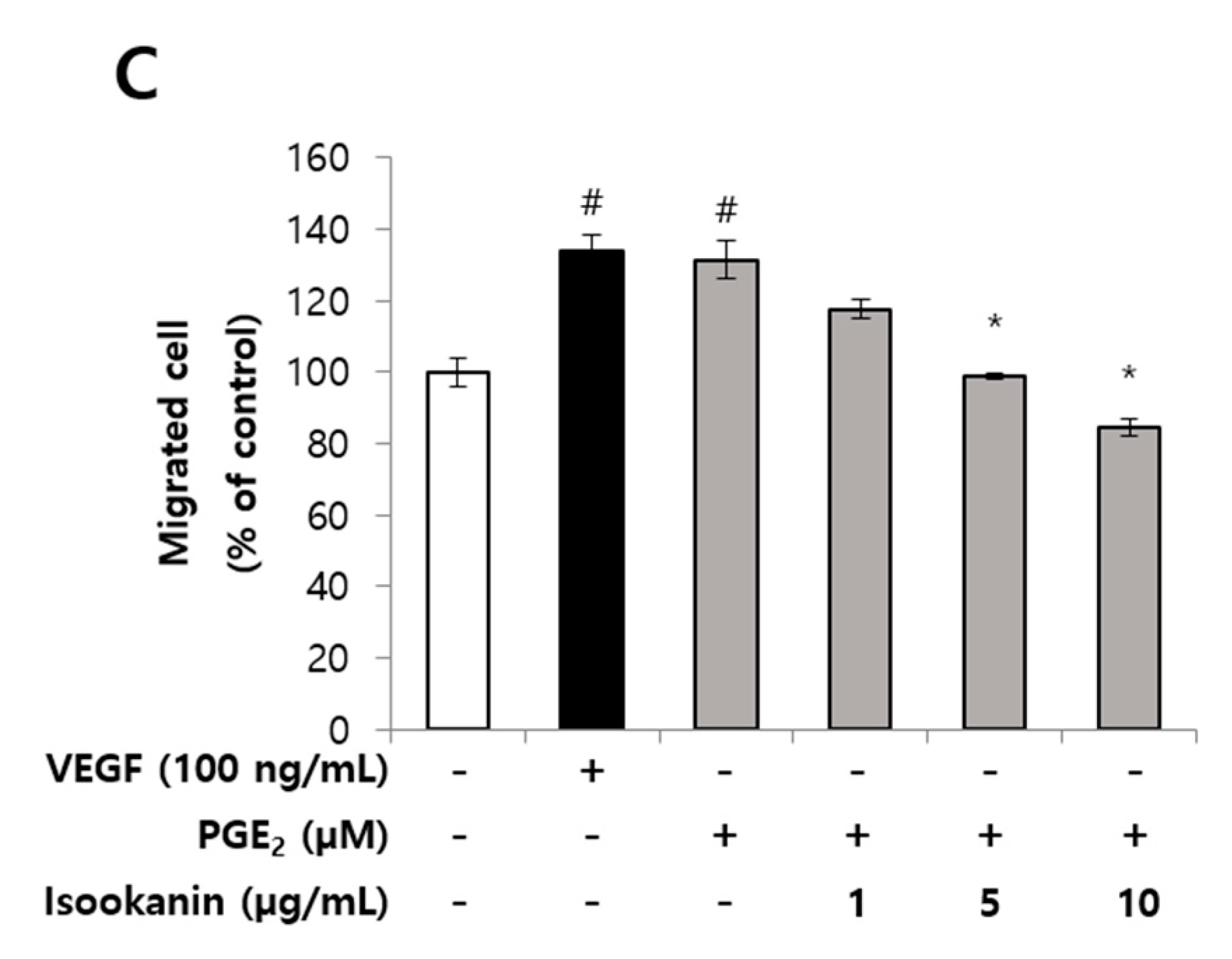
2.2. Effects of Isookanin on PGE2-Induced Endothelial Cell Migration
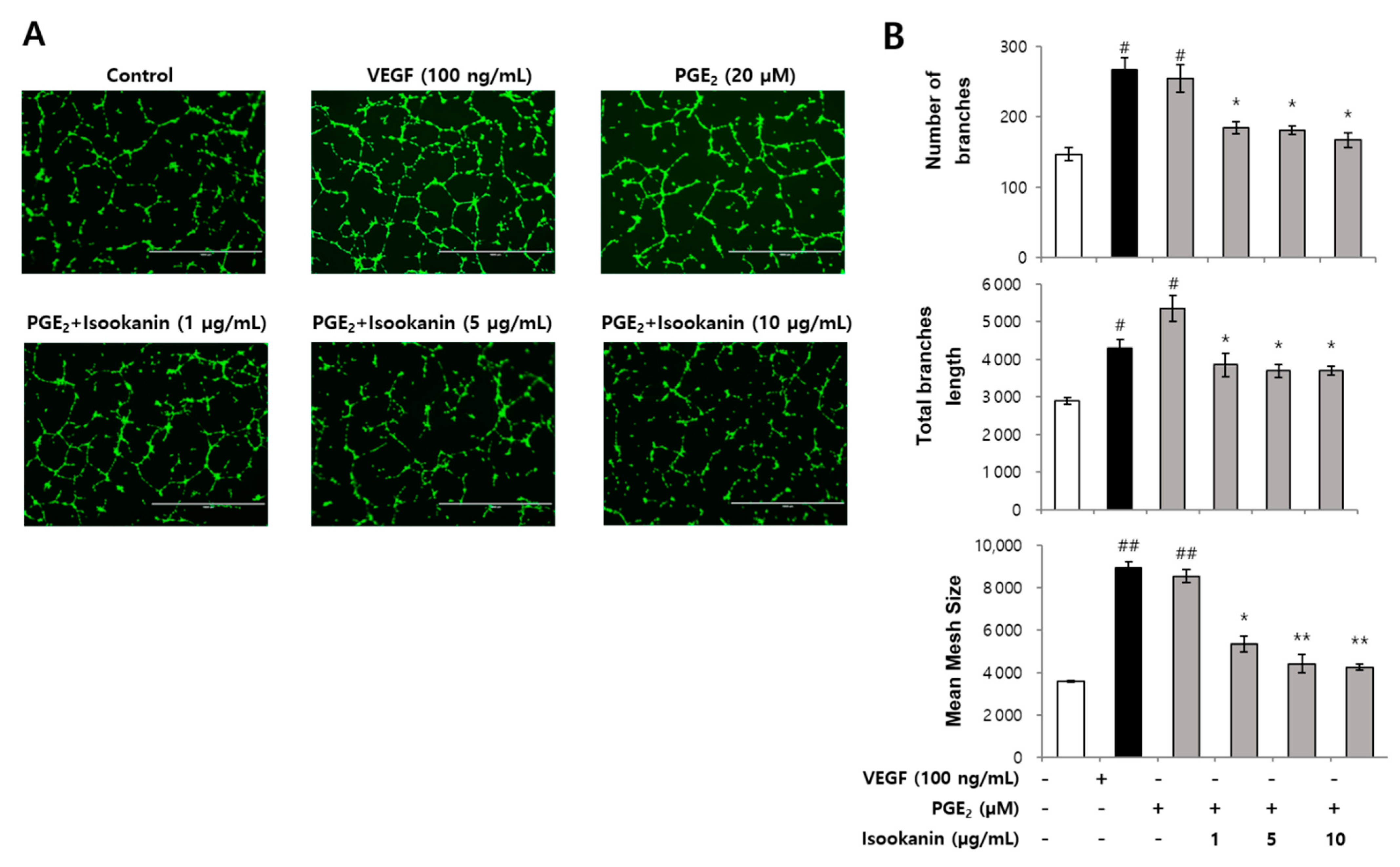
2.3. Effect of Isookanin on PGE2-Induced Endothelial Cell Tube Formation
2.4. Effect of Isookanin on HMEC-1 Proliferation Inhibition
2.5. Effects of Isookanin on the Regulation of Cell Cycle and the Expression Level of Cell Cycle Regulatory Proteins
2.6. Effects of Isookanin on Phophorylation of ERK1/2 and CREB in PGE2-Induced HMEC-1 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Lactate Dehydrogenase (LDH) Cytotoxicity Assay
4.5. Scratch Migration Assay
4.6. Transwell Migration Assay
4.7. Matrigel Endothelial Cell Tube Formation Assay
4.8. Flow Cytometry Analysis of Edu Cell Proliferation Assay and Cell Cycle Arrest
4.9. Western Blotting
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adair, T.H.; Montani, J.-P. Angiogenesis, Colloquium Series on Integrated Systems Physiology: From Molecule to Function; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010; pp. 1–84. [Google Scholar]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Velasco, P.; Lange-Asschenfeldt, B. Dermatological aspects of angiogenesis. Br. J. Dermatol. 2002, 147, 841–852. [Google Scholar] [CrossRef] [PubMed]
- van Hinsbergh, V.W.; Collen, A.; Koolwijk, P. Angiogenesis and anti-angiogenesis: Perspectives for the treatment of solid tumors. Ann. Oncol. 1999, 10, S60–S63. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.; Folkman, J. Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2002, 2, 727–739. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. Cyclooxygenase pathways. Acta Biochim. Pol. 2014, 61. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Atertio. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Dey, I.; Lejeune, M.; Chadee, K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br. J. Pharmacol. 2006, 149, 611–623. [Google Scholar] [CrossRef]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Bruckner, M.; Uetz-von Allmen, E.; Krause, P. Prostaglandin E2 at new glance: Novel insights in functional diversity offer therapeutic chances. Int. J. Biochem. Cell Biol. 2010, 42, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Daaka, Y. PGE2 promotes angiogenesis through EP4 and PKA Cγ pathway. Blood J. Am. Soc. Hematol. 2011, 118, 5355–5364. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef]
- Hatazawa, R.; Tanigami, M.; Izumi, N.; Kamei, K.; Tanaka, A.; Takeuchi, K. Prostaglandin E 2 stimulates VEGF expression in primary rat gastric fibroblasts through EP4 receptors. Inflammopharmacology 2007, 15, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Redha, R.; Macias-Perez, I.; Su, Y.; Hao, C.; Zent, R.; Breyer, M.D.; Pozzi, A. Prostaglandin E2-EP4 receptor promotes endothelial cell migration via ERK activation and angiogenesis in vivo. J. Biol. Chem. 2007, 282, 16959–16968. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Sales, K.J.; Katz, A.A.; Jabbour, H.N. Seminal plasma promotes the expression of tumorigenic and angiogenic genes in cervical adenocarcinoma cells via the E-series prostanoid 4 receptor. Endocrinology 2006, 147, 3356–3365. [Google Scholar] [CrossRef] [PubMed]
- Yanni, S.E.; Barnett, J.M.; Clark, M.L.; Penn, J.S. The role of PGE2 receptor EP4 in pathologic ocular angiogenesis. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5479–5486. [Google Scholar] [CrossRef]
- Olivieri, D.; Chetta, A. Therapeutic perspectives in vascular remodeling in asthma and chronic obstructive pulmonary disease. Angiogenesis Lymphangiogenesis Clin. Implic. 2014, 99, 216–225. [Google Scholar]
- Zhang, W.; Li, F.; Gao, W. Tripterygium wilfordii inhibiting angiogenesis for rheumatoid arthritis treatment. J. Natl. Med. Assoc. 2017, 109, 142–148. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Govindarajan, B.; Battle, T.E.; Lynch, R.; Frank, D.A.; Ushio-Fukai, M.; Perry, B.N.; Stern, D.F.; Bowden, G.T.; Liu, A. Carbazole is a naturally occurring inhibitor of angiogenesis and inflammation isolated from antipsoriatic coal tar. J. Investig. Dermatol. 2006, 126, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.X.; Cleck, J.N. Adverse effects of anticancer agents that target the VEGF pathway. Nat. Rev. Clin. Oncol. 2009, 6, 465. [Google Scholar] [CrossRef] [PubMed]
- Des Guetz, G.; Uzzan, B.; Chouahnia, K.; Morère, J.-F. Cardiovascular toxicity of anti-angiogenic drugs. Target. Oncol. 2011, 6, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Rajasekar, J.; Perumal, M.K.; Vallikannan, B. A critical review on anti-angiogenic property of phytochemicals. J. Nutr. Biochem. 2019, 71, 1–15. [Google Scholar] [CrossRef]
- Yang, X.-W.; Huang, M.-Z.; Jin, Y.-S.; Sun, L.-N.; Song, Y.; Chen, H.-S. Phenolics from Bidens bipinnata and their amylase inhibitory properties. Fitoterapia 2012, 83, 1169–1175. [Google Scholar] [CrossRef]
- Ahmed, D.; Kumar, V.; Sharma, M.; Verma, A. Target guided isolation, in-vitro antidiabetic, antioxidant activity and molecular docking studies of some flavonoids from Albizzia Lebbeck Benth. bark. BMC Complement. Altern. Med. 2014, 14, 155. [Google Scholar] [CrossRef]
- Wang, W.; Chen, W.; Yang, Y.; Liu, T.; Yang, H.; Xin, Z. New phenolic compounds from Coreopsis tinctoria Nutt. and their antioxidant and angiotensin I-converting enzyme inhibitory activities. J. Agric. Food Chem. 2015, 63, 200–207. [Google Scholar] [CrossRef]
- Xin, Y.-J.; Choi, S.; Roh, K.-B.; Cho, E.; Ji, H.; Weon, J.B.; Park, D.; Whang, W.K.; Jung, E. Anti-Inflammatory Activity and Mechanism of Isookanin, Isolated by Bioassay-Guided Fractionation from Bidens pilosa L. Molecules 2021, 26, 255. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, Y.; Xing, D. Low-power laser irradiation (LPLI) promotes VEGF expression and vascular endothelial cell proliferation through the activation of ERK/Sp1 pathway. Cell. Signal. 2012, 24, 1116–1125. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Montesano, R.; Orci, L.; Vassalli, P. In vitro rapid organization of endothelial cells into capillary-like networks is promoted by collagen matrices. J. Cell Biol. 1983, 97, 1648–1652. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, H.; Wan, H.; Zou, X.; Ma, X.; Gao, G. Harmine suppresses the proliferation and migration of human ovarian cancer cells through inhibiting ERK/CREB pathway. Oncol. Rep. 2017, 38, 2927–2934. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Kim, S.-W.; Cheon, K.; Tabassam, F.H.; Yoon, J.-H.; Koo, J.S. Nonclassical action of retinoic acid on the activation of the cAMP response element-binding protein in normal human bronchial epithelial cells. Mol. Biol. Cell. 2006, 17, 566–575. [Google Scholar] [CrossRef] [PubMed]
- New, D.C.; Wong, Y.H. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2007, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Koch, A.E. Mechanisms of disease: Angiogenesis in inflammatory diseases. Nat. Clin. Pract. Rheumatol. 2007, 3, 635–643. [Google Scholar] [CrossRef]
- Kofler, S.; Nickel, T.; Weis, M. Role of cytokines in cardiovascular diseases: A focus on endothelial responses to inflammation. Clin. Sci. 2005, 108, 205–213. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The role of angiogenesis in cancer treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Gáspár, L.; Koch, A.E. Angiogenesis in rheumatoid arthritis. Front Biosci 2005, 10, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Pousa, I.; Maté, J.; Gisbert, J. Angiogenesis in inflammatory bowel disease. Eur. J. Clin. Investig. 2008, 38, 73–81. [Google Scholar] [CrossRef]
- EL-Meghawry, E.; Rahman, H.; Abdelkarim, G.; Najda, A. Natural products against cancer angiogenesis. Tumor Biol. 2016, 37, 14513–14536. [Google Scholar]
- Ji, H.F.; Li, X.J.; Zhang, H.Y. Natural products and drug discovery: Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Rep. 2009, 10, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Klemm, D.J.; Watson, P.A.; Frid, M.G.; Dempsey, E.C.; Schaack, J.; Colton, L.A.; Nesterova, A.; Stenmark, K.R.; Reusch, J.E.-B. cAMP response element-binding protein content is a molecular determinant of smooth muscle cell proliferation and migration. J. Biol. Chem. 2001, 276, 46132–46141. [Google Scholar] [CrossRef] [PubMed]
- Reusch, J.E.; Colton, L.A.; Klemm, D.J. CREB activation induces adipogenesis in 3T3-L1 cells. Mol. Cell. Biol. 2000, 20, 1008–1020. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Filiz, G.; Brown, D.; Hollande, F.; Gonzales, M.; D’Abaco, G.; Papalexis, N.; Phillips, W.; Malaterre, J.; Ramsay, R.G. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis 2014, 3, e108. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Kessler, K.M.; Pincheira, R.; Warren, R.S.; Donner, D.B. Vascular endothelial cell growth factor activates CRE-binding protein by signaling through the KDR receptor tyrosine kinase. J. Biol. Chem. 2001, 276, 25184–25189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-J.; Liu, C.; Wang, Y.-X.; Zhu, N.; Hu, Z.-Y.; Liao, D.-F.; Qin, L. Long non-coding RNA-SRA promotes neointimal hyperplasia and vascular smooth muscle cells proliferation via MEK-ERK-CREB pathway. Vascul. Pharmacol. 2019, 116, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta BBA Mol. Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Pozzi, A.; Yan, X.; Macias-Perez, I.; Wei, S.; Hata, A.N.; Breyer, R.M.; Morrow, J.D.; Capdevila, J.H. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J. Biol. Chem. 2004, 279, 29797–29804. [Google Scholar] [CrossRef]
- Kim, G.D. Hesperetin inhibits vascular formation by suppressing of the PI3K/AKT, ERK, and p38 MAPK signaling pathways. Prev. Nutr. Food Sci. 2014, 19, 299. [Google Scholar] [CrossRef]
- Song, Y.; Dai, F.; Zhai, D.; Dong, Y.; Zhang, J.; Lu, B.; Luo, J.; Liu, M.; Yi, Z. Usnic acid inhibits breast tumor angiogenesis and growth by suppressing VEGFR2-mediated AKT and ERK1/2 signaling pathways. Angiogenesis 2012, 15, 421–432. [Google Scholar] [CrossRef]
- Ansari, K.M.; Rundhaug, J.E.; Fischer, S.M. Multiple Signaling Pathways Are Responsible for Prostaglandin E2–Induced Murine Keratinocyte Proliferation. Mol. Cancer Res. 2008, 6, 1003–1016. [Google Scholar] [CrossRef]
- Bradbury, D.; Clarke, D.; Seedhouse, C.; Corbett, L.; Stocks, J.; Knox, A. Vascular endothelial growth factor induction by prostaglandin E2 in human airway smooth muscle cells is mediated by E prostanoid EP2/EP4 receptors and SP-1 transcription factor binding sites. J. Biol. Chem. 2005, 280, 29993–30000. [Google Scholar] [CrossRef] [PubMed]
- Battersby, S.; Sales, K.; Williams, A.; Anderson, R.; Gardner, S.; Jabbour, H. Seminal plasma and prostaglandin E2 up-regulate fibroblast growth factor 2 expression in endometrial adenocarcinoma cells via E-series prostanoid-2 receptor-mediated transactivation of the epidermal growth factor receptor and extracellular signal-regulated kinase pathway. Hum. Reprod. 2007, 22, 36–44. [Google Scholar] [PubMed]
- Wang, D.; Wang, H.; Brown, J.; Daikoku, T.; Ning, W.; Shi, Q.; Richmond, A.; Strieter, R.; Dey, S.K.; DuBois, R.N. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J. Exp. Med. 2006, 203, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Konya, V.; Marsche, G.; Schuligoi, R.; Heinemann, A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol. Ther. 2013, 138, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Chell, S.D.; Witherden, I.R.; Dobson, R.R.; Moorghen, M.; Herman, A.A.; Qualtrough, D.; Williams, A.C.; Paraskeva, C. Increased EP4 receptor expression in colorectal cancer progression promotes cell growth and anchorage independence. Cancer Res. 2006, 66, 3106–3113. [Google Scholar] [CrossRef]
- Musser, M.L.; Viall, A.K.; Phillips, R.L.; Hostetter, J.M.; Johannes, C.M. Gene expression of prostaglandin EP4 receptor in three canine carcinomas. BMC Vet. Res. 2020, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Prostaglandin E2 primes the angiogenic switch via a synergic interaction with the fibroblast growth factor-2 pathway. Circ. Res. 2009, 105, 657–666. [Google Scholar] [CrossRef]
- Tsuji, T.; Yamaguchi, K.; Kikuchi, R.; Itoh, M.; Nakamura, H.; Nagai, A.; Aoshiba, K. Promotion of adipogenesis by an EP2 receptor agonist via stimulation of angiogenesis in pulmonary emphysema. Prostaglandins Other Lipid Mediat. 2014, 112, 9–15. [Google Scholar] [CrossRef]
- Rafiee, P.; Heidemann, J.; Ogawa, H.; Johnson, N.A.; Fisher, P.J.; Li, M.S.; Otterson, M.F.; Johnson, C.P.; Binion, D.G. Cyclosporin A differentially inhibits multiple steps in VEGF induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell Commun. Signal. 2004, 2, 1–22. [Google Scholar] [CrossRef][Green Version]
- Koyama, S.; Takagi, H.; Otani, A.; Suzuma, K.; Nishimura, K.; Honda, Y. Tranilast inhibits protein kinase C-dependent signalling pathway linked to angiogenic activities and gene expression of retinal microcapillary endothelial cells. Br. J. Pharmacol. 1999, 127, 537–545. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, Y.; Roh, K.; Cho, E.; Park, D.; Whang, W.; Jung, E. Isookanin Inhibits PGE2-Mediated Angiogenesis by Inducing Cell Arrest through Inhibiting the Phosphorylation of ERK1/2 and CREB in HMEC-1 Cells. Int. J. Mol. Sci. 2021, 22, 6466. https://doi.org/10.3390/ijms22126466
Xin Y, Roh K, Cho E, Park D, Whang W, Jung E. Isookanin Inhibits PGE2-Mediated Angiogenesis by Inducing Cell Arrest through Inhibiting the Phosphorylation of ERK1/2 and CREB in HMEC-1 Cells. International Journal of Molecular Sciences. 2021; 22(12):6466. https://doi.org/10.3390/ijms22126466
Chicago/Turabian StyleXin, Yingji, Kyungbaeg Roh, Eunae Cho, Deokhoon Park, Wankyunn Whang, and Eunsun Jung. 2021. "Isookanin Inhibits PGE2-Mediated Angiogenesis by Inducing Cell Arrest through Inhibiting the Phosphorylation of ERK1/2 and CREB in HMEC-1 Cells" International Journal of Molecular Sciences 22, no. 12: 6466. https://doi.org/10.3390/ijms22126466
APA StyleXin, Y., Roh, K., Cho, E., Park, D., Whang, W., & Jung, E. (2021). Isookanin Inhibits PGE2-Mediated Angiogenesis by Inducing Cell Arrest through Inhibiting the Phosphorylation of ERK1/2 and CREB in HMEC-1 Cells. International Journal of Molecular Sciences, 22(12), 6466. https://doi.org/10.3390/ijms22126466