
Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances



Abstract
1. Introduction
2. Aetiology
2.1. Ultraviolet Radiation
2.2. Skin Phototype
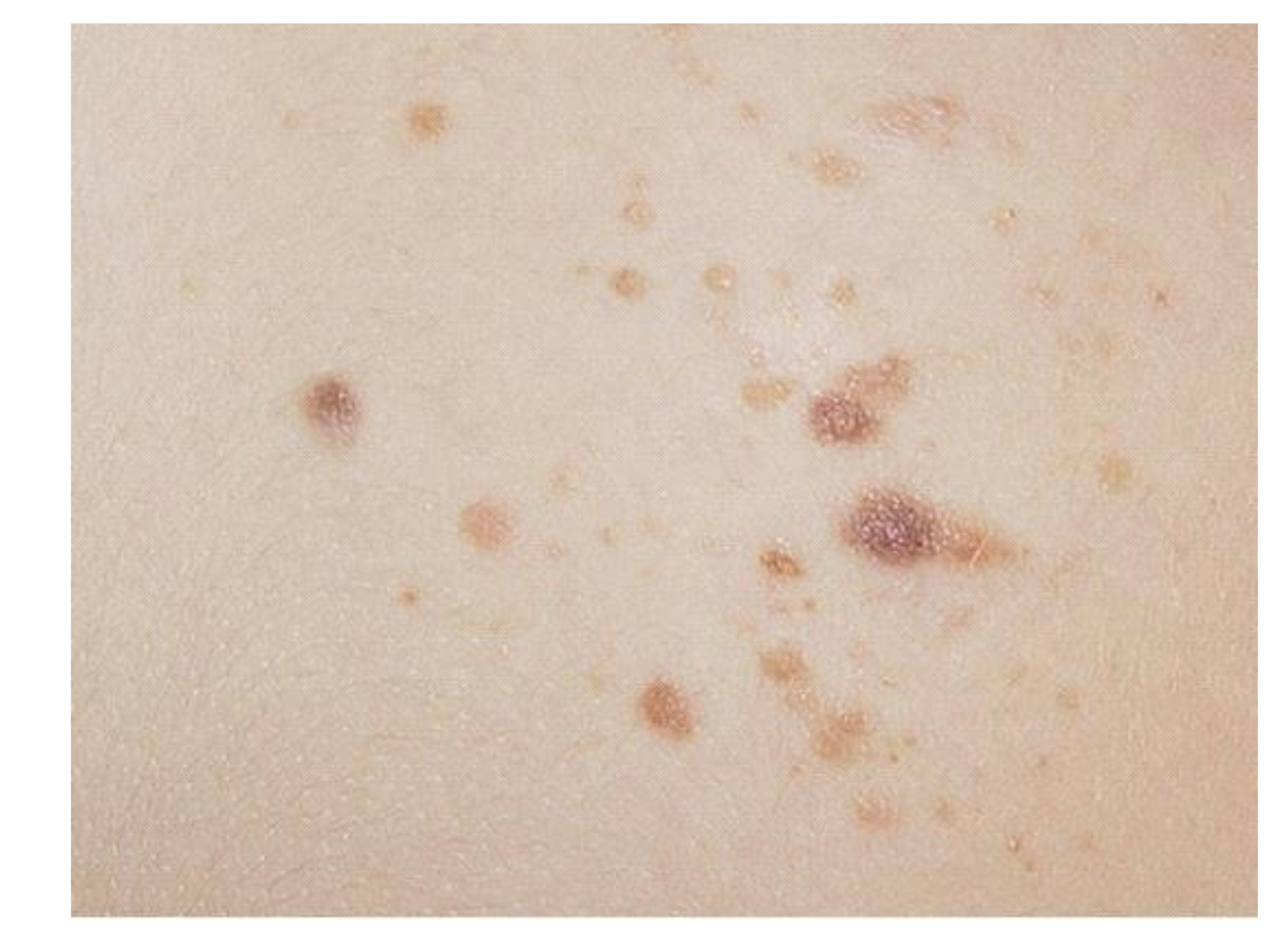
2.3. Pigmented Nevi
2.4. Use of Pesticides
2.5. Prolonged Sun Exposure and Sunburn
2.6. Geographical Location
2.7. Genetic Factors
2.8. Heredity
2.9. Immunosuppressive Conditions
2.10. Non-Melanoma Skin Cancer
3. Pathogenesis of Cutaneous Melanoma
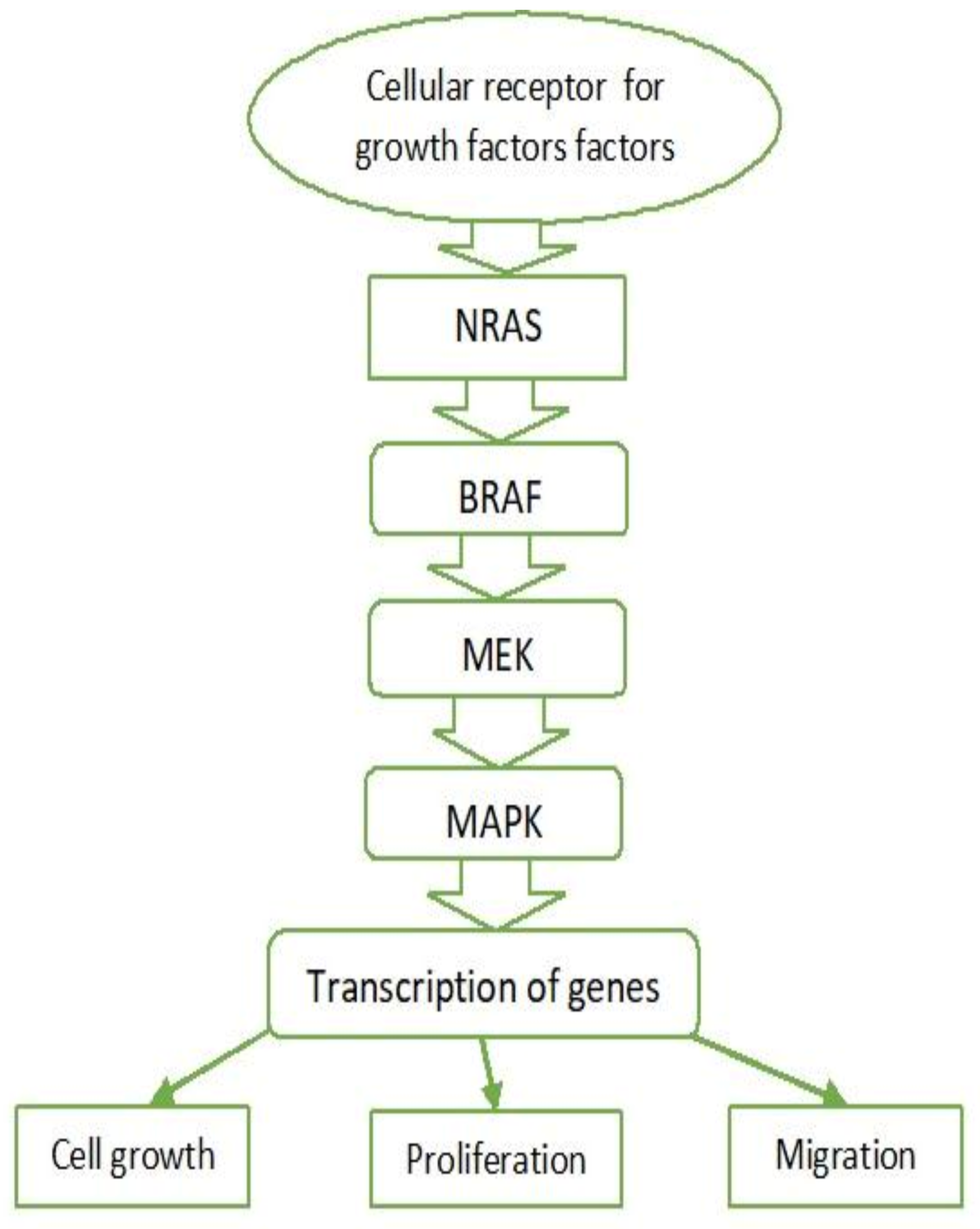
3.1. MAPK Pathway
3.2. BRAF
3.3. RAS
3.4. c-KIT
3.5. c-MET and HGF
3.6. Other Factors
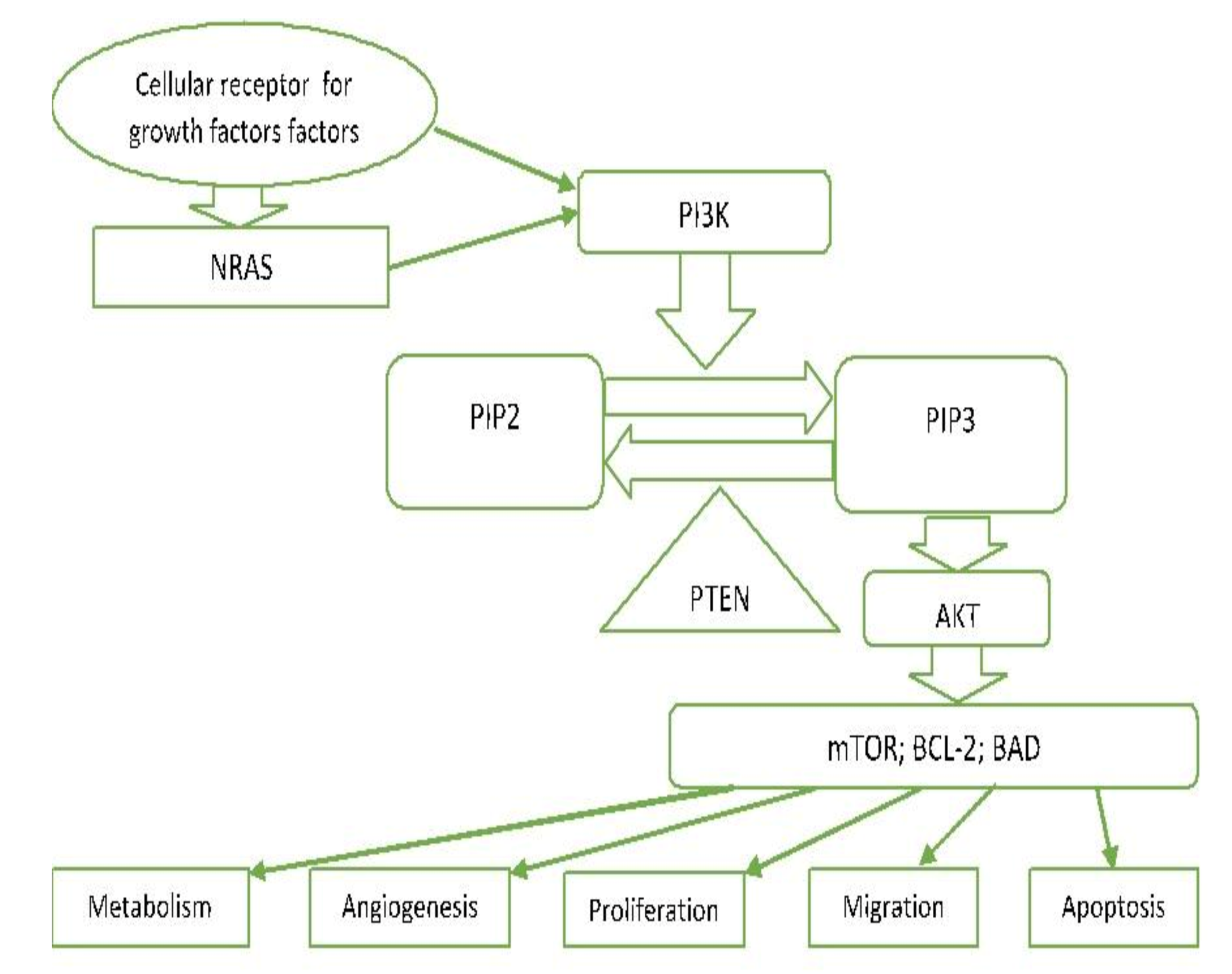
3.7. PI3K/PTEN/AKT Pathway (Phosphatidylinositol 3-kinase Pathway)
3.8. MITF Signalling
3.9. p53
3.10. Hypoxia-Induced Factor (HIF)
3.11. Notch Signalling Pathway
3.12. Other Factors Important in the Pathogenesis of Melanoma
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chin, L.; Merlino, G.; Depinho, R.A. Malignant melanoma: Modern black plague and genetic black box. Genes Dev. 1998, 12, 3467–3481. [Google Scholar] [CrossRef]
- Sayan, M.; Mamidanna, S.; Oncel, D.; Jan, I.; Vergalasova, I.; Weiner, J.; Ohri, N.; Acikalin, B.; Chundury, A. Clinical management of uveal melanoma: A comprehensive review with a treatment algorithm. Radiat. Oncol. J. 2020, 38, 162–169. [Google Scholar] [CrossRef]
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal melanoma. Nat. Rev. Dis. Primers 2020, 6, 1–25. [Google Scholar] [CrossRef]
- Zheng, Y.; Cong, C.; Su, C.; Sun, Y.; Xing, L. Epidemiology and survival outcomes of primary gastrointestinal melanoma: A SEER-based population study. Int. J. Clin. Oncol. 2020, 25, 1951–1959. [Google Scholar] [CrossRef]
- Kahl, A.R.; Gao, X.; Chioreso, C.; Goffredo, P.; Hassan, I.; Charlton, M.E.; Lin, C. Presentation, Management, and Prognosis of Primary Gastrointestinal Melanoma: A Population-Based Study. J. Surg. Res. 2021, 260, 46–55. [Google Scholar] [CrossRef]
- Wohlmuth, C.; Wohlmuth-Wieser, I.; May, T.; Vicus, D.; Gien, L.T.; LaFramboise, S. Malignant Melanoma of the Vulva and Vagina: A US Population-Based Study of 1863 Patients. Am. J. Clin. Dermatol. 2019, 21, 285–295. [Google Scholar] [CrossRef]
- Sun, X.; Gu, Y.; Xie, J.; Wang, L.; Zhou, Q. Melanoma of female genital tract: A clinicopathological analysis of 5 cases. Zhonghua Bing Li Xue Za Zhi 2020, 49, 834–836. [Google Scholar] [PubMed]
- Acikalin, A.; Bagir, E.; Karim, S.; Bisgin, A.; Izol, V.; Erdogan, S. Primary melanoma of the urinary tract; Clinicopathologic and molecular review of a case series. Pathol. Res. Pract. 2020, 216, 153095. [Google Scholar] [CrossRef] [PubMed]
- Kaboré, F.A.; Ouédraogo, B.; Ido, F.A.H.A.; Hafing, T.; Karama, H.; Traoré, O. Primary malignant melanoma of the urethra in women: About a case. Urol. Case Rep. 2021, 35, 101542. [Google Scholar] [CrossRef]
- Machado, A.K.L.P.; Nunes, D.B.C.; Carneiro, F.R.O.; Mendes, A.M.D. Primary melanoma of leptomeninge in a patient with giant congenital melanocytic nevus. An. Bras. Dermatol. 2020, 95, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, Y.-B.; Tan, X.-Y.; Cui, Y.-H.; Zhao, G. Multifocal primary amelanotic meningeal melanomas mimicking lymphoma: A case report and literature review. Br. J. Neurosurg. 2020, 15, 1–5. [Google Scholar] [CrossRef]
- Karimkhani, C.; Green, A.; Nijsten, T.; Weinstock, M.; Dellavalle, R.; Naghavi, M.; Fitzmaurice, C. The global burden of melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Kozmin, S.; Slezak, G.; Reynaud-Angelin, A.; Elie, C.; De Rycke, Y.; Boiteux, S.; Sage, E. Ultraviolet A radiation is highly mutagenic in cells that are unable to repair 7,8-dihydro-8-oxoguanine in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2005, 102, 538–543. [Google Scholar] [CrossRef]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar] [CrossRef]
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and Risk Factors of Melanoma. Surg. Clin. North Am. 2020, 100, 1–12. [Google Scholar] [CrossRef]
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612. [Google Scholar] [CrossRef] [PubMed]
- Douki, T. Oxidative Stress and Genotoxicity in Melanoma Induction: Impact on Repair Rather Than Formation of DNA Damage? Photochem. Photobiol. 2020, 96, 962–972. [Google Scholar] [CrossRef]
- Khan, A.Q.; Travers, J.B.; Kemp, M.G. Roles of UVA radiation and DNA damage responses in melanoma pathogenesis. Environ. Mol. Mutagenesis. 2018, 59, 438–460. [Google Scholar] [CrossRef]
- Trucco, L.D.; Mundra, P.A.; Hogan, K.; Garcia-Martinez, P.; Viros, A.; Mandal, A.; Macagno, N.; Gaudy-Marqueste, C.; Allan, D.; Baenke, F.; et al. Ultraviolet radiation–induced DNA damage is prognostic for outcome in melanoma. Nat. Med. 2019, 25, 221–224. [Google Scholar] [CrossRef]
- Bishop, J. Lentigos, Melanocytic Naevi and Melanoma. In Rook’s Textbook of Dermatology, 8th ed.; Blackwell publishing: Oxford, UK, 2010; Volume 54, pp. 32–56. [Google Scholar]
- Trakatelli, M.; Bylaite-Bucinskiene, M.; Correia, O.; Cozzio, A.; De Vries, E.; Medenica, L.; Nagore, E.; Paoli, J.; Stratigos, A.J.; Del Marmol, V.; et al. Clinical assessment of skin phototypes: Watch your words! Eur. J. Dermatol. 2017, 27, 615–619. [Google Scholar] [CrossRef]
- Huang, J.M.; Chikeka, I.; Hornyak, T.J. Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence. Dermatol. Clin. 2017, 35, 85–93. [Google Scholar] [CrossRef]
- Damsky, W.; Bosenberg, M. Melanocytic nevi and melanoma: Unraveling a complex relationship. Oncogene 2017, 36, 5771–5792. [Google Scholar] [CrossRef] [PubMed]
- Loria, D.; Matos, E. Risk factors for cutaneous melanoma: A case-control study in Argentina. Int. J. Dermatol. 2001, 40, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Stanganelli, I.; De Felici, M.B.; Mandel, V.D.; Caini, S.; Raimondi, S.; Corso, F.; Bellerba, F.; Quaglino, P.; Sanlorenzo, M.; Ribero, S.; et al. The association between pesticide use and cutaneous melanoma: A systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 691–708. [Google Scholar] [CrossRef] [PubMed]
- Hausauer, A.; Swetter, S.; Cockburn, M.; Clarke, C. Increases in melanoma among adolescent girls and young women in California: Trends by socioeconomic status and UV radiation exposure. Arch. Dermatol. 2011, 147, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Suppa, M.; Gandini, S. Melanoma Epidemiology and Sun Exposure. Acta Derm. Venereol. 2020, 100, 250–258. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.; Bray, F.; Coebergh, J.; Parkin, D. Changing epidemiology of malignant cutaneous melanoma in Europe 1953-1997: Rising trends in incidence and mortality but recent stabilizations in western Europe and decreases in Scandinavia. Int. J. Cancer 2003, 107, 119–126. [Google Scholar] [CrossRef]
- Dahl, C.; Guldberg, P. The genome and epigenome of malignant melanoma. Apmis 2007, 115, 1161–1176. [Google Scholar] [CrossRef]
- Sample, A.; He, Y.-Y. Mechanisms and prevention of UV-induced melanoma. Photodermatol. Photoimmunol. Photomed. 2018, 34, 13–24. [Google Scholar] [CrossRef]
- Ko, J.M.; Vélez, N.F.; Tsao, H. Pathways to Melanoma. Semin. Cutan. Med. Surg. 2010, 29, 210–217. [Google Scholar]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Vélez, C.O.; Soberino-García, J.; Perez-Garcia, J.M. NRAS-mutant melanoma: Current challenges and future prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Widlund, H.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central Role of p53 in the Suntan Response and Pathologic Hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar] [CrossRef]
- Pons, M.; Quintanilla, M. Molecular biology of malignant melanoma and other cutaneous tumors. Clin. Transl. Oncol. 2006, 8, 466–474. [Google Scholar] [CrossRef]
- Hima, P.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar]
- Kim, M. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Small GTPases 2010, 1, 6222–6232. [Google Scholar] [CrossRef][Green Version]
- Romano, C.; Schepis, C. PTEN Gene: A Model for Genetic Diseases in Dermatology. Sci. World J. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Abdo, J.F.; Sharma, A.; Sharma, R. Role of Heredity in Melanoma Susceptibility: A Primer for the Practicing Surgeon. Surg. Clin. North Am. 2020, 100, 13–28. [Google Scholar] [CrossRef]
- Soura, E.; Eliades, P.; Shannon, K.; Stratigos, A.; Tsao, H. Hereditary Melanoma: Update on Syndromes and Management—Genetics of familial atypical multiple mole melanoma syndrome. J. Am. Acad. Dermatol. 2016, 74, 395–407. [Google Scholar] [CrossRef]
- Demierre, M.-F.; Sondak, V.K. Cutaneous melanoma: Pathogenesis and rationale for chemoprevention. Crit. Rev. Oncol. 2005, 53, 225–239. [Google Scholar] [CrossRef]
- Nikolaou, V.; Kang, X.; Stratigos, A.; Gogas, H.; Latorre, M.; Gabree, M.; Plaka, M.; Njauw, C.; Kypreou, K.; Mirmigi, I.; et al. Comprehensive mutational analysis of CDKN2A and CDK4 in Greek patients with cutaneous melanoma. Br. J. Dermatol. 2011, 165, 1219–1222. [Google Scholar] [CrossRef]
- Rebecca, V.; Sondak, V.; Smalleya, K. A brief history of melanoma: From mummies to mutations. Melanoma Res. 2012, 22, 114–122. [Google Scholar] [CrossRef]
- Sargen, M.; Merrill, S.; Chu, E.; Nathanson, K. CDKN2A mutations with p14 loss predisposing to multiple nerve sheath tumours, melanoma, dysplastic naevi and internal malignancies: A case series and review of the literature. Br. J. Dermatol. 2016, 175, 785–789. [Google Scholar] [CrossRef]
- Read, J.; Wadt, K.; Hayward, N. Melanoma genetics. J. Med. Genet. 2016, 53, 1–14. [Google Scholar] [CrossRef]
- Leachman, S.; Lucero, O.; Sampson, J. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017, 36, 77–90. [Google Scholar] [CrossRef]
- Newton-Bishop, J.; Bishop, D.; Harland, M. Melanoma Genomics. Acta Derm Venereol. 2020. [Google Scholar] [CrossRef]
- Dalmasso, B.; Ghiorzo, P. Evolution of approaches to identify melanoma missing heritability. Expert. Rev. Mol. Diagn. 2020. [Google Scholar] [CrossRef]
- Kearney, L.; Hogan, D.; Conlon, P.; Roche, M.; O’Neill, J.; O’Sullivan, J. High-risk cutaneous malignancies and immunosuppression: Challenges for the reconstructive surgeon in the renal transplant population. J. Plast. Reconstr. Aesthetic Surg. 2017, 70, 922–930. [Google Scholar] [CrossRef]
- Kubica, A.W.; Brewer, J.D. Melanoma in Immunosuppressed Patients. Mayo Clin. Proc. 2012, 87, 991–1003. [Google Scholar] [CrossRef]
- Konsoulova, A. Principles of cancer immunobiology and immunotherapy of solid tumors. In Immunopathology and Immunomodulation; Metodiev, K., Ed.; 2015; pp. 77–100. [Google Scholar]
- Ricci, F.; Paradisi, A.; Fania, L.; Pallotta, S.; Di Lella, G.; Sobrino, L.; Panebianco, A.; Abeni, D. High melanoma risk in non-melanoma skin cancer patients under age 40: A large retrospective cohort study. G. Ital. Dermatol. Venereol. 2019. [Google Scholar] [CrossRef]
- Banan, P.; Marvi, S.K.; McMeniman, E.; De’Ambrosis, B. An Australian cohort of 210 patients with multiple invasive squamous cell carcinomas: Risk factors and associated increased risk of melanoma and internal malignancies. Australas. J. Dermatol. 2015, 57, 29–32. [Google Scholar] [CrossRef]
- Sarangarajan, R.; Apte, S.P. The polymerization of melanin: A poorly understood phenomenon with egregious biological implications. Melanoma Res. 2006, 16, 3–10. [Google Scholar] [CrossRef]
- Meyskens, F.; Farmer, P.; Anton-Culver, H. Etiologic pathogenesis of melanoma: A unifying hypothesis for the missing attributable risk. Clin. Cancer Res. 2004, 15, 2581–2583. [Google Scholar]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef]
- Garbe, C.; Bauer, J. Melanoma. In Dermatology, 3rd ed.; Bolognia, J., Jorizzo, J., Schaffer, J., Eds.; Elsevier: Amsterdam, The Netherlands; Saunders: Philadelphia, PA, USA, 2012; Volume 2, pp. 1885–1914. [Google Scholar]
- Davies, M.; Garraway, L. Molecular biology of cutaneous melanoma. In Principles and Practice of Oncology; Devita, V., Hellman, T., Rosenberg, S., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2015; pp. 1337–1345. [Google Scholar]
- Cohen, J.; Sullivan, R. Developments in the space of new MAPK pathway inhibitors for BRAF-mutant melanoma. Clin. Cancer Res. 2019, 25, 5735–5742. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy. Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar]
- Lo, J.; Fisher, D. Melanoma pathogenesis. In BRAF Targets in Melanoma; Sullivan, R., Ed.; Springer: New York, NY, USA, 2015; pp. 25–45. [Google Scholar]
- Sullivan, R.; Fisher, D. The Molecular Biology of Melanoma 2017. Available online: https://www.uptodate.com/contents/ (accessed on 12 June 2021).
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef]
- Meng, D.; Carvajal, R.D. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am. J. Clin. Dermatol. 2019, 20, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844. [Google Scholar] [CrossRef]
- Williams, E.A.; Montesion, M.; Shah, N.; Sharaf, R.; Pavlick, D.C.; Sokol, E.S.; Alexander, B.; Venstrom, J.; Elvin, J.A.; Ross, J.S.; et al. Melanoma with in-frame deletion of MAP2K1: A distinct molecular subtype of cutaneous melanoma mutually exclusive from BRAF, NRAS, and NF1 mutations. Mod. Pathol. 2020, 33, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- Roesch, A.; Volkenandt, M. Melanoma. In Dermatology, 3rd ed.; Braun-Falco, O., Plewig, G., Wolf, H., Burgdorf, W.H.C., Eds.; Springer: Berlin, Germany, 2009; pp. 1416–1432. [Google Scholar]
- Ballesteros-Álvarez, J.; Dilshat, R.; Fock, V.; Möller, K.; Karl, L.; Larue, L.; Ögmundsdóttir, M.H.; Steingrímsson, E. MITF and TFEB cross-regulation in melanoma cells. PLoS ONE 2020, 15, e0238546. [Google Scholar]
- Garraway, L.A.; Widlund, H.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, J.D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nat. Cell Biol. 2005, 436, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Houben, R.; Schrama, D.; Voigt, H.; Zapatka, M.; Schadendorf, D.; Bröcker, E.B.; Becker, J.C. Microphthalmia-Associated Transcription Factor Gene Amplification in Metastatic Melanoma Is a Prognostic Marker for Patient Survival, But Not a Predictive Marker for Chemosensitivity and Chemotherapy Response. Clin. Cancer Res. 2007, 13, 6344–6350. [Google Scholar] [CrossRef]
- Du, J.; Widlund, H.; Horstmann, M.A.; Ramaswamy, S.; Ross, K.; Huber, W.E.; Nishimura, E.K.; Golub, T.R.; Fisher, D.E. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell 2004, 6, 565–576. [Google Scholar] [CrossRef]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Principles of Tumor Suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Bowden, N.A.; Croft, A.J.; Scurr, L.L.; Kairupan, C.F.; Ashton, K.A.; Talseth-Palmer, B.A.; Rizos, H.; Zhang, X.D.; Scott, R.J.; et al. P53 in human melanoma fails to regulate target genes associated with apoptosis and the cell cycle and may contribute to proliferation. BMC Cancer 2011, 11, 1–17. [Google Scholar] [CrossRef]
- Sirigu, P.; Piras, F.; Minerba, L.; Murtas, D.; Maxia, C.; Colombari, R.; Corbu, A.; Perra, M.T.; Ugalde, J. Prognostic prediction of the immunohistochemical expression of p16 and p53 in cutaneous melanoma: A comparison of two populations from different geographical regions. Eur. J. Histochem. 2006, 50, 191–198. [Google Scholar]
- Ragnarsson-Olding, B.; Platz, A.; Olding, L.; Ringborg, U. p53 protein expression and TP53 mutations in malignant melanomas of sun-sheltered mucosal membranes versus chronically sun-exposed skin. Melanoma Res. 2004, 14, 395–401. [Google Scholar] [CrossRef]
- Smalley, K.S.; Contractor, R.; Haass, N.K.; Kulp, A.N.; Atilla-Gokcumen, G.E.; Williams, U.S.; Bregman, H.; Flaherty, K.T.; Soengas, M.S.; Meggers, E.; et al. An Organometallic Protein Kinase Inhibitor Pharmacologically Activates p53 and Induces Apoptosis in Human Melanoma Cells. Cancer Res. 2007, 67, 209–217. [Google Scholar] [CrossRef]
- Onder, T.; Gupta, P.B.; Mani, S.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-Cadherin Promotes Metastasis via Multiple Downstream Transcriptional Pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef]
- Bachmann, I.; Ladstein, R.; Straume, O.; Naumov, G.; Akslen, L. Tumor necrosis is associated with increased alphavbeta3 integrin expression and poor prognosis in nodular cutaneous melanomas. BMC Cancer 2008, 8, 1–10. [Google Scholar] [CrossRef]
- Chang, S.-H.; Worley, L.A.; Onken, M.; Harbour, J.W. Prognostic biomarkers in uveal melanoma: Evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008, 18, 191–200. [Google Scholar] [CrossRef]
- Hurlbut, G.D.; Kankel, M.W.; Lake, R.J.; Artavanis-Tsakonas, S. Crossing paths with Notch in the hyper-network. Curr. Opin. Cell Biol. 2007, 19, 166–175. [Google Scholar] [CrossRef]
- Balint, K.; Xiao, M.; Pinnix, C.C.; Soma, A.; Veres, I.; Juhasz, I.; Brown, E.J.; Capobianco, A.J.; Herlyn, M.; Liu, Z.-J. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J. Clin. Investig. 2005, 115, 3166–3176. [Google Scholar] [CrossRef]
- Hoek, K.; Rimm, D.L.; Williams, K.R.; Zhao, H.; Ariyan, S.; Lin, A.; Kluger, H.M.; Berger, A.J.; Cheng, E.; Trombetta, E.S.; et al. Expression Profiling Reveals Novel Pathways in the Transformation of Melanocytes to Melanomas. Cancer Res. 2004, 64, 5270–5282. [Google Scholar] [CrossRef]
- Pinnix, C.C.; Lee, J.T.; Liu, Z.-J.; McDaid, R.; Balint, K.; Beverly, L.J.; Brafford, P.A.; Xiao, M.; Himes, B.; Zabierowski, S.E.; et al. Active Notch1 Confers a Transformed Phenotype to Primary Human Melanocytes. Cancer Res. 2009, 69, 5312–5320. [Google Scholar] [CrossRef]
- Bedogni, B.; Warneke, J.A.; Nickoloff, B.J.; Giaccia, A.J.; Powell, M.B. Notch1 is an effector of Akt and hypoxia in melanoma development. J. Clin. Investig. 2008, 118, 3660–3670. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.P.; Pincus, L.B.; Ruben, B.S.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar]
- Schadendorf, D.; Kochs, C.; Livingstone, E. Introduction to cutaneous melanoma. In Handbook of Cutaneous Melanoma—A Guide to Diagnosis and Treatment; Schadendorf, D., Kochs, C., Livingstone, E., Eds.; Springer Healthcare: New York, NY, USA, 2013; pp. 1–12. [Google Scholar]
- Lugović, L.; Situm, M.; Kos, L. Malignant melanoma--future prospects. Acta Dermatovenerol. Croat. ADC 2005, 13, 36–43. [Google Scholar] [PubMed]
- Sullivan, R.J.; Atkins, M.B.; Kirkwood, J.M.; Agarwala, S.S.; Clark, J.I.; Ernstoff, M.S.; Fecher, L.; Gajewski, T.F.; Gastman, B.; Lawson, D.H.; et al. An update on the Society for Immunotherapy of Cancer consensus statement on tumor immunotherapy for the treatment of cutaneous melanoma: Version 2.0. J. Immunother. Cancer 2018, 6, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef] [PubMed]
- Lade-Keller, J.; Riber-Hansen, R.; Guldberg, P.; Schmidt, H.; Hamilton-Dutoit, S.; Steiniche, T. E- to N-cadherin switch in melanoma is associated with decreased expression of phosphatase and tensin homolog and cancer progression. Br. J. Dermatol. 2013, 169, 618–628. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Risk Factors Related to Human | Risk Factors Related to the Environment |
|---|---|
| Skin phototype Pigmented nevi Genetic factors Heredity Immunosuppressive conditions Non-melanoma skin cancer | Ultraviolet radiation Use of pesticides Prolonged sun exposure and sunburn Geographical location |
| Melanomas Typically Associated with Cumulative Solar Damage | |||
|---|---|---|---|
| Pathway | I SSM/ low-CSD melanoma | II LMM/ high-CSD melanoma | III Desmoplastic melanoma |
| Benign Neoplasms (nevi) | Nevus | IMP (?) | IMP (?) |
| Intermediate Neoplasms (low-grade dysplasias and melanocytomas) | 1 Nevus with low-grade dysplasia; 2 BIN; 3 DPN | IAMP/dysplasia (?) | IAMP/dysplasia (?) |
| Intermediate neoplasms (high-grade dysplasias and melanocytomas) | 1 Nevus with high-grade dysplasia/ MIS; 2 BAP1- inactivated melanocytoma/ MELTUMP; 3 Deep penetrating melanocytoma/ MELTUMP; 4 PEM/ MELTUMP | Lentigo maligna (MIS) | MIS |
| Malignant neoplasms | 1 SSM(VGP); 2 Melanoma in BIN(rare); 3 Melanoma in DPN(rare); 4 Melanoma in PEM(rare); | LMM (VGP) | Desmoplastic melanoma |
| Common mutations | 1 BRAF p.V600 b; NRAS b; TERT d; CDKN2A a; TP53 a; PTEN a2 BRAF b; NRAS b + BAP1 a; 3 BRAF b; MAP2K1 b; NRAS b + CTNNB1 b; APC a;4 BRAF b +PRKAR1A a; RKCA c | NRAS b; BRAF non-p.V600E b; KIT b; NF1 a; TERT d; CDKN2A a; TP53 a; PTEN a; RAC1 b | NF1 a; ERBB2 e; MAP2K1 e; MAP3K1 e; BRAF e; EGFR e; MET e; TERT d; NFKBIE d; NRAS b; PIK3CA b; PTPN11 b |
| Melanomas Not Consistently Associated with Cumulative Solar Damage | |||||
|---|---|---|---|---|---|
| Pathway | IV Spitz melanoma | V Acral melanoma | VI Mucosal melanoma | VII Melanomas arising in congenital nevi | VIII Melanomas arising in blue nevi |
| Benign neoplasms (nevi) | Spitz nevus | Acral nevus (?) | Melanosis (?) | Congenital nevus | Blue nevus |
| Intermediate neoplasms (low-grade dysplasias and melanocytomas) | Atypica Spitz tumor melanocytoma) | IAMPUS/dysplasia | Atypical melanosis /dysplasia/IAMPUS | Nodul in congenital nevus (melanocytoma) | (Atypical) cellular blue nevus (melanocytoma) |
| Intermediate neoplasms (high -grade dysplasias and melanocytomas) | STUMP/ MELTUMP | Acral MIS | Mucosal MIS | MIS in congenital nevus | Atypical cellular blue nevus |
| Malignant neoplasms | Malignant Spitz tumor/Spitz melanoma (tumorigenic) | Acral melanoma (VGP) | Mucosal lentiginous melanoma (VGP) | Melanoma in congenital nevus (tumorigenic) | Melanoma in blue nevus (tumorigenic) |
| Common mutations | HRAS b; ALK e; ROS1 e; RET e; NTRK1 e; NTRK3 e; BRAF e; MET e; CDKN2A a | KIT b; NRAS b; BRAF b; HRAS b; KRAS b; NTRK3 e; ALK e; NF1 a; CDKN2A a; TERT f; CCND1 d; GAB2 d | KIT b; NRAS b; KRAS b; BRAF b; NF1 b; CDKN2A a; SF3B1 a; CCND1 d; CDK4 d; MDM2 d | NRAS b; BRAF p.V600E b (small lesions); BRAF e | GNAQ b; GNA11 b; CYSLTR2 b; BAP1 a; EIF1AX c; SF3B1c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strashilov, S.; Yordanov, A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. Int. J. Mol. Sci. 2021, 22, 6395. https://doi.org/10.3390/ijms22126395
Strashilov S, Yordanov A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. International Journal of Molecular Sciences. 2021; 22(12):6395. https://doi.org/10.3390/ijms22126395
Chicago/Turabian StyleStrashilov, Strahil, and Angel Yordanov. 2021. "Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances" International Journal of Molecular Sciences 22, no. 12: 6395. https://doi.org/10.3390/ijms22126395
APA StyleStrashilov, S., & Yordanov, A. (2021). Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. International Journal of Molecular Sciences, 22(12), 6395. https://doi.org/10.3390/ijms22126395