
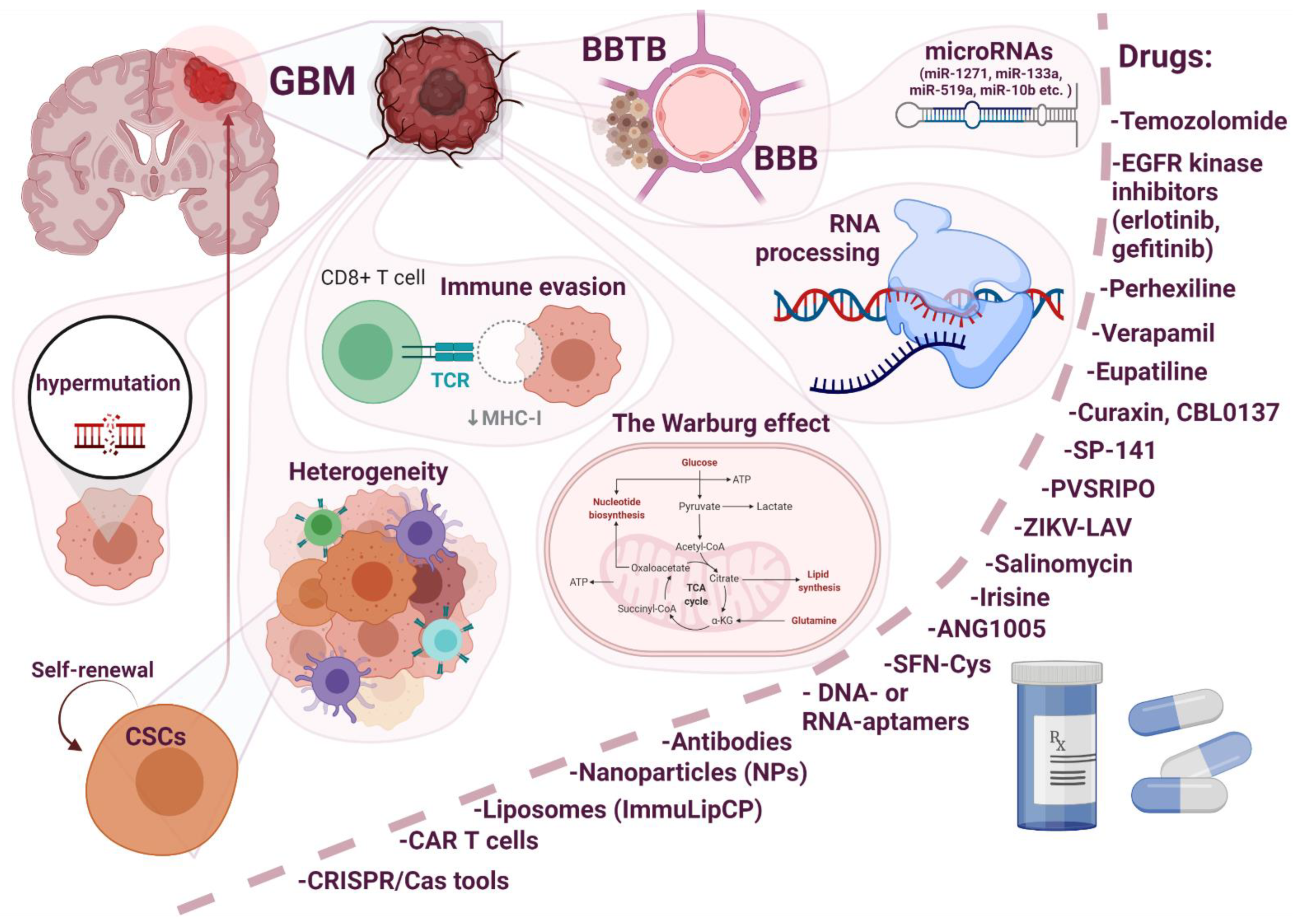
Molecular Mechanisms of Drug Resistance in Glioblastoma


{kind=link}
Abstract
1. Introduction
2. The Diffuse Infiltrative Growth of GBM
3. Barriers in Brain and Advances in Drug Delivery across the Blood–Brain Barrier
4. Molecular Features of Glioblastoma Cells Which Promote Chemoresistance
4.1. Heterogeneity
4.2. Hypermutation
4.3. The Warburg Effect
4.4. Immune Evasion
4.5. Oncologically Activated Alternative Splicing Pathways
4.6. Role of microRNAs in the Resistance of Glioblastoma
5. Current Drugs for the Treatment of GBM and Possible Mechanisms of Resistance to Them
5.1. Temozolomide (TMZ)
5.2. Other Drugs
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma Incidence of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; p. 14. [Google Scholar]
- Nuzzo, S.; Brancato, V.; Affinito, A.; Salvatore, M.; Cavaliere, C.; Condorelli, G. The role of RNA and DNA aptamers in glioblastoma diagnosis and therapy: A systematic review of the literature. Cancers 2020, 12, 2173. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef]
- Seystahl, K.; Network, T.G.G.; Hentschel, B.; Loew, S.; Gramatzki, D.; Felsberg, J.; Herrlinger, U.; Westphal, M.; Schackert, G.; Thon, N.; et al. Bevacizumab versus alkylating chemotherapy in recurrent glioblastoma. J. Cancer Res. Clin. Oncol. 2019, 146, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Güç, E.; Kapourani, C.-A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470.e26. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. The 2016 WHO Classification of Tumours of the Central Nervous System: The Major Points of Revision. Neurol. Med. Chir. 2017, 57, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Bastola, S.; Pavlyukov, M.S.; Yamashita, D.; Ghosh, S.; Cho, H.; Kagaya, N.; Zhang, Z.; Minata, M.; Lee, Y.; Sadahiro, H.; et al. Glioma-initiating cells at tumor edge gain signals from tumor core cells to promote their malignancy. Nat. Commun. 2020, 11, 4660. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed]
- de Souza, C.F.; Sabedot, T.S.; Malta, T.M.; Stetson, L.; Morozova, O.; Sokolov, A.; Laird, P.W.; Wiznerowicz, M.; Iavarone, A.; Snyder, J.; et al. A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep. 2018, 23, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O6-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2020, 9, 1547. [Google Scholar] [CrossRef]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hänggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro-Oncology 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Solecki, G.; Wick, W.; Winkler, F. A malignant cellular network in gliomas: Potential clinical implications. Neuro-Oncology 2016, 18, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Pinto, G.; Saenz-De-Santa-Maria, I.; Chastagner, P.; Perthame, E.; Delmas, C.; Toulas, C.; Moyal-Jonathan-Cohen, E.; Brou, C.; Zurzolo, C. Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem. J. 2021, 478, 21–39. [Google Scholar] [CrossRef]
- Wang, D.; Wang, C.; Wang, L.; Chen, Y. A comprehensive review in improving delivery of small-molecule chemotherapeutic agents overcoming the blood-brain/brain tumor barriers for glioblastoma treatment. Drug Deliv. 2019, 26, 551–565. [Google Scholar] [CrossRef]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2020, 165-166, 1–14. [Google Scholar] [CrossRef]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; A Banks, W.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Papadopoulos, M.; Saadoun, S.; Binder, D.; Manley, G.; Krishna, S.; Verkman, A. Molecular mechanisms of brain tumor edema. Neuroscience 2004, 129, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.C. Blood-brain barrier breakdown in septic encephalopathy and brain tumours*. J. Anat. 2002, 200, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Fischmann, A.; Rascher, G.; Duffner, F.; Grote, E.-H.; Kalbacher, H.; Wolburg, H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000, 100, 323–331. [Google Scholar] [CrossRef]
- Wolburg, H.; Warth, A. Redistribution of aquaporin-4 in human glioblastoma correlates with loss of agrin immunoreactivity from brain capillary basal laminae. Acta Neuropathol. 2004, 107, 311–318. [Google Scholar] [CrossRef]
- E Golde, T. Open questions for Alzheimer’s disease immunotherapy. Alzheimer’s Res. Ther. 2014, 6, 3. [Google Scholar] [CrossRef]
- Debinski, W.; Tatter, S.B. Convection-enhanced delivery for the treatment of brain tumors. Expert Rev. Neurother. 2009, 9, 1519–1527. [Google Scholar] [CrossRef]
- Hersh, D.; Wadajkar, A.S.; Roberts, N.B.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Nam, L.; Coll, C.; Erthal, L.C.S.; De La Torre, C.; Serrano, D.; Martínez-Máñez, R.; Santos-Martínez, M.J.; Ruiz-Hernández, E. Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials 2018, 11, 779. [Google Scholar] [CrossRef] [PubMed]
- Vigani, B.; Valentino, C.; Sandri, G.; Listro, R.; Fagiani, F.; Collina, S.; Lanni, C.; Bonferoni, M.; Caramella, C.; Rossi, S.; et al. A Composite Nanosystem as a Potential Tool for the Local Treatment of Glioblastoma: Chitosan-Coated Solid Lipid Nanoparticles Embedded in Electrospun Nanofibers. Polymers 2021, 13, 1371. [Google Scholar] [CrossRef]
- Straehla, J.P.; Warren, K.E. Pharmacokinetic Principles and Their Application to Central Nervous System Tumors. Pharmaceutics 2020, 12, 948. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood–Brain Barrier Transport of Therapeutics via Receptor-Mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef]
- Stanimirovic, D.B.; Sandhu, J.K.; Costain, W.J. Emerging Technologies for Delivery of Biotherapeutics and Gene Therapy across the Blood–Brain Barrier. BioDrugs 2018, 32, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.; He, M. Unlocking the Power of Exosomes for Crossing Biological Barriers in Drug Delivery. Pharmaceutics 2021, 13, 122. [Google Scholar] [CrossRef]
- Hanson, L.R.; Frey, W.H. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008, 9, S5. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, N.D.; Muldoon, L.L.; Culp, A.Y.; Neuwelt, E.A. Delivery of chemotherapeutics across the blood-brain barrier: Challenges and advances. Adv. Pharmacol. 2014, 71, 203–243. [Google Scholar] [CrossRef]
- Chen, K.-T.; Chai, W.-Y.; Lin, Y.-J.; Lin, C.-J.; Chen, P.-Y.; Tsai, H.-C.; Huang, C.-Y.; Kuo, J.S.; Liu, H.-L.; Wei, K.-C. Neuronavigation-guided focused ultrasound for transcranial blood-brain barrier opening and immunostimulation in brain tumors. Sci. Adv. 2021, 7, eabd0772. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- Régina, A.; Demeule, M.; Ché, C.; Lavallée, I.; Poirier, J.; Gabathuler, R.; Béliveau, R.; Castaigne, J.-P. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br. J. Pharmacol. 2008, 155, 185–197. [Google Scholar] [CrossRef]
- Michiue, H.; Sakurai, Y.; Kondo, N.; Kitamatsu, M.; Bin, F.; Nakajima, K.; Hirota, Y.; Kawabata, S.; Nishiki, T.-I.; Ohmori, I.; et al. The acceleration of boron neutron capture therapy using multi-linked mercaptoundecahydrododecaborate (BSH) fused cell-penetrating peptide. Biomaterials 2014, 35, 3396–3405. [Google Scholar] [CrossRef]
- Iguchi, Y.; Michiue, H.; Kitamatsu, M.; Hayashi, Y.; Takenaka, F.; Nishiki, T.-I.; Matsui, H. Tumor-specific delivery of BSH-3R for boron neutron capture therapy and positron emission tomography imaging in a mouse brain tumor model. Biomaterials 2015, 56, 10–17. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Wang, Y.; Hays, E.; Rama, M.; Bonavida, B. Cell-mediated immune resistance in cancer. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell Plasticity and Heterogeneity in Cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Fontana, F.; Raimondi, M.; Limonta, P. Cancer stem cells-key players in tumor relapse. Cancers 2021, 13, 376. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Multiforme Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, V.; Arnotskaya, N.; Korneyko, M.; Zaytsev, S.; Khotimchenko, Y.; Sharma, H.; Bryukhovetskiy, I. Proteins of the Wnt signaling pathway as targets for the regulation of CD133+ cancer stem cells in glioblastoma. Oncol. Rep. 2019, 41, 3080–3088. [Google Scholar] [CrossRef]
- Bhuvanalakshmi, G.; Gamit, N.; Patil, M.; Arfuso, F.; Sethi, G.; Dharmarajan, A.; Kumar, A.P.; Warrier, S. Stemness, Pluripotentiality, and Wnt Antagonism: sFRP4, a Wnt antagonist Mediates Pluripotency and Stemness in Glioblastoma. Cancers 2018, 11, 25. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Wallenborn, M.; Xu, L.-X.; Kirsten, H.; Rohani, L.; Rudolf, D.; Ahnert, P.; Schmidt, C.; Schulz, R.M.; Richter, M.; Krupp, W.; et al. Molecular analyses of glioblastoma stem-like cells and glioblastoma tissue. PLoS ONE 2020, 15, e0234986. [Google Scholar] [CrossRef]
- Cruickshanks, N.; Zhang, Y.; Yuan, F.; Pahuski, M.; Gibert, M.; Abounader, R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9, 87. [Google Scholar] [CrossRef]
- Liu, F.; Mischel, P.S. Targeting epidermal growth factor receptor co-dependent signaling pathways in glioblastoma. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1398. [Google Scholar] [CrossRef]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef]
- Li, H.; Yan, R.; Chen, W.; Ding, X.; Liu, J.; Chen, G.; Zhao, Q.; Tang, Y.; Lv, S.; Liu, S.; et al. Long non coding RNA SLC26A4-AS1 exerts antiangiogenic effects in human glioma by upregulating NPTX1 via NFKB1 transcriptional factor. FEBS J. 2021, 288, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liao, T.; Liu, H.; Yuan, H.; Ouyang, T.; Wang, J.; Chai, S.; Li, J.; Chen, J.; Li, X.; et al. Hypoxic glioma stem cell-derived exosomes containing Linc01060 promote progression of glioma by regulating the MZF1/c-Myc/HIF-1α. Cancer Res. 2020, 81. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nat. Cell Biol. 2020, 580, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Sabri, S.; Chaddad, A.; Meehan, B.; Jean-Claude, B.; Rak, J.; Abdulkarim, B.S. Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Front. Oncol. 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.E.; Hoey, B.; Margison, G.P. Ionizing radiation induces O6-alkylguanine-DNA-alkyltransferase mRNA and activity in mouse tissues. Carcinogenesis 1993, 14, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Quick, Q.A.; Gewirtz, D.A. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J. Neurosurg. 2006, 105, 111–118. [Google Scholar] [CrossRef] [PubMed]
- McFarland, C.D.; Yaglom, J.A.; Wojtkowiak, J.W.; Scott, J.G.; Morse, D.L.; Sherman, M.Y.; Mirny, L.A. The Damaging Effect of Passenger Mutations on Cancer Progression. Cancer Res. 2017, 77, 4763–4772. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41. [Google Scholar] [CrossRef]
- Duraj, T.; García-Romero, N.; Carrión-Navarro, J.; Madurga, R.; Mendivil, A.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef]
- Chen, X.S.; Li, L.Y.; Guan, Y.; Di Yang, J.M.; Cheng, Y. Anticancer strategies based on the metabolic profile of tumor cells: Therapeutic targeting of the Warburg effect. Acta Pharmacol. Sin. 2016, 37. [Google Scholar] [CrossRef]
- Negi, N.; Das, B.K. CNS: Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int. Rev. Immunol. 2018, 37, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.-Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, L.; Ye, M.; Kang, C.; You, H. Treatment Progress of Immune Checkpoint Blockade Therapy for Glioblastoma. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, S.; Zhou, Q. The Resistance Mechanisms of Lung Cancer Immunotherapy. Front. Oncol. 2020, 10, 568059. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Harjunpää, H.; Asens, M.L.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Marincola, F.M.; Ferrone, S.; Abken, H. The complex role of B7 molecules in tumor immunology. Trends Mol. Med. 2008, 14, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, L.L.P.; De Souza, P.O.; Dal Prá, M.; Falchetti, M.; De Abreu, A.M.; Azambuja, J.H.; Bertoni, A.; Paz, A.; Araújo, A.; Visioli, F.; et al. TLR4 expression and functionality are downregulated in glioblastoma cells and in tumor-associated macrophages: A new mechanism of immune evasion? Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 166155. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The Role of Cytokines in Breast Cancer Development and Progression. J. Interf. Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological Characterization of Cancer Stem Cells Isolated from Glioblastoma Patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef]
- Wei, J.; Barr, J.; Kong, L.-Y.; Wang, Y.; Wu, A.; Sharma, A.K.; Gumin, J.; Henry, V.; Colman, H.; Sawaya, R.; et al. Glioma-Associated Cancer-Initiating Cells Induce Immunosuppression. Clin. Cancer Res. 2010, 16, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Pellegatta, S.; Poliani, P.L.; Corno, D.; Menghi, F.; Ghielmetti, F.; Suarez-Merino, B.; Caldera, V.; Nava, S.; Ravanini, M.; Facchetti, F.; et al. Neurospheres Enriched in Cancer Stem–Like Cells Are Highly Effective in Eliciting a Dendritic Cell–Mediated Immune Response against Malignant Gliomas. Cancer Res. 2006, 66, 10247–10252. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, G.; Yuan, X.; Xu, M.; Wang, H.; Ji, J.; Konda, B.; Black, K.L.; Yu, J.S. Antigen-Specific T-Cell Response from Dendritic Cell Vaccination Using Cancer Stem-Like Cell-Associated Antigens. Stem Cells 2009, 27, 1734–1740. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; A Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Sarkar, S.; Döring, A.; Zemp, F.J.; Da Silva, C.L.; Lun, X.; Wang, X.; Kelly, J.; Hader, W.J.; Hamilton, M.; Mercier, P.; et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 2014, 17, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Meliso, F.M.; Hubert, C.G.; Galante, P.A.F.; Penalva, L.O. RNA processing as an alternative route to attack glioblastoma. Qual. Life Res. 2017, 136, 1129–1141. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef]
- Correa, B.R.; De Araujo, P.R.; Qiao, M.; Burns, S.C.; Chen, C.; Schlegel, R.; Agarwal, S.; Galante, P.A.F.; Penalva, L.O.F. Functional genomics analyses of RNA-binding proteins reveal the splicing regulator SNRPB as an oncogenic candidate in glioblastoma. Genome Biol. 2016, 17, 125. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Li, Y.-J.; Zeng, C.-C. Chemo-resistance of A172 glioblastoma cells is controlled by miR-1271-regulated Bcl-2. Biomed. Pharmacother. 2018, 108, 734–740. [Google Scholar] [CrossRef]
- Wang, S.-S.; Feng, L.; Hu, B.-G.; Lu, Y.-F.; Wang, W.-M.; Guo, W.; Suen, C.-W.; Jiao, B.-H.; Pang, J.-X.; Fu, W.-M.; et al. miR-133a Promotes TRAIL Resistance in Glioblastoma via Suppressing Death Receptor 5 and Activating NF-κB Signaling. Mol. Ther. Nucleic Acids 2017, 8, 482–492. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Li, J.-J.; Zhou, Q.; Huang, A.; Liu, W.-W.; Wang, K.; Gao, L.; Qi, S.-T.; Lu, Y.-T. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J. Hematol. Oncol. 2018, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Margison, G.P.; Koref, M.F.S.; Povey, A.C. Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine. Mutagenesis 2002, 17, 483–487. [Google Scholar] [CrossRef]
- Karran, P.; Marinus, M.G. Mismatch correction at O6-methylguanine residues in E. coli DNA. Nat. Cell Biol. 1982, 296, 868–869. [Google Scholar] [CrossRef] [PubMed]
- Pepponi, R.; Marra, G.; Fuggetta, M.P.; Falcinelli, S.; Pagani, E.; Bonmassar, E.; Jiricny, J.; D’Atri, S. The Effect ofO6-Alkylguanine-DNA Alkyltransferase and Mismatch Repair Activities on the Sensitivity of Human Melanoma Cells to Temozolomide, 1,3-bis(2-Chloroethyl)1-nitrosourea, and Cisplatin. J. Pharmacol. Exp. Ther. 2003, 304, 661–668. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-H.; Shen, W.-L.; Shih, C.-M.; Ho, K.-H.; Cheng, C.-H.; Lin, C.-W.; Lee, C.-C.; Liu, A.-J.; Chen, K.-C. The CHAC1-inhibited Notch3 pathway is involved in temozolomide-induced glioma cytotoxicity. Neuropharmacology 2017, 116, 300–314. [Google Scholar] [CrossRef]
- Haas, B.; Klinger, V.; Keksel, C.; Bonigut, V.; Kiefer, D.; Caspers, J.; Walther, J.; Wos-Maganga, M.; Weickhardt, S.; Röhn, G.; et al. Inhibition of the PI3K but not the MEK/ERK pathway sensitizes human glioma cells to alkylating drugs. Cancer Cell Int. 2018, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, J.; Zhao, C.; Ren, H.; Yuan, Z.; Zhang, B.; Zhuang, J.; Wang, J.; Feng, B. MiR-181b-5p modulates chemosensitivity of glioma cells to temozolomide by targeting Bcl-2. Biomed. Pharmacother. 2019, 109, 2192–2202. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Hoadley, K.; Kushwaha, D.; Ramakrishnan, V.; Li, S.; Kang, C.; You, Y.; Jiang, C.; Song, S.W.; et al. miR-181d: A predictive glioblastoma biomarker that downregulates MGMT expression. Neuro-Oncology 2012, 14, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.-X.; Yin, W.-B.; Wang, Z.-Y. MicroRNA-132 induces temozolomide resistance and promotes the formation of cancer stem cell phenotypes by targeting tumor suppressor candidate 3 in glioblastoma. Int. J. Mol. Med. 2017, 40, 1307–1314. [Google Scholar] [CrossRef]
- Ho, K.-H.; Cheng, C.-H.; Chou, C.-M.; Chen, P.-H.; Liu, A.-J.; Lin, C.-W.; Shih, C.-M.; Chen, K.-C. miR-140 targeting CTSB signaling suppresses the mesenchymal transition and enhances temozolomide cytotoxicity in glioblastoma multiforme. Pharmacol. Res. 2019, 147, 104390. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.R.; Wu, M.Y.; Dai, L.J.; Huang, Y.; Shan, M.Y.; Ma, S.N.; Wang, J.; Peng, H.; Ding, Y.; Zhang, Q.F.; et al. Nuclear FAM289-Galectin-1 interaction controls FAM289-mediated tumor promotion in malignant glioma. J. Exp. Clin. Cancer Res. 2019, 38, 1–19. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, B.; Feng, L.; Sun, B.; He, S.; Yang, Y.; Wu, G.; E, G.; Liu, C.; Gao, Y.; et al. Targeting JUN, CEBPB, and HDAC3: A Novel Strategy to Overcome Drug Resistance in Hypoxic Glioblastoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Sarker, D.; Plummer, R.; Meyer, T.; Sodergren, M.H.; Basu, B.; Chee, C.E.; Huang, K.-W.; Palmer, D.H.; Ma, Y.T.; Evans, T.J.; et al. MTL-CEBPA, a Small Activating RNA Therapeutic Upregulating C/EBP-α, in Patients with Advanced Liver Cancer: A First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin. Cancer Res. 2020, 26, 3936–3946. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.-C.; Chen, K.-Y.; Chang, C.-K.; Chuang, J.-Y.; Liu, C.-C.; Liu, S.-H.; Chen, R.-M. Improved effects of honokiol on temozolomide-induced autophagy and apoptosis of drug-sensitive and -tolerant glioma cells. BMC Cancer 2018, 18, 379. [Google Scholar] [CrossRef]
- Luo, S.; Lei, K.; Xiang, D.; Ye, K. NQO1 Is Regulated by PTEN in Glioblastoma, Mediating Cell Proliferation and Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 9146528. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Kesarwani, P.; Guastella, A.R.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Perhexiline Demonstrates FYN-mediated Antitumor Activity in Glioblastoma. Mol. Cancer Ther. 2020, 19, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.W.; Jeon, H.-Y.; Kim, H. Verapamil augments carmustine- and irradiation-induced senescence in glioma cells by reducing intracellular reactive oxygen species and calcium ion levels. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Żesławska, E.; Kucwaj-Brysz, K.; Kincses, A.; Spengler, G.; Szymańska, E.; Czopek, A.; Marć, M.A.; Kaczor, A.; Nitek, W.; Domínguez-Álvarez, E.; et al. An insight into the structure of 5-spiro aromatic derivatives of imidazolidine-2,4-dione, a new group of very potent inhibitors of tumor multidrug resistance in T-lymphoma cells. Bioorg. Chem. 2021, 109, 104735. [Google Scholar] [CrossRef]
- Chung, Y.H.; Cai, H.; Steinmetz, N.F. Viral nanoparticles for drug delivery, imaging, immunotherapy, and theranostic applications. Adv. Drug Deliv. Rev. 2020, 156, 214–235. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, J.; Ye, Q.; Ma, F.; Zhu, Q.; Wu, Y.; Shan, C.; Xie, X.; Li, D.; Zhan, X.; et al. Treatment of Human Glioblastoma with a Live Attenuated Zika Virus Vaccine Candidate. mBio 2018, 9, e01683-18. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle disease virus enhances the growth-inhibiting and proapoptotic effects of temozolomide on glioblastoma cells in vitro and in vivo. Sci. Rep. 2018, 8, 11470. [Google Scholar] [CrossRef]
- Martin-Hijano, L.; Sainz, B.J. The Interactions Between Cancer Stem Cells and the Innate Interferon Signaling Pathway. Front. Immunol. 2020, 11, 526. [Google Scholar] [CrossRef]
- Martikainen, M.; Essand, M. Virus-Based Immunotherapy of Glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Punganuru, S.R.; Artula, V.; Zhao, W.; Rajaei, M.; Deokar, H.; Zhang, R.; Buolamwini, J.K.; Srivenugopal, K.S.; Wang, W. Targeted Brain Tumor Therapy by Inhibiting the MDM2 Oncogene: In Vitro and In Vivo Antitumor Activity and Mechanism of Action. Cells 2020, 9, 1592. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Wang, J.; Chen, C.; Ding, B.; Fu, X.; Chen, W.; Wang, C.; Xu, R. Eupatilin inhibits glioma proliferation, migration, and invasion by arresting cell cycle at G1/S phase and disrupting the cytoskeletal structure. Cancer Manag. Res. 2019, 11, 4781–4796. [Google Scholar] [CrossRef]
- Barone, T.A.; Burkhart, C.A.; Safina, A.; Haderski, G.; Gurova, K.V.; Purmal, A.A.; Gudkov, A.V.; Plunkett, R.J. Anticancer drug candidate CBL0137, which inhibits histone chaperone FACT, is efficacious in preclinical orthotopic models of temozolomide-responsive and -resistant glioblastoma. Neuro-Oncology 2016, 19, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-N.; Kim, S.-H.; Kim, K.-Y.; Ji, J.-H.; Seo, Y.-K.; Yu, H.S.; Ahn, S.-C. Salinomycin induces endoplasmic reticulum stress-mediated autophagy and apoptosis through generation of reactive oxygen species in human glioma U87MG cells. Oncol. Rep. 2017, 37, 3321–3328. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, C.; Chang, Y.; Lee, H.; Wu, J.; Huang, J.; Chung, Y.; Hsu, S.; Chow, L.; Wei, K.; Huang, F. Irisin, an exercise myokine, potently suppresses tumor proliferation, invasion, and growth in glioma. FASEB J. 2020, 34, 9678–9693. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhou, Y.; Yang, G.; Tian, H.; Geng, Y.; Hu, Y.; Lin, K.; Wu, W. Sulforaphane-cysteine induces apoptosis by sustained activation of ERK1/2 and caspase 3 in human glioblastoma U373MG and U87MG cells. Oncol. Rep. 2017, 37, 2829–2838. [Google Scholar] [CrossRef]
- Amero, P.; Khatua, S.; Rodriguez-Aguayo, C.; Lopez-Berestein, G. Aptamers: Novel therapeutics and potential role in neuro-oncology. Cancers 2020, 12, 2889. [Google Scholar] [CrossRef]
- Razpotnik, R.; Novak, N.; Šerbec, V.Č.; Rajcevic, U. Targeting Malignant Brain Tumors with Antibodies. Front. Immunol. 2017, 8, 1181. [Google Scholar] [CrossRef]
- Wankhede, M.; Bouras, A.; Kaluzova, M.; Hadjipanayis, C.G. Magnetic nanoparticles: An emerging technology for malignant brain tumor imaging and therapy. Expert Rev. Clin. Pharmacol. 2012, 5, 173–186. [Google Scholar] [CrossRef]
- Revia, R.A.; Stephen, Z.; Zhang, M. Theranostic Nanoparticles for RNA-Based Cancer Treatment. Acc. Chem. Res. 2019, 52, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Duro-Castano, A.; Leite, D.M.; Forth, J.; Deng, Y.; Matias, D.; Jesus, C.N.; Battaglia, G. Designing peptide nanoparticles for efficient brain delivery. Adv. Drug Deliv. Rev. 2020, 160, 52–77. [Google Scholar] [CrossRef]
- Jose, G.; Lu, Y.-J.; Hung, J.-T.; Yu, A.; Chen, J.-P. Co-Delivery of CPT-11 and Panobinostat with Anti-GD2 Antibody Conjugated Immunoliposomes for Targeted Combination Chemotherapy. Cancers 2020, 12, 3211. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.L.; Marqués-Torrejón, M.-A.; Morrison, G.M.; Pollard, S.M. Experimental models and tools to tackle glioblastoma. Dis. Model. Mech. 2019, 12, 12. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dymova, M.A.; Kuligina, E.V.; Richter, V.A. Molecular Mechanisms of Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6385. https://doi.org/10.3390/ijms22126385
Dymova MA, Kuligina EV, Richter VA. Molecular Mechanisms of Drug Resistance in Glioblastoma. International Journal of Molecular Sciences. 2021; 22(12):6385. https://doi.org/10.3390/ijms22126385
Chicago/Turabian StyleDymova, Maya A., Elena V. Kuligina, and Vladimir A. Richter. 2021. "Molecular Mechanisms of Drug Resistance in Glioblastoma" International Journal of Molecular Sciences 22, no. 12: 6385. https://doi.org/10.3390/ijms22126385
APA StyleDymova, M. A., Kuligina, E. V., & Richter, V. A. (2021). Molecular Mechanisms of Drug Resistance in Glioblastoma. International Journal of Molecular Sciences, 22(12), 6385. https://doi.org/10.3390/ijms22126385