
A Mathematical Description of the Bone Marrow Dynamics during CAR T-Cell Therapy in B-Cell Childhood Acute Lymphoblastic Leukemia

 ,
,  ,
,  , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Mathematical Model
2.1. Main Elements of the Model
2.2. Hematopoietic and Leukemic Compartments
2.3. CAR T-Cell Compartment
2.4. Parameter Estimation
2.5. Computational Details
3. Results
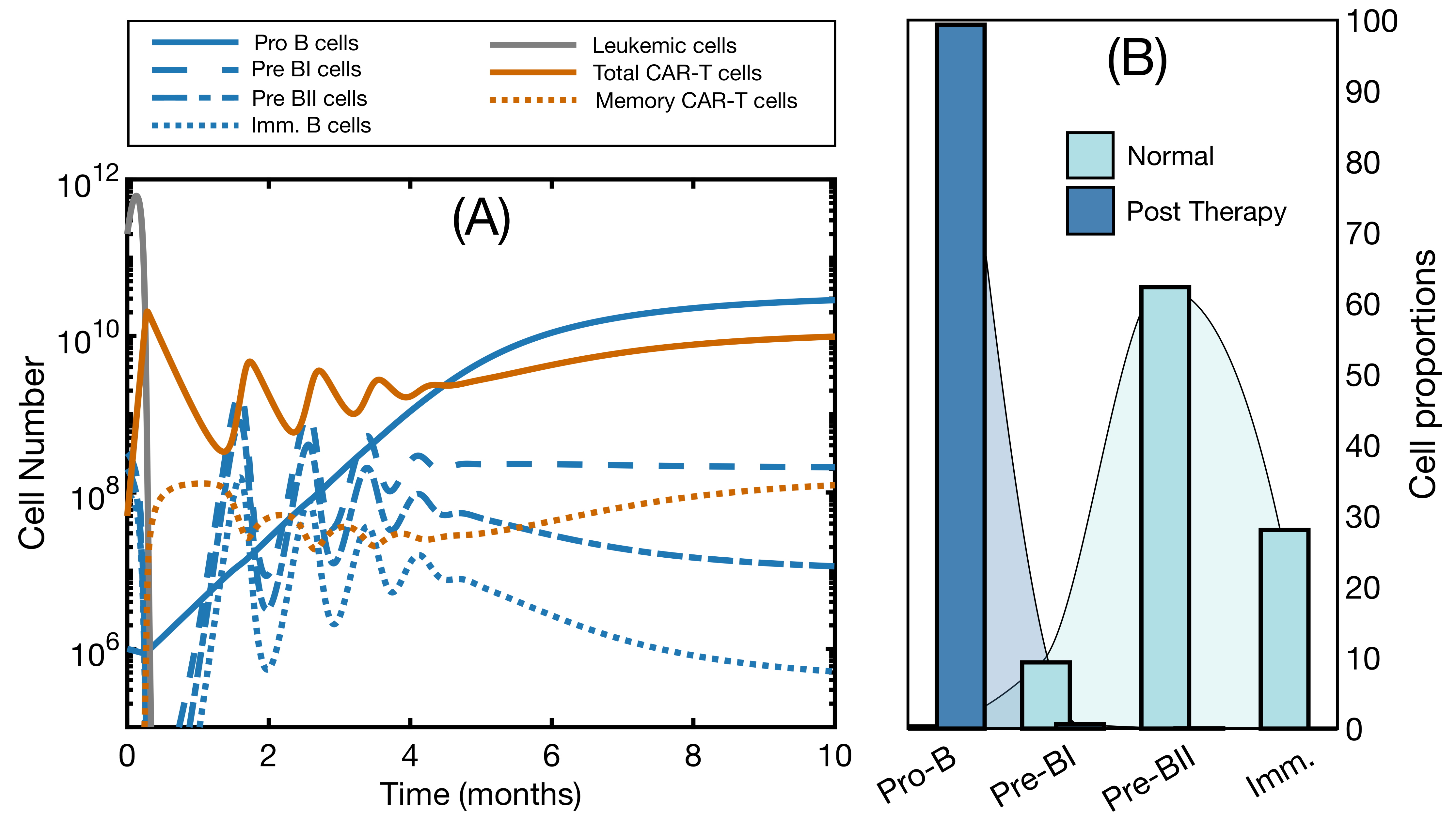
3.1. The Mathematical Model without B-Cell Development Reproduces Clinical Data
3.2. Effector and Memory CAR T-Cells Are Able to Control the Disease
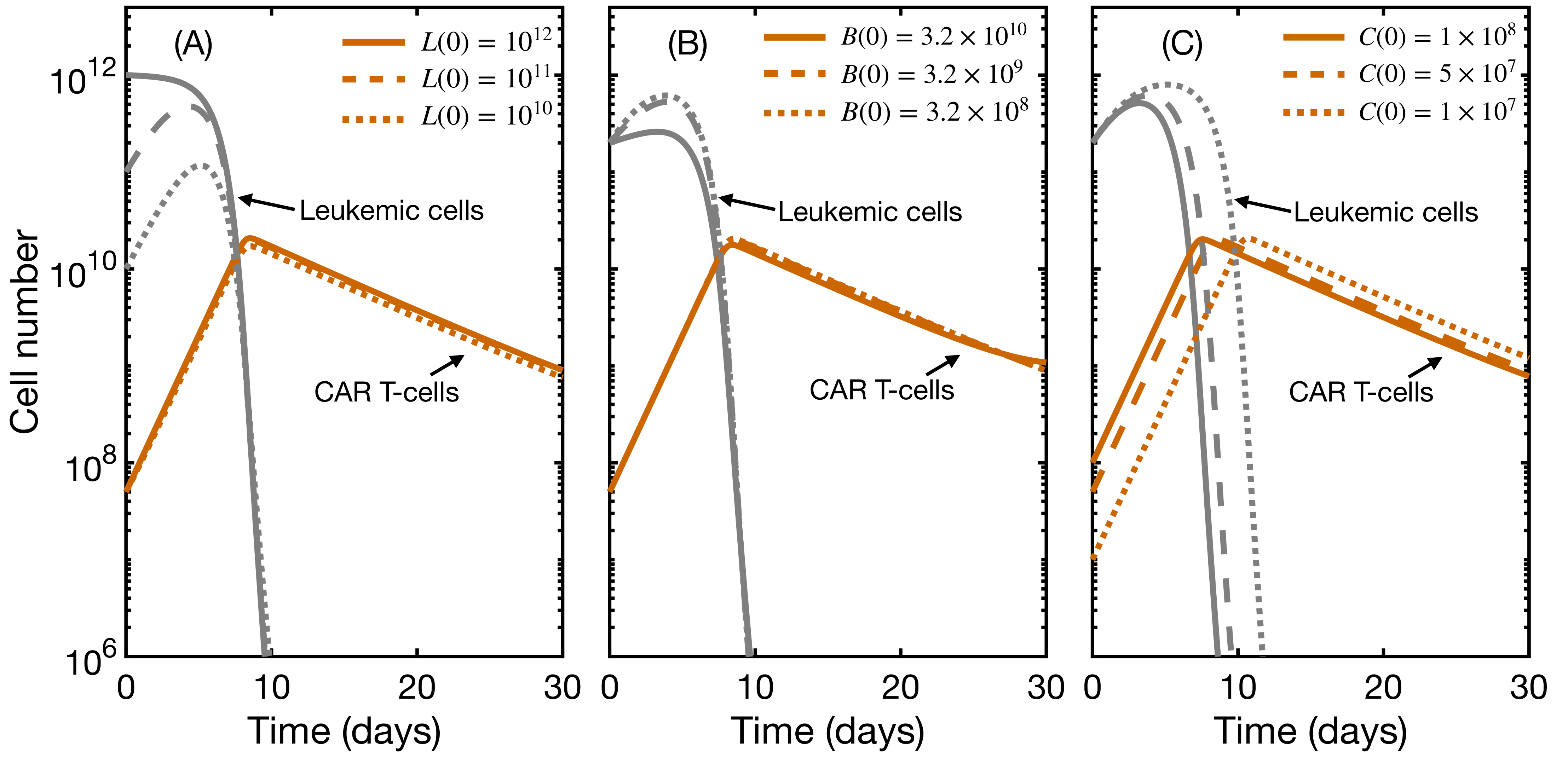
3.3. Initial State Does Not Affect CAR T Expansion and Outcome
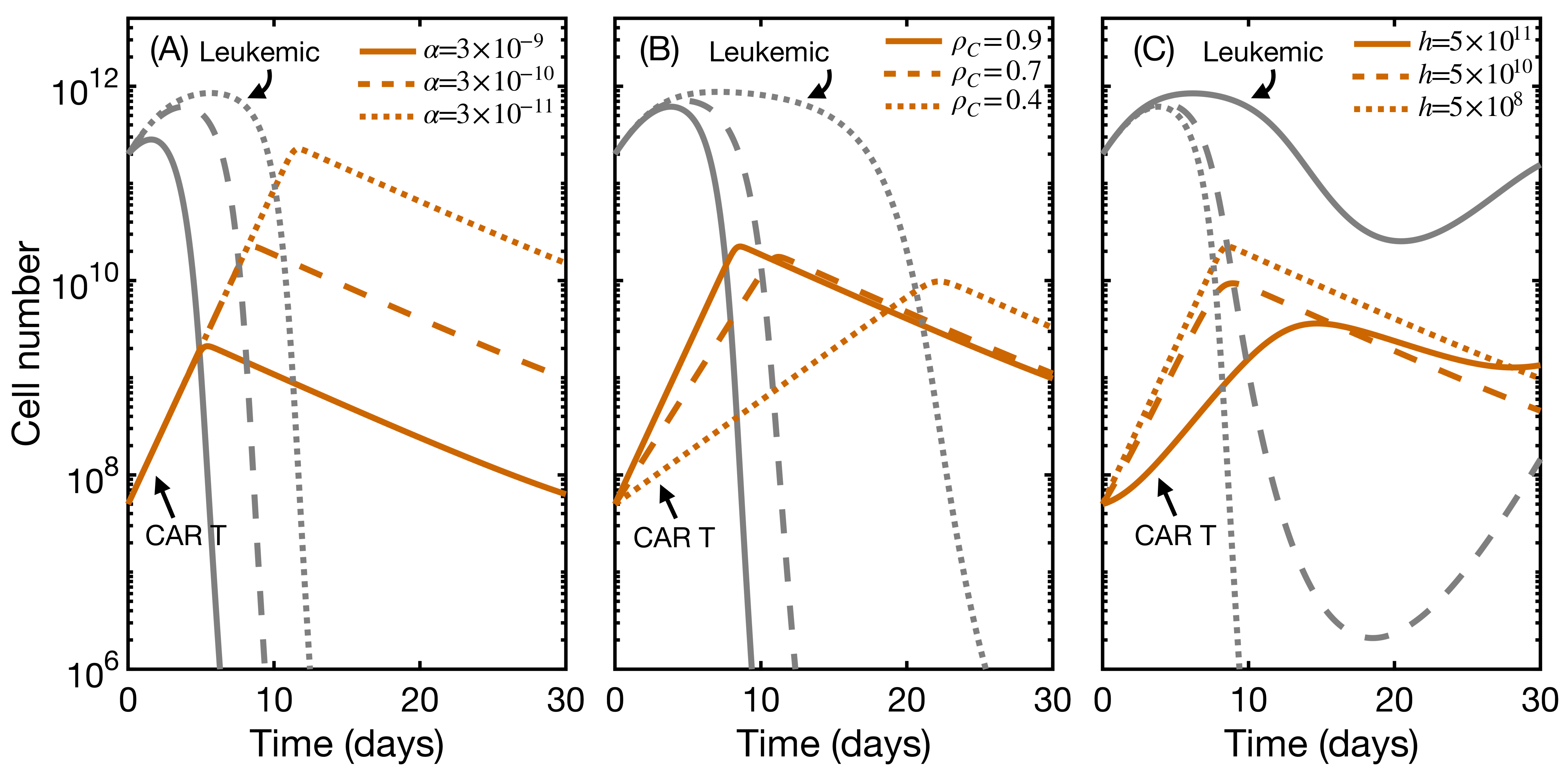
3.4. CAR T Product Characteristics Determine Therapy Success or Failure
3.5. Second Infusion in Non-Responding Patients May Improve the Therapy Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR | Chimeric Antigen Receptor |
| ALL | Acute Lymphoblastic Leukemia |
| CD | Cluster of Differentiation |
| BCA | B-Cell Aplasia |
| FDA | Federal Drug Administration |
References
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global manufacturing of CAR T cell therapy. Mol. Ther.-Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T-cells—Challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Mackall, C.L. CAR T-cell therapy: Inroads to response and resistance. Nat. Rev. Immunol. 2019, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M. CAR therapy: The CD19 paradigm. J. Clin. Investig. 2015, 125, 3392–3400. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA approval summary: Tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef]
- Bouchkouj, N.; Kasamon, Y.L.; de Claro, R.A.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA approval summary: Axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef]
- Wartenberg, D.; Groves, F.D.; Adelman, A.S. Acute lymphoblastic leukemia: Epidemiology and etiology. In Acute Leukemias; Springer: Berlin, Germany, 2008; pp. 77–93. [Google Scholar]
- Pui, C.H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood acute lymphoblastic leukemia: Progress through collaboration. J. Clin. Oncol. 2015, 33, 2938. [Google Scholar] [CrossRef]
- Bhojwani, D.; Pui, C.H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013, 14, e205–e217. [Google Scholar] [CrossRef]
- Martino, M.; Alati, C.; Canale, F.A.; Musuraca, G.; Martinelli, G.; Cerchione, C. A Review of Clinical Outcomes of CAR T-cell Therapies for B-Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 2150. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T-cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T-cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T-cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T-cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T-cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guang, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the mechanisms of resistance to CAR T-cell therapy in malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Turtle, C.J. Toxicities of CD19 CAR-T-cell immunotherapy. Am. J. Hematol. 2019, 94, S42–S49. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 2017, 130, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.T.; Waldron, E.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.J.; Hamilton, J.; Awasthi, R.; et al. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2018, 24, 6175–6184. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.M.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.J.; Levine, B.L.; Tomassian, L.; Shah, S.; et al. Tisagenlecleucel Model-Based Cellular Kinetic Analysis of Chimeric Antigen Receptor–T-cells. CPT Pharmacometrics Syst. Pharmacol. 2019, 8, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Zhu, X.; Chu, L.; Goliaei, A.; June, C.H.; Kearns, J.D.; Stein, A.M. Chimeric Antigen Receptor T-cell Therapies: A Review of Cellular Kinetic-Pharmacodynamic Modeling Approaches. J. Clin. Pharmacol. 2020, 60, S147–S159. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, P.; Yang, X.; Abler, D.; Maestrini, D.; Adhikarla, V.; Frankhouser, D.; Cho, H.; Machuca, V.; Wang, D.; Barish, M.; et al. Mathematical deconvolution of CAR T-cell proliferation and exhaustion from real-time killing assay data. J. R. Soc. Interface 2020, 17, 20190734. [Google Scholar] [CrossRef] [PubMed]
- León-Triana, O.; Pérez-Martínez, A.; Ramírez-Orellana, M.; Pérez-García, V.M. Dual-target CAR-Ts with on-and off-tumour activity may override immune suppression in solid cancers: A mathematical proof of concept. Cancers 2021, 13, 703. [Google Scholar] [CrossRef]
- Barros, L.R.C.; de Jesus Rodrigues, B.; Almeida, R.C. CAR-T-cell Goes on a Mathematical Model. J. Cell. Immunol. 2020, 2, 31–37. [Google Scholar]
- Khatun, M.S.; Biswas, M.H.A. Modeling the effect of adoptive T-cell therapy for the treatment of leukemia. Comput. Math. Methods 2020, 2, e1069. [Google Scholar] [CrossRef]
- Owens, K.; Bozic, I. Modeling CAR T-cell Therapy with Patient Preconditioning. Bull. Math. Biol. 2021, 83, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.R.C.; Paixão, E.A.; Valli, A.M.P.; Naozuka, G.T.; Fassoni, A.C.; Almeida, R.C.C. CARTmath-A Mathematical Model of CAR-T Immunotherapy in Preclinical Models. Preprints 2021. [Google Scholar] [CrossRef]
- Kimmel, G.J.; Locke, F.L.; Altrock, P.M. The roles of T-cell competition and stochastic extinction events in chimeric antigen receptor T-cell therapy. Proc. R. Soc. B 2021, 288, 20210229. [Google Scholar] [CrossRef] [PubMed]
- Hardiansyah, D.; Ng, C.M. Quantitative Systems Pharmacology Model of Chimeric Antigen Receptor T-cell Therapy. Clin. Transl. Sci. 2019, 12, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, V.M.; León-Triana, O.; Rosa, M.; Pérez-Martínez, A. CAR T-cells for T-cell leukemias: Insights from mathematical models. Commun. Nonlinear Sci. Numer. Simul. 2021, 96, 105684. [Google Scholar] [CrossRef]
- Mostolizadeh, R.; Afsharnezhad, Z.; Marciniak-Czochra, A. Mathematical model of chimeric anti-gene receptor (CAR) T-cell therapy with presence of cytokine. Numer. Algebr. Control Optim. 2018, 8, 63. [Google Scholar] [CrossRef]
- León-Triana, O.; Sabir, S.; Calvo, G.F.; Belmonte-Beitia, J.; Chulián, S.; Martínez-Rubio, Á.; Rosa, M.; Pérez-Martínez, A.; Ramirez-Orellana, M.; Pérez-García, V.M. CAR T-cell therapy in B-cell acute lymphoblastic leukaemia: Insights from mathematical models. Commun. Nonlinear Sci. Numer. Simul. 2021, 94, 105570. [Google Scholar] [CrossRef]
- Murphy, K.; Weaver, C. Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2016. [Google Scholar]
- Lucio, P.; Parreira, A.; Van den Beemd, M.; Van Lochem, E.; Van Wering, E.; Baars, E.; Porwit-MacDonald, A.; Bjorklund, E.; Gaipa, G.; Biondi, A.; et al. Flow cytometric analysis of normal B cell differentiation: A frame of reference for the detection of minimal residual disease in precursor-B-ALL. Leukemia 1999, 13, 419–427. [Google Scholar] [CrossRef]
- Bevan, M.J. Helping the CD8+ T-cell response. Nat. Rev. Immunol. 2004, 4, 595–602. [Google Scholar] [CrossRef]
- Chulián, S.; Martínez-Rubio, Á.; Marciniak-Czochra, A.; Stiehl, T.; Goñi, C.B.; Rodríguez Gutiérrez, J.F.; Orellana, M.R.; Robleda, A.C.; Pérez-García, V.M.; Rosa, M. Dynamical properties of feedback signalling in B lymphopoiesis: A mathematical modelling approach. J. Theor. Biol. 2021, 522, 110685. [Google Scholar] [CrossRef]
- Chulián, S.; Rubio, Á.M.; Rosa, M.; Pérez-García, V.M. Mathematical models of Leukaemia and its treatment: A review. arXiv 2020, arXiv:2011.05881. [Google Scholar]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- De Boer, R.J.; Perelson, A.S. Quantifying T lymphocyte turnover. J. Theor. Biol. 2013, 327, 45–87. [Google Scholar] [CrossRef]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef]
- Ahmed, R.; Bevan, M.J.; Reiner, S.L.; Fearon, D.T. The precursors of memory: Models and controversies. Nat. Rev. Immunol. 2009, 9, 662–668. [Google Scholar] [CrossRef]
- De Boer, R.J.; Oprea, M.; Antia, R.; Murali-Krishna, K.; Ahmed, R.; Perelson, A.S. Recruitment times, proliferation, and apoptosis rates during the CD8+ T-cell response to lymphocytic choriomeningitis virus. J. Virol. 2001, 75, 10663–10669. [Google Scholar] [CrossRef]
- De Boer, R.J.; Perelson, A.S. Towards a general function describing T-cell proliferation. J. Theor. Biol. 1995, 175, 567–576. [Google Scholar] [CrossRef]
- Grossman, Z.; Min, B.; Meier-Schellersheim, M.; Paul, W.E. Concomitant regulation of T-cell activation and homeostasis. Nat. Rev. Immunol. 2004, 4, 387–395. [Google Scholar] [CrossRef]
- Ganusov, V.V.; Barber, D.L.; De Boer, R.J. Killing of targets by CD8+ T-cells in the mouse spleen follows the law of mass action. PLoS ONE 2011, 6, e15959. [Google Scholar] [CrossRef]
- Kaech, S.M.; Ahmed, R. Memory CD8+ T-cell differentiation: Initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2001, 2, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Pamer, E.G. Cutting edge: Antigen-independent CD8 T-cell proliferation. J. Immunol. 2001, 166, 5864–5868. [Google Scholar] [CrossRef] [PubMed]
- Saltelli, A.; Annoni, P.; Azzini, I.; Campolongo, F.; Ratto, M.; Tarantola, S. Variance based sensitivity analysis of model output. Design and estimator for the total sensitivity index. Comput. Phys. Commun. 2010, 181, 259–270. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T-cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Mikhailova, E.; Semchenkova, A.; Illarionova, O.; Kashpor, S.; Brilliantova, V.; Zakharova, E.; Zerkalenkova, E.; Zangrando, A.; Bocharova, N.; Shelikhova, L.; et al. Relative expansion of CD19-negative very-early normal B-cell precursors in children with acute lymphoblastic leukaemia after CD19 targeting by blinatumomab and CAR-T-cell therapy: Implications for flow cytometric detection of minimal residual disease. Br. J. Haematol. 2021, 192, 602–612. [Google Scholar] [CrossRef]
- Awasthi, R.; Mueller, K.T.; Yanik, G.A.; Tam, C.S.; Rives, S.; McGuirk, J.P.; Pulsipher, M.A.; Boyer, M.W.; Jaeger, U.; Baruchel, A.; et al. Evaluation of in vivo CAR transgene levels in relapsed/refractory pediatric and young adult ALL and adult DLBCL tisagenlecleucel-treated patients. Blood 2018, 132, 899. [Google Scholar] [CrossRef]
- Wudhikarn, K.; Park, J.H. Dissecting factors influencing response to CAR T-cell therapy in B lymphoid hematologic malignancies: From basic to practice. Leuk. Lymphoma 2020, 61, 1–11. [Google Scholar] [CrossRef]
- Awasthi, R.; Mueller, K.T.; Yanik, G.A.; Tam, C.S.L.; Rives, S.; McGuirk, J.; Boyer, M.W.; Jäger, U.; Baruchel, A.; Myers, G.D.; et al. Considerations for tisagenlecleucel dosing rationale. J. Clin. Oncol. 2018, 36, e15056. [Google Scholar] [CrossRef]
- Finney, O.C.; Brakke, H.; Rawlings-Rhea, S.; Hicks, R.; Doolittle, D.; Lopez, M.; Futrell, B.; Orentas, R.J.; Li, D.; Gardner, R.; et al. CD19 CAR T-cell product and disease attributes predict leukemia remission durability. J. Clin. Investig. 2019, 129, 2123–2132. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T-cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T-cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T-cells of defined CD4+: CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Bezerra, E.D.; Hirayama, A.V.; Fiorenza, S.; Sheih, A.; Chou, C.K.; Kimble, E.L.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell malignancies. Blood 2021, 137, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Lu, W.; Chen, D.; Tu, H.; Guo, Z.; Zhou, X.; Li, M.; Tu, S.; Li, Y. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark. Res. 2020, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Annesley, C.; Gardner, R.; Wilson, A.; Summers, C.; Lamble, A.J.; Rivers, J.; Wu, Q.V.; Brand, A.; Johnson, A.; Spratt, K.; et al. Novel CD19t T-Antigen Presenting Cells Expand CD19 CAR T-cells in Vivo. Blood 2019, 134, 223. [Google Scholar] [CrossRef]
- Ma, F.; Ho, J.Y.; Du, H.; Xuan, F.; Wu, X.; Wang, Q.; Wang, L.; Liu, Y.; Ba, M.; Wang, Y.; et al. Evidence of long-lasting anti-CD19 activity of engrafted CD19 chimeric antigen receptor–modified T cells in a phase I study targeting pediatrics with acute lymphoblastic leukemia. Hematol. Oncol. 2019, 37, 601–608. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Rontiers Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef]
- Wherry, E.J. T-cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef]
- Curran, K.J.; Margossian, S.P.; Kernan, N.A.; Silverman, L.B.; Williams, D.A.; Shukla, N.; Kobos, R.; Forlenza, C.J.; Steinherz, P.; Prockop, S.; et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 2019, 134, 2361–2368. [Google Scholar] [CrossRef]
- Greenbaum, U.; Mahadeo, K.M.; Kebriaei, P.; Shpall, E.J.; Saini, N.Y. Chimeric Antigen Receptor T-cells in B-Acute Lymphoblastic Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1594. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Meaning | Value | Units |
|---|---|---|---|
| Pro-B proliferation rate | /8 | day | |
| Pre-BI proliferation rate | /1 | day | |
| Pre-BII proliferation rate | /1.5 | day | |
| Transition rate: Pro-B to Pre-BI | day | ||
| Transition rate: Pre-BI to Pre-BII | day | ||
| Transition rate: Pre-BII to Immature | day | ||
| Blood exit rate | dayt | ||
| k | Signal intensity | cell | |
| Leukemic cell proliferation rate | day | ||
| Leukemic cell carrying capacity | cell | ||
| Leukemic cell blood exit rate | day | ||
| Activated CAR T killing capacity | – | day· cell | |
| Activated CAR T proliferation rate | day | ||
| Activated CAR T mean lifetime | day | ||
| Activated to memory transition rate | day | ||
| Memory to activated transition rate | day | ||
| Memory CAR T mean lifetime | 300 | day | |
| h | CAR T activation threshold | – | cell |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Rubio, Á.; Chulián, S.; Blázquez Goñi, C.; Ramírez Orellana, M.; Pérez Martínez, A.; Navarro-Zapata, A.; Ferreras, C.; Pérez-García, V.M.; Rosa, M. A Mathematical Description of the Bone Marrow Dynamics during CAR T-Cell Therapy in B-Cell Childhood Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 6371. https://doi.org/10.3390/ijms22126371
Martínez-Rubio Á, Chulián S, Blázquez Goñi C, Ramírez Orellana M, Pérez Martínez A, Navarro-Zapata A, Ferreras C, Pérez-García VM, Rosa M. A Mathematical Description of the Bone Marrow Dynamics during CAR T-Cell Therapy in B-Cell Childhood Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2021; 22(12):6371. https://doi.org/10.3390/ijms22126371
Chicago/Turabian StyleMartínez-Rubio, Álvaro, Salvador Chulián, Cristina Blázquez Goñi, Manuel Ramírez Orellana, Antonio Pérez Martínez, Alfonso Navarro-Zapata, Cristina Ferreras, Victor M. Pérez-García, and María Rosa. 2021. "A Mathematical Description of the Bone Marrow Dynamics during CAR T-Cell Therapy in B-Cell Childhood Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 22, no. 12: 6371. https://doi.org/10.3390/ijms22126371
APA StyleMartínez-Rubio, Á., Chulián, S., Blázquez Goñi, C., Ramírez Orellana, M., Pérez Martínez, A., Navarro-Zapata, A., Ferreras, C., Pérez-García, V. M., & Rosa, M. (2021). A Mathematical Description of the Bone Marrow Dynamics during CAR T-Cell Therapy in B-Cell Childhood Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 22(12), 6371. https://doi.org/10.3390/ijms22126371