
The BMP Pathway in Blood Vessel and Lymphatic Vessel Biology


Abstract
1. Introduction
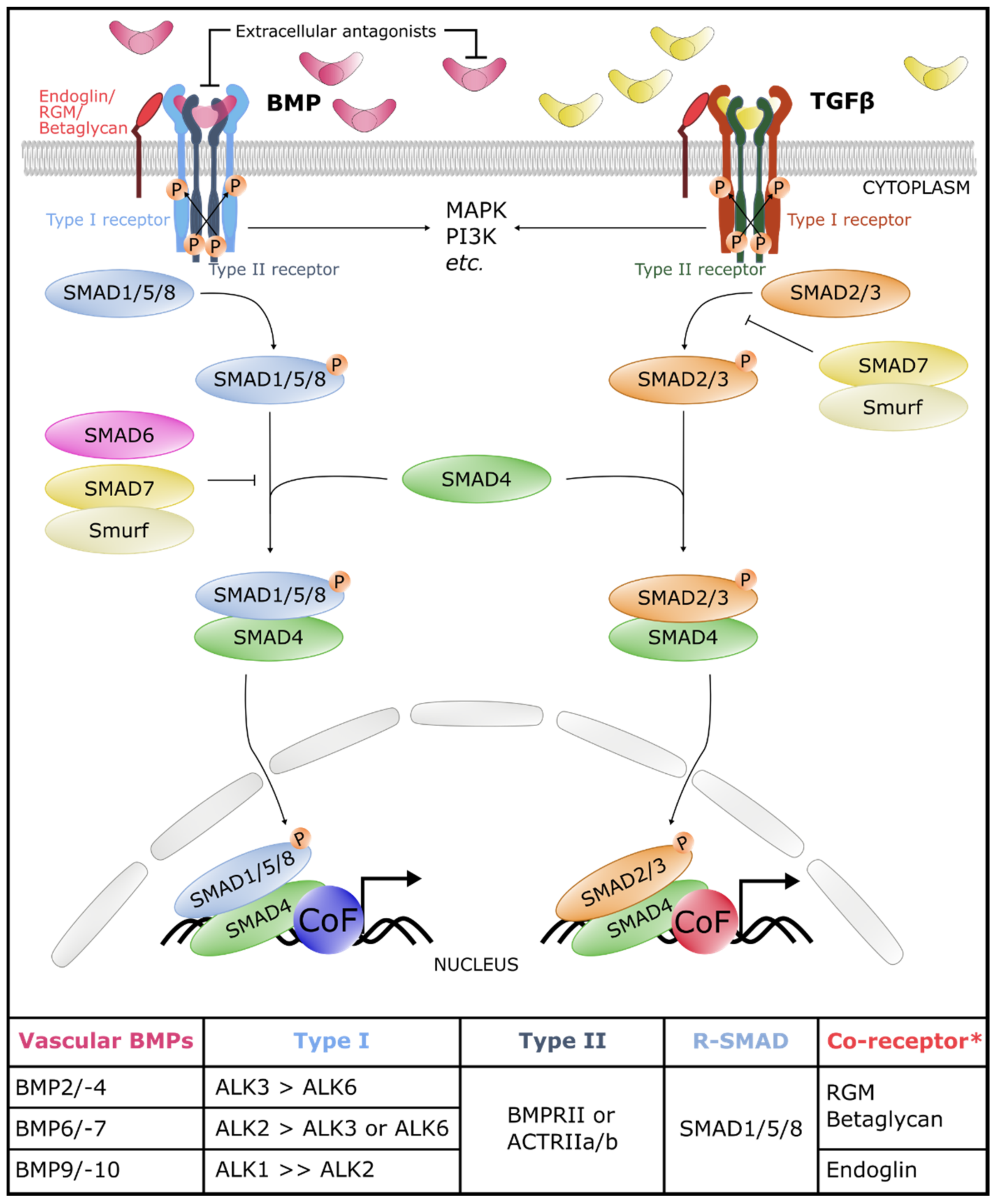
2. The Core of the BMP Signaling Pathway
2.1. BMP Ligands
2.2. BMP Receptors
2.3. Canonical and Non-Canonical Signaling Cascades
3. The Core BMP Signaling Pathway in Blood and Lymphatic Vasculature
3.1. BMP Ligands in Blood and Lymphatic Vasculature
3.2. BMP Receptors in Blood and Lymphatic Vasculature
3.3. Canonical and Non-Canonical Signaling Cascades
4. Fine-Tuning Mechanisms of BMP Signaling in the Vasculature
4.1. Ligand Activation
4.2. Co-Receptors
4.3. Antagonists
4.4. Mechano-Transduction
4.5. Inhibitory SMADs (I-SMADs)
4.6. SMAD Interacting Proteins (SIPs)
4.7. Interplay between BMP and TGFβ Signaling
4.8. Interplay between BMP and VEGF Signaling
4.9. Interplay between BMP and Notch Signaling
4.10. Interplay between BMP and WNT Signaling
5. BMP-Linked Vascular Pathologies
5.1. Hereditary Hemorrhagic Telangiectasia (HHT)
5.2. Pulmonary Arterial Hypertension (PAH)
5.3. Cerebral Cavernous Malformation (CCM)
5.4. Bicuspid Aortic Valve (BAV) with Thoracic Aortic Aneurysm (TAA) and Aortic Valve Stenosis (AOVD2)
5.5. Atherosclerosis and Peripheral Arterial Disease
5.6. Fibrodysplasia Ossificans Progressiva (FOP)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- García de Vinuesa, A.; Abdelilah-Seyfried, S.; Knaus, P.; Zwijsen, A.; Bailly, S. BMP signaling in vascular biology and dysfunction. Cytokine Growth Factor Rev. 2015. [Google Scholar] [CrossRef]
- Goumans, M.-J.; Zwijsen, A.; ten Dijke, P.; Bailly, S. Bone morphogenetic proteins in vascular homeostasis and disease. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Morrell, N.W.; Bloch, D.B.; Ten Dijke, P.; Goumans, M.J.T.H.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signalling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.I.; Magnusson, P.U.; Dejana, E.; Lampugnani, M.G. Deregulated TGF-β/BMP signaling in vascular malformations. Circ. Res. 2017, 121, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Urist, M.R. Bone: Formation by autoinduction. New Ser. 1965, 150, 893–899. [Google Scholar] [CrossRef]
- Urist, M.R.; Strates, B.S. Bone morphogenetic protein. J. Dent. Res. 1971, 50, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Sampath, T.K.; Reddi, A.H. Discovery of bone morphogenetic proteins—A historical perspective. Bone 2020, 140. [Google Scholar] [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-b family signaling. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Economou, A.D.; Hill, C.S. Temporal dynamics in the formation and interpretation of Nodal and BMP morphogen gradients. Curr. Top. Dev. Biol. 2020, 137, 363–389. [Google Scholar] [CrossRef]
- Miyazono, K. TGF-β signaling by Smad proteins. Cytokine Growth Factor Rev. 2000, 11, 15–22. [Google Scholar] [CrossRef]
- Huang, T.; Hinck, A.P. Production, isolation, and structural analysis of ligands and receptors of the TGF-β superfamily. Methods Mol. Biol. 2016, 1344, 63–92. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; Ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.-J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Benn, A.; Hiepen, C.; Osterland, M.; Schütte, C.; Zwijsen, A.; Knaus, P. Role of bone morphogenetic proteins in sprouting angiogenesis: Differential BMP receptor-dependent signaling pathways balance stalk vs. tip cell competence. FASEB J. 2017, 31, 4720–4733. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.D.; Nickel, J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012, 586, 1846–1859. [Google Scholar] [CrossRef]
- Nohe, A.; Hassel, S.; Ehrlich, M.; Neubauer, F.; Sebald, W.; Henis, Y.I.; Knaus, P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem. 2002, 277, 5330–5338. [Google Scholar] [CrossRef]
- Gilboa, L.; Nohe, A.; Geissendörfer, T.; Sebald, W.; Henis, Y.I.; Knaus, P. Bone morphogenetic protein receptor complexes on the surface of live cells: A new oligomerization mode for serine/threonine kinase receptors. Mol. Biol. Cell 2000, 11, 1023–1035. [Google Scholar] [CrossRef]
- Ehrlich, M.; Horbelt, D.; Marom, B.; Knaus, P.; Henis, Y.I. Homomeric and heteromeric complexes among TGF-Β and BMP receptors and their roles in signaling. Cell. Signal. 2011, 23, 1424–1432. [Google Scholar] [CrossRef]
- Park, S.Y.; DiMaio, T.A.; Liu, W.; Wang, S.; Sorenson, C.M.; Sheibani, N. Endoglin regulates the activation and quiescence of endothelium by participating in canonical and non-canonical TGF-β signaling pathways. J. Cell Sci. 2013, 126, 1392–1405. [Google Scholar] [CrossRef]
- Siebold, C.; Yamashita, T.; Monnier, P.P.; Mueller, B.K.; Pasterkamp, R.J. RGMs: Structural insights, molecular regulation, and downstream signaling. Trends Cell Biol. 2017, 27, 365–378. [Google Scholar] [CrossRef]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massagué, J. The TGFβ receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Guzman, A.; Zelman-Femiak, M.; Boergermann, J.H.; Paschkowsky, S.; Kreuzaler, P.A.; Fratzl, P.; Harms, G.S.; Knaus, P. SMAD versus non-SMAD signaling is determined by lateral mobility of bone morphogenetic protein (BMP) receptors. J. Biol. Chem. 2012, 287, 39492–39504. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Gaarenstroom, T.; Hill, C.S. TGF-β signaling to chromatin: How Smads regulate transcription during self-renewal and differentiation. Semin. Cell Dev. Biol. 2014, 32, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, G.; Alarcón, C.; Spagnoli, F.M.; Brivanlou, A.H.; Massagué, J. Balancing BMP Signaling through Integrated Inputs into the Smad1 Linker. Mol. Cell 2007, 25, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, H.B.; Kamato, D.; Ansari, G.; Osman, N.; Little, P.J. Cell biology of Smad2/3 linker region phosphorylation in vascular smooth muscle. Clin. Exp. Pharmacol. Physiol. 2012, 39, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Cheung, E.; Petrakis, T.G.; Howell, M.; Kraus, W.L.; Hill, C.S. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006, 25, 4490–4502. [Google Scholar] [CrossRef]
- Drake, K.M.; Zygmunt, D.; Mavrakis, L.; Harbor, P.; Wang, L.; Comhair, S.A.; Erzurum, S.C.; Aldred, M.A. Altered MicroRNA processing in heritable pulmonary arterial hypertension: An important role for Smad-8. Am. J. Respir. Crit. Care Med. 2011, 184, 1400–1408. [Google Scholar] [CrossRef]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.C.; Van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef]
- Hiepen, C.; Mendez, P.-L.; Knaus, P. It takes two to tango: Endothelial TGFβ/BMP Signaling crosstalk with mechanobiology. Cells 2020, 9, 1965. [Google Scholar] [CrossRef]
- Dunworth, W.P.; Cardona-Costa, J.; Bozkulak, E.C.; Kim, J.D.; Meadows, S.; Fischer, J.C.; Wang, Y.; Cleaver, O.; Qyang, Y.; Ober, E.A.; et al. Bone morphogenetic protein 2 signaling negatively modulates lymphatic development in vertebrate embryos. Circ. Res. 2014, 114, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Levet, S.; Ciais, D.; Merdzhanova, G.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Navarro, F.P.; Texier, I.; Feige, J.J.; Bailly, S.; et al. Bone morphogenetic protein 9 (BMP9) controls lymphatic vessel maturation and valve formation. Blood 2013, 122, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Lee, Y.G.; Akatsu, Y.; Taguchi, L.; Suzuki, H.I.; Cunha, S.I.; Maruyama, K.; Suzuki, Y.; Yamazaki, T.; Katsura, A.; et al. Bone morphogenetic protein-9 inhibits lymphatic vessel formation via activin receptor-like kinase 1 during development and cancer progression. Proc. Natl. Acad. Sci. USA 2013, 110, 18940–18945. [Google Scholar] [CrossRef]
- Subileau, M.; Merdzhanova, G.; Ciais, D.; Collin-Faure, V.; Feige, J.-J.; Bailly, S.; Vittet, D. Bone morphogenetic protein 9 regulates early lymphatic-specified endothelial cell expansion during mouse embryonic stem cell differentiation. Stem Cell Rep. 2019, 12, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Zhang, G.; Ridgway, J.B.; Chen, H.; Yan, M. ALK1 signaling regulates early postnatal lymphatic vessel development. Blood 2010, 115, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.D.; Kim, J. Alk3/Alk3b and Smad5 mediate BMP signaling during lymphatic development in zebrafish. Mol. Cells 2014, 37, 270–274. [Google Scholar] [CrossRef]
- Moon, E.H.; Kim, Y.H.; Vu, P.N.; Yoo, H.; Hong, K.; Lee, Y.J.; Oh, S.P. TMEM100 is a key factor for specification of lymphatic endothelial progenitors. Angiogenesis 2020, 23, 339–355. [Google Scholar] [CrossRef]
- Rothhammer, T.; Bataille, F.; Spruss, T.; Eissner, G.; Bosserhoff, A.K. Functional implication of BMP4 expression on angiogenesis in malignant melanoma. Oncogene 2007, 26, 4158–4170. [Google Scholar] [CrossRef]
- Langenfeld, E.M.; Langenfeld, J. Bone Morphogenetic Protein-2 stimulates angiogenesis in developing tumors. Mol. Cancer Res. 2004, 2, 141–149. [Google Scholar] [PubMed]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Löwik, C.W.G.M.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Salmon, R.M.; Guo, J.; Wood, J.H.; Tong, Z.; Beech, J.S.; Lawera, A.; Yu, M.; Grainger, D.J.; Reckless, J.; Morrell, N.W.; et al. Molecular basis of ALK1-mediated signalling by BMP9/BMP10 and their prodomain-bound forms. Nat. Commun. 2020, 11, 1621. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Dagenais, S.L.; Erickson, R.P.; Arlt, M.F.; Glynn, M.W.; Gorski, J.L.; Seaver, L.H.; Glover, T.W. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am. J. Hum. Genet. 2000, 67, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ridgway, J.B.; Sai, T.; Lai, J.; Warming, S.; Chen, H.; Roose-Girma, M.; Zhang, G.; Shou, W.; Yan, M. Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc. Natl. Acad. Sci. USA 2013, 110, 11887–11892. [Google Scholar] [CrossRef]
- Xiang, M.; Grosso, R.A.; Takeda, A.; Pan, J.; Bekkhus, T.; Brulois, K.; Dermadi, D.; Nordling, S.; Vanlandewijck, M.; Jalkanen, S.; et al. A single-cell transcriptional roadmap of the mouse and human lymph node lymphatic vasculature. Front. Cardiovasc. Med. 2020, 7. [Google Scholar] [CrossRef]
- Moya, I.M.; Umans, L.; Maas, E.; Pereira, P.G.; Beets, K.; Francis, A.; Sents, W.; Robertson, E.J.; Mummery, C.L.; Huylebroeck, D.; et al. Stalk cell phenotype depends on integration of notch and Smad1/5 signaling cascades. Dev. Cell 2012, 22. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, D.; Ihida-Stansbury, K.; Jones, P.L.; Martin, J.F. Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum. Mol. Genet. 2009, 18, 2791–2801. [Google Scholar] [CrossRef]
- Drake, K.M.; Comhair, S.A.; Erzurum, S.C.; Tuder, R.M.; Aldred, M.A. Endothelial chromosome 13 deletion in congenital heart disease-associated pulmonary arterial hypertension dysregulates SMAD9 signaling. Am. J. Respir. Crit. Care Med. 2015, 191, 850–854. [Google Scholar] [CrossRef]
- Wang, Z.P.; Mu, X.Y.; Guo, M.; Wang, Y.J.; Teng, Z.; Mao, G.P.; Niu, W.B.; Feng, L.Z.; Zhao, L.H.; Xia, G.L. Transforming growth factor-β signaling participates in the maintenance of the primordial follicle pool in the mouse ovary. J. Biol. Chem. 2014. [Google Scholar] [CrossRef]
- Li, Y.; Luo, W.; Yang, W. Nuclear transport and accumulation of smad proteins studied by single-molecule microscopy. Biophys. J. 2018, 114, 2243–2251. [Google Scholar] [CrossRef]
- Richter, A.; Alexdottir, M.S.; Magnus, S.H.; Richter, T.R.; Morikawa, M.; Zwijsen, A.; Valdimarsdottir, G. EGFL7 mediates BMP9-induced sprouting angiogenesis of endothelial cells derived from human embryonic stem cells. Stem Cell Rep. 2019. [Google Scholar] [CrossRef]
- Morikawa, M.; Mitani, Y.; Holmborn, K.; Kato, T.; Koinuma, D.; Maruyama, J.; Vasilaki, E.; Sawada, H.; Kobayashi, M.; Ozawa, T.; et al. The ALK-1/SMAD/ATOH8 axis attenuates hypoxic responses and protects against the development of pulmonary arterial hypertension. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Somekawa, S.; Imagawa, K.; Hayashi, H.; Sakabe, M.; Ioka, T.; Sato, G.E.; Inada, K.; Iwamoto, T.; Mori, T.; Uemura, S.; et al. Tmem100, an ALK1 receptor signaling-dependent gene essential for arterial endothelium differentiation and vascular morphogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 12064–12069. [Google Scholar] [CrossRef]
- Valdimarsdottir, G.; Goumans, M.J.; Rosendahl, A.; Brugman, M.; Itoh, S.; Lebrin, F.; Sideras, P.; Ten Dijke, P. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation 2002, 106, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.M.; de Sousa Lopes, S.M.C.; Bialecka, M.; de Boer, S.; Zwijsen, A.; Mummery, C.L. Real time monitoring of BMP smads transcriptional activity during mouse development. Genesis 2008, 46, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.M.; de Sousa Lopes, S.M.C.; Korchynskyi, O.; ten Dijke, P.; Mummery, C.L. Spatio-temporal activation of Smad1 and Smad5 in vivo: Monitoring transcriptional activity of Smad proteins. J. Cell Sci. 2004, 117, 4653. [Google Scholar] [CrossRef]
- Collery, R.F.; Link, B.A. Dynamic smad-mediated BMP signaling revealed through transgenic zebrafish. Dev. Dyn. 2011, 240, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Javier, A.L.; Doan, L.T.; Luong, M.; Reyes de Mochel, N.S.; Sun, A.; Monuki, E.S.; Cho, K.W.Y. Bmp Indicator Mice Reveal Dynamic Regulation of Transcriptional Response. PLoS ONE 2012, 7, e42566. [Google Scholar] [CrossRef]
- Laux, D.W.; Febbo, J.A.; Roman, B.L. Dynamic analysis of BMP-responsive smad activity in live zebrafish embryos. Dev. Dyn. 2011, 240, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Leeuwis, J.W.; Nguyen, T.Q.; Chuva De Sousa Lopes, S.M.; Van Der Giezen, D.M.; Van Der Ven, K.; Rouw, P.J.H.; Offerhaus, G.J.A.; Mummery, C.L.; Goldschmeding, R. Direct visualization of Smad1/5/8-mediated transcriptional activity identifies podocytes and collecting ducts as major targets of BMP signalling in healthy and diseased kidneys. J. Pathol. 2011, 224, 121–132. [Google Scholar] [CrossRef]
- Beets, K.; Staring, M.W.; Criem, N.; Maas, E.; Schellinx, N.; de Sousa Lopes, S.M.C.; Umans, L.; Zwijsen, A. BMP-SMAD signalling output is highly regionalized in cardiovascular and lymphatic endothelial networks. BMC Dev. Biol. 2016, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Constam, D.B. Regulation of TGFβ and related signals by precursor processing. Semin. Cell Dev. Biol. 2014, 32, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.A.; Al-Musawi, S.L.; Walton, K.L. Prodomains regulate the synthesis, extracellular localisation and activity of TGF-β superfamily ligands. Growth Factors 2011, 29, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Sengle, G.; Ono, R.N.; Sasaki, T.; Sakai, L.Y. Prodomains of transforming growth factor β (TGFβ) Superfamily members specify different functions: Extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 2011, 286, 5087–5099. [Google Scholar] [CrossRef]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin αvβ6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Watabe, T. Bone morphogenetic proteins. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Sengle, G.; Ono, R.N.; Lyons, K.M.; Bächinger, H.P.; Sakai, L.Y. A New Model for Growth Factor Activation: Type II Receptors Compete with the Prodomain for BMP-7. J. Mol. Biol. 2008, 381, 1025–1039. [Google Scholar] [CrossRef]
- Mi, L.Z.; Brown, C.T.; Gao, Y.; Tian, Y.; Le, V.Q.; Walz, T.; Springer, T.A. Structure of bone morphogenetic protein 9 procomplex. Proc. Natl. Acad. Sci. USA 2015, 112, 3710–3715. [Google Scholar] [CrossRef]
- Ramirez, F.; Rifkin, D.B. Extracellular microfibrils: Contextual platforms for TGFβ and BMP signaling. Curr. Opin. Cell Biol. 2009, 21, 616–622. [Google Scholar] [CrossRef]
- Sengle, G.; Charbonneau, N.L.; Ono, R.N.; Sasaki, T.; Alvarez, J.; Keene, D.R.; Bächinger, H.P.; Sakai, L.Y. Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J. Biol. Chem. 2008, 283, 13874–13888. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. Prediction of heparin binding sites in bone morphogenetic proteins (BMPs). Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 1374–1381. [Google Scholar] [CrossRef]
- Choi, Y.J.; Lee, J.Y.; Park, J.H.; Park, J.B.; Suh, J.S.; Choi, Y.S.; Lee, S.J.; Chung, C.P.; Park, Y.J. The identification of a heparin binding domain peptide from bone morphogenetic protein-4 and its role on osteogenesis. Biomaterials 2010, 31, 7226–7238. [Google Scholar] [CrossRef]
- De Laporte, L.; Rice, J.J.; Tortelli, F.; Hubbell, J.A. Tenascin C promiscuously binds growth factors via its fifth fibronectin type III-like domain. PLoS ONE 2013, 8, e62076. [Google Scholar] [CrossRef]
- Von Offenberg Sweeney, N.; Cummins, P.M.; Cotter, E.J.; Fitzpatrick, P.A.; Birney, Y.A.; Redmond, E.M.; Cahill, P.A. Cyclic strain-mediated regulation of vascular endothelial cell migration and tube formation. Biochem. Biophys. Res. Commun. 2005, 329, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Von Offenberg Sweeney, N.; Cummins, P.M.; Birney, Y.A.; Cullen, J.P.; Redmond, E.M.; Cahill, P.A. Cyclic strain-mediated regulation of endothelial matrix metalloproteinase-2 expression and activity. Cardiovasc. Res. 2004, 63, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Little, S.C.; Mullins, M.C. Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes to pattern the dorsoventral axis. Nat. Cell Biol. 2009, 11, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Tajer, B.; Dutko, J.A.; Little, S.C.; Mullins, M.C. BMP heterodimers signal via distinct type I receptor class functions. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Neugebauer, J.; McKnite, A.; Tilak, A.; Christian, J.L. BMP7 functions predominantly as a heterodimer with BMP2 or BMP4 during mammalian embryogenesis. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Danussi, C.; Spessotto, P.; Petrucco, A.; Wassermann, B.; Sabatelli, P.; Montesi, M.; Doliana, R.; Bressan, G.M.; Colombatti, A. Emilin1 deficiency causes structural and functional defects of lymphatic vasculature. Mol. Cell. Biol. 2008, 28, 4026–4039. [Google Scholar] [CrossRef] [PubMed]
- Leak, L.V.; Burke, J.F. Ultrastructural studies on the lymphatic anchoring filaments. J. Cell Biol. 1968, 36, 129–149. [Google Scholar] [CrossRef]
- Nickel, J.; Ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Barbara, N.P.; Wrana, J.L.; Letarte, M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-β superfamily. J. Biol. Chem. 1999, 274, 584–594. [Google Scholar] [CrossRef]
- Schoonderwoerd, M.J.A.; Goumans, M.J.T.H.; Hawinkels, L.J.A.C. Endoglin: Beyond the endothelium. Biomolecules 2020, 10, 289. [Google Scholar] [CrossRef]
- Alt, A.; Miguel-Romero, L.; Donderis, J.; Aristorena, M.; Blanco, F.J.; Round, A.; Rubio, V.; Bernabeu, C.; Marina, A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE 2012, 7, e29948. [Google Scholar] [CrossRef]
- Tian, H.; Mythreye, K.; Golzio, C.; Katsanis, N.; Blobe, G.C. Endoglin mediates fibronectin/α5β1 integrin and TGF-β pathway crosstalk in endothelial cells. EMBO J. 2012, 31, 3885–3900. [Google Scholar] [CrossRef]
- Sieber, C.; Kopf, J.; Hiepen, C.; Knaus, P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev. 2009, 20, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Kirkbride, K.C.; Townsend, T.A.; Bruinsma, M.W.; Barnett, J.V.; Blobe, G.C. Bone morphogenetic proteins signal through the transforming growth factor-β type III receptor. J. Biol. Chem. 2008, 283, 7628–7637. [Google Scholar] [CrossRef] [PubMed]
- Healey, E.G.; Bishop, B.; Elegheert, J.; Bell, C.H.; Padilla-Parra, S.; Siebold, C. Repulsive guidance molecule is a structural bridge between neogenin and bone morphogenetic protein. Nat. Struct. Mol. Biol. 2015, 22, 458–465. [Google Scholar] [CrossRef]
- Halbrooks, P.J.; Ding, R.; Wozney, J.M.; Bain, G. Role of RGM coreceptors in bone morphogenetic protein signaling. J. Mol. Signal. 2007, 2. [Google Scholar] [CrossRef]
- Gipson, G.R.; Goebel, E.J.; Hart, K.N.; Kappes, E.C.; Kattamuri, C.; McCoy, J.C.; Thompson, T.B. Structural perspective of BMP ligands and signaling. Bone 2020, 140. [Google Scholar] [CrossRef]
- Aspalter, I.M.; Gordon, E.; Dubrac, A.; Ragab, A.; Narloch, J.; Vizán, P.; Geudens, I.; Collins, R.T.; Franco, C.A.; Abrahams, C.L.; et al. Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Clements, T.P.; Tang, L.K.; Morales, J.E.; Lee, H.S.; Oh, S.P.; Rivera, G.M.; Wagner, D.S.; McCarty, J.H. Neuropilin 1 balances β8 integrin-activated TGFβ signaling to control sprouting angiogenesis in the brain. Dev. 2015, 142, 4363–4373. [Google Scholar] [CrossRef]
- Glinka, Y.; Stoilova, S.; Mohammed, N.; Prud’homme, G.J. Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis 2011, 32, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Orsenigo, F.; Gagliani, M.C.; Tacchetti, C.; Dejana, E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 2006, 174, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Rudini, N.; Felici, A.; Giampietro, C.; Lampugnani, M.; Corada, M.; Swirsding, K.; Garrè, M.; Liebner, S.; Letarte, M.; Ten Dijke, P.; et al. VE-cadherin is a critical endothelial regulator of TGF-β signalling. EMBO J. 2008, 27, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Benn, A.; Bredow, C.; Casanova, I.; Vukičević, S.; Knaus, P. VE-cadherin facilitates BMP-induced endothelial cell permeability and signaling. J. Cell Sci. 2016, 129, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Ouahoud, S.; Hardwick, J.C.H.; Hawinkels, L.J.A.C. Extracellular bmp antagonists, multifaceted orchestrators in the tumor and its microenvironment. Int. J. Mol. Sci. 2020, 21, 3888. [Google Scholar] [CrossRef]
- Groppe, J.; Greenwald, J.; Wiater, E.; Rodriguez-Leon, J.; Economides, A.N.; Kwiatkowski, W.; Affolter, M.; Vale, W.W.; Izpisua Belmonte, J.C.; Choe, S. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature 2002, 420, 636–642. [Google Scholar] [CrossRef]
- Snider, P.; Simmons, O.; Wang, J.; Hoang, C.; Conway, S. Ectopic Noggin in a Population of Nfatc1 Lineage Endocardial Progenitors Induces Embryonic Lethality. J. Cardiovasc. Dev. Dis. 2014, 1, 214–236. [Google Scholar] [CrossRef]
- Nimmagadda, S.; Loganathan, P.G.; Huang, R.; Scaal, M.; Schmidt, C.; Christ, B. BMP4 and noggin control embryonic blood vessel formation by antagonistic regulation of VEGFR-2 (Quek1) expression. Dev. Biol. 2005, 280, 100–110. [Google Scholar] [CrossRef]
- Ramasamy, S.K.; Kusumbe, A.P.; Wang, L.; Adams, R.H. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 2014, 507, 376–380. [Google Scholar] [CrossRef]
- Hsu, D.R.; Economides, A.N.; Wang, X.; Eimon, P.M.; Harland, R.M. The Xenopus dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol. Cell 1998, 1, 673–683. [Google Scholar] [CrossRef]
- Cahill, E.; Costello, C.M.; Rowan, S.C.; Harkin, S.; Howell, K.; Leonard, M.O.; Southwood, M.; Cummins, E.P.; Fitzpatrick, S.F.; Taylor, C.T.; et al. Gremlin plays a key role in the pathogenesis of pulmonary hypertension. Circulation 2012, 125, 920–930. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Xie, W.; Wan, J.; Tao, X.; Liu, M.; Zhen, Y.; Lin, F.; Wu, B.; Zhai, Z.; et al. Gremlin-1 is a key regulator of endothelial-to-mesenchymal transition in human pulmonary artery endothelial cells. Exp. Cell Res. 2020, 390. [Google Scholar] [CrossRef]
- Mitola, S.; Ravelli, C.; Moroni, E.; Salvi, V.; Leali, D.; Ballmer-Hofer, K.; Zammataro, L.; Presta, M. Gremlin is a novel agonist of the major proangiogenic receptor VEGFR2. Blood 2010, 116, 3677–3680. [Google Scholar] [CrossRef] [PubMed]
- Grillo, E.; Ravelli, C.; Corsini, M.; Ballmer-Hofer, K.; Zammataro, L.; Oreste, P.; Zoppetti, G.; Tobia, C.; Ronca, R.; Presta, M.; et al. Monomeric gremlin is a novel vascular endothelial growth factor receptor-2 antagonist. Oncotarget 2016, 7, 35353–35368. [Google Scholar] [CrossRef] [PubMed]
- Stabile, H.; Mitola, S.; Moroni, E.; Belleri, M.; Nicoli, S.; Coltrini, D.; Peri, F.; Pessi, A.; Orsatti, L.; Talamo, F.; et al. Bone morphogenic protein antagonist Drm/gremlin is a novel proangiogenic factor. Blood 2007, 109, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.; Ren, R.; Pi, X.; Wu, Y.; Moreno, I.; Willis, M.; Moser, M.; Ross, M.; Podkowa, M.; Attisano, L.; et al. A concentration-dependent endocytic trap and sink mechanism converts Bmper from an activator to an inhibitor of Bmp signaling. J. Cell Biol. 2009, 184, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Miralles, I.; Ren, R.; Moser, M.; Hartnett, M.E.; Patterson, C. Bone morphogenetic protein endothelial cell precursor-derived regulator regulates retinal angiogenesis in vivo in a mouse model of oxygen-induced retinopathy. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Heinke, J.; Juschkat, M.; Charlet, A.; Mnich, L.; Helbing, T.; Bode, C.; Moser, M.; Patterson, C. Antagonism and synergy between extracellular bmp modulators tsg and bmper balance blood vessel formation. J. Cell Sci. 2013, 126, 3082–3094. [Google Scholar] [CrossRef] [PubMed]
- Lockhart-Cairns, M.P.; Lim, K.T.W.; Zuk, A.; Godwin, A.R.F.; Cain, S.A.; Sengle, G.; Baldock, C. Internal cleavage and synergy with twisted gastrulation enhance BMP inhibition by BMPER. Matrix Biol. 2019, 77, 73–86. [Google Scholar] [CrossRef]
- Esser, J.S.; Steiner, R.E.; Deckler, M.; Schmitt, H.; Engert, B.; Link, S.; Charlet, A.; Patterson, C.; Bode, C.; Zhou, Q.; et al. Extracellular bone morphogenetic protein modulator BMPER and twisted gastrulation homolog 1 preserve arterial-venous specification in zebrafish blood vessel development and regulate Notch signaling in endothelial cells. FEBS J. 2018, 285, 1419–1436. [Google Scholar] [CrossRef] [PubMed]
- Helbing, T.; Rothweiler, R.; Ketterer, E.; Goetz, L.; Heinke, J.; Grundmann, S.; Duerschmied, D.; Patterson, C.; Bode, C.; Moser, M. BMP activity controlled by BMPER regulates the proinflammatory phenotype of endothelium. Blood 2011, 118, 5040–5049. [Google Scholar] [CrossRef]
- Guillot, N.; Kollins, D.; Badimon, J.J.; Schlondorff, D.; Hutter, R. Accelerated reendothelialization, increased neovascularization and erythrocyte extravasation after arterial injury in BAMBI-/- Mice. PLoS ONE 2013, 8, e58550. [Google Scholar] [CrossRef]
- Okumura, N.; Nakamura, T.; Kay, E.D.P.; Nakahara, M.; Kinoshita, S.; Koizumi, N. R-spondin1 regulates cell proliferation of corneal endothelial cells via the Wnt3a/β-catenin pathway. Invest. Ophthalmol. Vis. Sci. 2014, 55, 6861–6869. [Google Scholar] [CrossRef]
- Ogasawara, R.; Hashimoto, D.; Kimura, S.; Hayase, E.; Ara, T.; Takahashi, S.; Ohigashi, H.; Yoshioka, K.; Tateno, T.; Yokoyama, E.; et al. Intestinal Lymphatic Endothelial Cells Produce R-Spondin3. Sci. Rep. 2018, 8, 10719. [Google Scholar] [CrossRef]
- Lee, H.; Seidl, C.; Sun, R.; Glinka, A.; Niehrs, C. R-spondins are BMP receptor antagonists in Xenopus early embryonic development. Nat. Commun. 2020, 11, 5570. [Google Scholar] [CrossRef]
- Tanaka, K.; Joshi, D.; Timalsina, S.; Schwartz, M.A. Early events in endothelial flow sensing. Cytoskeleton 2021. [Google Scholar] [CrossRef]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef]
- Herbert, S.P.; Stainier, D.Y.R. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef]
- Sorescu, G.P.; Sykes, M.; Weiss, D.; Platt, M.O.; Saha, A.; Hwang, J.; Boyd, N.; Boo, Y.C.; Vega, J.D.; Taylor, W.R.; et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress stimulates an inflammatory response. J. Biol. Chem. 2003, 278, 31128–31135. [Google Scholar] [CrossRef] [PubMed]
- Ruter, D.L.; Liu, Z.; Ngo, K.M.; Shaka, X.; Marvin, A.; Buglak, D.B.; Kidder, E.J.; Bautch, V.L. SMAD6 transduces endothelial cell flow responses required for blood vessel homeostasis. Angiogenesis 2021. [Google Scholar] [CrossRef]
- Ramirez, F.; Sakai, L.Y.; Rifkin, D.B.; Dietz, H.C. Extracellular microfibrils in development and disease. Cell. Mol. Life Sci. 2007, 64, 2437–2446. [Google Scholar] [CrossRef] [PubMed]
- Keski-Oja, J.; Koli, K.; Von Melchner, H. TGF-β activation by traction? Trends Cell Biol. 2004, 14, 657–659. [Google Scholar] [CrossRef] [PubMed]
- Arteaga-Solis, E.; Gayraud, B.; Lee, S.Y.; Shum, L.; Sakai, L.; Ramirez, F. Regulation of limb patterning by extracellular microfibrils. J. Cell Biol. 2001, 154, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Kniazeva, E.; Putnam, A.J. Endothelial cell traction and ECM density influence both capillary morphogenesis and maintenance in 3-D. Am. J. Physiol. Cell Physiol. 2009, 297. [Google Scholar] [CrossRef] [PubMed]
- Dalfino, G.; Simone, S.; Porreca, S.; Cosola, C.; Balestra, C.; Manno, C.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Bone morphogenetic protein-2 may represent the molecular link between oxidative stress and vascular stiffness in chronic kidney disease. Atherosclerosis 2010, 211, 418–423. [Google Scholar] [CrossRef]
- Zhong, A.; Mirzaei, Z.; Simmons, C.A. The Roles of Matrix Stiffness and ß-Catenin Signaling in Endothelial-to-Mesenchymal Transition of Aortic Valve Endothelial Cells. Cardiovasc. Eng. Technol. 2018, 9, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Ren, R.; Kelley, R.; Zhang, C.; Moser, M.; Bohil, A.B.; DiVito, M.; Cheney, R.E.; Patterson, C. Sequential roles for myosin-X in BMP6-dependent filopodial extension, migration, and activation of BMP receptors. J. Cell Biol. 2007, 179, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- Friedland, J.C.; Lee, M.H.; Boettiger, D. Mechanically activated integrin switch controls α5β 1 function. Science 2009, 323, 642–644. [Google Scholar] [CrossRef]
- Zhou, J.; Lee, P.L.; Lee, C.I.; Wei, S.Y.; Lim, S.H.; Lin, T.E.; Chien, S.; Chiu, J.J. BMP receptor-integrin interaction mediates responses of vascular endothelial Smad1/5 and proliferation to disturbed flow. J. Thromb. Haemost. 2013, 11, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lee, P.L.; Tsai, C.S.; Lee, C.I.; Yang, T.L.; Chuang, H.S.; Lin, W.W.; Lin, T.E.; Lim, S.H.; Wei, S.Y.; et al. Force-specific activation of Smad1/5 regulates vascular endothelial cell cycle progression in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2012, 109, 7770–7775. [Google Scholar] [CrossRef] [PubMed]
- Peacock, H.M.; Tabibian, A.; Criem, N.; Caolo, V.; Hamard, L.; Deryckere, A.; Haefliger, J.-A.; Kwak, B.R.; Zwijsen, A.; Jones, E.A.V. Impaired SMAD1/5 Mechanotransduction and Cx37 (Connexin37) expression enable pathological vessel enlargement and shunting. Arterioscler. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Gkatzis, K.; Thalgott, J.; Dos-Santos-Luis, D.; Martin, S.; Lamandé, N.; Carette, M.F.; Disch, F.; Snijder, R.J.; Westermann, C.J.; Mager, J.J.; et al. Interaction between ALK1 signaling and Connexin40 in the development of arteriovenous malformations. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, N.; Larrivée, B.; Ola, R.; Hayward-Piatkowskyi, B.; Dubrac, A.; Huang, B.; Ross, T.D.; Coon, B.G.; Min, E.; Tsarfati, M.; et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 2016, 214. [Google Scholar] [CrossRef]
- Santos-Oliveira, P.; Correia, A.; Rodrigues, T.; Ribeiro-Rodrigues, T.M.; Matafome, P.; Rodríguez-Manzaneque, J.C.; Seiça, R.; Girão, H.; Travasso, R.D.M. The force at the tip—Modelling tension and proliferation in sprouting angiogenesis. PLoS Comput. Biol. 2015, 11. [Google Scholar] [CrossRef]
- Sabine, A.; Bovay, E.; Demir, C.S.; Kimura, W.; Jaquet, M.; Agalarov, Y.; Zangger, N.; Scallan, J.P.; Graber, W.; Gulpinar, E.; et al. FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Investig. 2015, 125, 3861–3877. [Google Scholar] [CrossRef] [PubMed]
- Sweet, D.T.; Jiménez, J.M.; Chang, J.; Hess, P.R.; Mericko-Ishizuka, P.; Fu, J.; Xia, L.; Davies, P.F.; Kahn, M.L. Lymph flow regulates collecting lymphatic vessel maturation in vivo. J. Clin. Investig. 2015, 125, 2995–3007. [Google Scholar] [CrossRef]
- Frye, M.; Taddei, A.; Dierkes, C.; Martinez-Corral, I.; Fielden, M.; Ortsäter, H.; Kazenwadel, J.; Calado, D.P.; Ostergaard, P.; Salminen, M.; et al. Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2-dependent transcriptional program. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Alderfer, L.; Russo, E.; Archilla, A.; Coe, B.; Hanjaya-Putra, D. Matrix stiffness primes lymphatic tube formation directed by vascular endothelial growth factor-C. FASEB J. 2021, 35, e21498. [Google Scholar] [CrossRef] [PubMed]
- Bazigou, E.; Xie, S.; Chen, C.; Weston, A.; Miura, N.; Sorokin, L.; Adams, R.; Muro, A.F.; Sheppard, D.; Makinen, T. Integrin-α9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev. Cell 2009, 17, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Planas-Paz, L.; Strilić, B.; Goedecke, A.; Breier, G.; Fässler, R.; Lammert, E. Mechanoinduction of lymph vessel expansion. EMBO J. 2012, 31, 788–804. [Google Scholar] [CrossRef]
- Planas-Paz, L.; Lammert, E. Mechanical forces in lymphatic vascular development and disease. Cell. Mol. Life Sci. 2013, 70, 4341–4354. [Google Scholar] [CrossRef]
- Galvagni, F.; Pennacchini, S.; Salameh, A.; Rocchigiani, M.; Neri, F.; Orlandini, M.; Petraglia, F.; Gotta, S.; Sardone, G.L.; Matteucci, G.; et al. Endothelial cell adhesion to the extracellular matrix induces c-Src-dependent VEGFR-3 phosphorylation without the activation of the receptor intrinsic kinase activity. Circ. Res. 2010, 106, 1839–1848. [Google Scholar] [CrossRef]
- Hadizadeh, M.; Mohaddes Ardebili, S.M.; Salehi, M.; Young, M.; Mokarian, F.; McClellan, J.; Xu, Q.; Kazemi, M.; Moazam, E.; Mahaki, B.; et al. GJA4/connexin 37 mutations correlate with secondary lymphedema following surgery in breast cancer patients. Biomedicines 2018, 6, 23. [Google Scholar] [CrossRef]
- Kanady, J.D.; Dellinger, M.T.; Munger, S.J.; Witte, M.H.; Simon, A.M. Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev. Biol. 2011, 354, 253–266. [Google Scholar] [CrossRef]
- Sabine, A.; Petrova, T. V Interplay of mechanotransduction, FOXC2, connexins, and calcineurin signaling in lymphatic valve formation. Adv. Anat. Embryol. Cell Biol. 2014, 214, 67–80. [Google Scholar] [CrossRef]
- Munger, S.J.; Davis, M.J.; Simon, A.M. Defective lymphatic valve development and chylothorax in mice with a lymphatic-specific deletion of Connexin43. Dev. Biol. 2017, 421, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Munger, S.J.; Geng, X.; Srinivasan, R.S.; Witte, M.H.; Paul, D.L.; Simon, A.M. Segregated Foxc2, NFATc1 and Connexin expression at normal developing venous valves, and Connexin-specific differences in the valve phenotypes of Cx37, Cx43, and Cx47 knockout mice. Dev. Biol. 2016, 412, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Meens, M.J.; Sabine, A.; Petrova, T.V.; Kwak, B.R. Connexins in lymphatic vessel physiology and disease. FEBS Lett. 2014, 588, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Kanady, J.D.; Munger, S.J.; Witte, M.H.; Simon, A.M. Combining Foxc2 and Connexin37 deletions in mice leads to severe defects in lymphatic vascular growth and remodeling. Dev. Biol. 2015, 405, 33–46. [Google Scholar] [CrossRef]
- Sabine, A.; Agalarov, Y.; Maby-El Hajjami, H.; Jaquet, M.; Hägerling, R.; Pollmann, C.; Bebber, D.; Pfenniger, A.; Miura, N.; Dormond, O.; et al. Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev. Cell 2012, 22, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.C.; Baluk, P.; Srinivasan, R.S.; Oliver, G.; McDonald, D.M. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 2012, 180, 2561–2575. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell 2009, 16, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Hägerling, R.; Hoppe, E.; Dierkes, C.; Stehling, M.; Makinen, T.; Butz, S.; Vestweber, D.; Kiefer, F. Distinct roles of VE -cadherin for development and maintenance of specific lymph vessel beds. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Yang, Y.; Cha, B.; Motawe, Z.Y.; Srinivasan, R.S.; Scallan, J.P. VE-cadherin is required for lymphatic valve formation and maintenance. Cell Rep. 2019, 28, 2397–2412.e4. [Google Scholar] [CrossRef]
- Zwijsen, A.; van Rooijen, M.; Goumans, M.; Dewulf, N.; Bosman, E.; ten Dijke, P.; Mummery, C.; Huylebroeck, D. Expression of the inhibitory Smad7 in early mouse development and upregulation during embryonic vasculogenesis. Dev. Dyn. 2000, 218. [Google Scholar] [CrossRef]
- Murayama, K.; Kato-Murayama, M.; Itoh, Y.; Miyazono, K.; Miyazawa, K.; Shirouzu, M. Structural basis for inhibitory effects of Smad7 on TGF-β family signaling. J. Struct. Biol. 2020, 212. [Google Scholar] [CrossRef]
- Goto, K.; Kamiya, Y.; Imamura, T.; Miyazono, K.; Miyazawa, K. Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J. Biol. Chem. 2007, 282, 20603–20611. [Google Scholar] [CrossRef] [PubMed]
- Wylie, L.A.; Mouillesseaux, K.P.; Chong, D.C.; Bautch, V.L. Developmental SMAD6 loss leads to blood vessel hemorrhage and disrupted endothelial cell junctions. Dev. Biol. 2018, 442, 199–209. [Google Scholar] [CrossRef]
- Murakami, G.; Watabe, T.; Takaoka, K.; Miyazono, K.; Imamura, T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell 2003, 14, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth Factor-β Type I receptor through smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef]
- De Ceuninck van Capelle, C.; Spit, M.; ten Dijke, P. Current perspectives on inhibitory SMAD7 in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 691–715. [Google Scholar] [CrossRef]
- Mouillesseaux, K.P.; Wiley, D.S.; Saunders, L.M.; Wylie, L.A.; Kushner, E.J.; Chong, D.C.; Citrin, K.M.; Barber, A.T.; Park, Y.; Kim, J.D.; et al. Notch regulates BMP responsiveness and lateral branching in vessel networks via SMAD6. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Su, F.; Li, X.; You, K.; Chen, M.; Xiao, J.; Zhang, Y.; Ma, J.; Liu, B. Expression of VEGF-D, SMAD4, and SMAD7 and their relationship with lymphangiogenesis and prognosis in colon cancer. J. Gastrointest. Surg. 2016, 20, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Conidi, A.; Cazzola, S.; Beets, K.; Coddens, K.; Collart, C.; Cornelis, F.; Cox, L.; Joke, D.; Dobreva, M.P.; Dries, R.; et al. Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGFβ/BMP signaling in vivo. Cytokine Growth Factor Rev. 2011, 22, 287–300. [Google Scholar] [CrossRef] [PubMed]
- De Haan, W.; Øie, C.; Benkheil, M.; Dheedene, W.; Vinckier, S.; Coppiello, G.; Aranguren, X.L.; Beerens, M.; Jaekers, J.; Topal, B.; et al. Unraveling the transcriptional determinants of liver sinusoidal endothelial cell specialization. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G803–G815. [Google Scholar] [CrossRef]
- De Haan, W.; Dheedene, W.; Apelt, K.; Décombas-Deschamps, S.; Vinckier, S.; Verhulst, S.; Conidi, A.; Deffieux, T.; Staring, M.W.; Vandervoort, P.; et al. Endothelial Zeb2 preserves the hepatic angioarchitecture and protects against liver fibrosis. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Gronroos, E.; Kingston, I.J.; Ramachandran, A.; Randall, R.A.; Vizan, P.; Hill, C.S. Transforming growth factor inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol. Cell. Biol. 2012, 32, 2904–2916. [Google Scholar] [CrossRef]
- Yuan, G.; Zhan, Y.; Gou, X.; Chen, Y.; Yang, G. TGF-β signaling inhibits canonical BMP signaling pathway during palate development. Cell Tissue Res. 2018, 371, 283–291. [Google Scholar] [CrossRef] [PubMed]
- James, J.M.; Nalbandian, A.; Mukouyama, Y. suke TGFβ signaling is required for sprouting lymphangiogenesis during lymphatic network development in the skin. Development 2013, 140, 3903–3914. [Google Scholar] [CrossRef]
- Vittet, D.; Merdzhanova, G.; Prandini, M.H.; Feige, J.J.; Bailly, S. TGFβ1 inhibits lymphatic endothelial cell differentiation from mouse embryonic stem cells. J. Cell. Physiol. 2012, 227, 3593–3602. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Iwata, C.; Suzuki, H.I.; Kiyono, K.; Morishita, Y.; Watabe, T.; Komuro, A.; Kano, M.R.; Miyazono, K. Inhibition of endogenous TGF-2 signaling enhances lymphangiogenesis. Blood 2008, 111, 4571–4579. [Google Scholar] [CrossRef]
- Leppänen, V.M.; Tvorogov, D.; Kisko, K.; Prota, A.E.; Jeltsch, M.; Anisimov, A.; Markovic-Mueller, S.; Stuttfeld, E.; Goldie, K.N.; Ballmer-Hofer, K.; et al. Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. Proc. Natl. Acad. Sci. USA 2013, 110, 12960–12965. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef]
- Krueger, J.; Liu, D.; Scholz, K.; Zimmer, A.; Shi, Y.; Klein, C.; Siekmann, A.; Schulte-Merker, S.; Cudmore, M.; Ahmed, A.; et al. Flt1 acts as a negative regulator of tip cell formation and branching morphogenesis in the zebrafish embryo. Development 2011, 138, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Pulkkinen, H.H.; Kiema, M.; Lappalainen, J.P.; Toropainen, A.; Beter, M.; Tirronen, A.; Holappa, L.; Niskanen, H.; Kaikkonen, M.U.; Ylä-Herttuala, S.; et al. BMP6/TAZ-Hippo signaling modulates angiogenesis and endothelial cell response to VEGF. Angiogenesis 2020, 24. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, J.; Morikawa, Y.; Bonilla-Claudio, M.; Klysik, E.; Martin, J.F. Bmp signaling represses vegfa to promote outflow tract cushion development. Development 2013, 140, 3395–3402. [Google Scholar] [CrossRef] [PubMed]
- Shao, E.S.; Lin, L.; Yao, Y.; Boström, K.I. Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood 2009, 114, 2197–2206. [Google Scholar] [CrossRef]
- Rezzola, S.; Di Somma, M.; Corsini, M.; Leali, D.; Ravelli, C.; Polli, V.A.B.; Grillo, E.; Presta, M.; Mitola, S. VEGFR2 activation mediates the pro-angiogenic activity of BMP4. Angiogenesis 2019, 22, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J.; Gutkind, J.S. VEGF Controls endothelial-cell permeability promoting β-arrestin-dependent Endocytosis VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Plein, A.; Fantin, A.; Ruhrberg, C. Neuropilin regulation of angiogenesis, arteriogenesis, and vascular permeability. Microcirculation 2014, 21, 315–323. [Google Scholar] [CrossRef]
- Becker, P.M.; Waltenberger, J.; Yachechko, R.; Mirzapoiazova, T.; Sham, J.S.K.; Lee, C.G.; Elias, J.A.; Verin, A.D. Neuropilin-1 regulates vascular endothelial growth factor-mediated endothelial permeability. Circ. Res. 2005, 96, 1257–1265. [Google Scholar] [CrossRef]
- Akla, N.; Viallard, C.; Popovic, N.; Gil, C.L.; Sapieha, P.; Larrivée, B. BMP9 (bone morphogenetic protein-9)/ALK1 (activin-like kinase receptor type I) signaling prevents hyperglycemia-induced vascular permeability. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1821–1836. [Google Scholar] [CrossRef]
- Haiko, P.; Makinen, T.; Keskitalo, S.; Taipale, J.; Karkkainen, M.J.; Baldwin, M.E.; Stacker, S.A.; Achen, M.G.; Alitalo, K. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol. Cell. Biol. 2008, 28, 4843–4850. [Google Scholar] [CrossRef]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74–80. [Google Scholar] [CrossRef]
- Alitalo, A.; Detmar, M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 2012, 31, 4499–4508. [Google Scholar] [CrossRef]
- Jeltsch, M.; Leppänen, V.M.; Saharinen, P.; Alitalo, K. Receptor tyrosine Kinase-Mediated angiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef]
- Gordon, K.; Schulte, D.; Brice, G.; Simpson, M.A.; Roukens, M.G.; Van Impel, A.; Connell, F.; Kalidas, K.; Jeffery, S.; Mortimer, P.S.; et al. Mutation in vascular endothelial growth factor-c, a ligand for vascular endothelial growth factor receptor-3, is associated with autosomal dominant milroy-like primary lymphedema. Circ. Res. 2013, 112, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Saaristo, A.; Jussila, L.; Karila, K.A.; Lawrence, E.C.; Pajusola, K.; Bueler, H.; Eichmann, A.; Kauppinen, R.; Kettunen, M.I.; et al. A model for gene therapy of human hereditary lymphedema. Proc. Natl. Acad. Sci. USA 2001, 98, 12677–12682. [Google Scholar] [CrossRef]
- Secker, G.A.; Harvey, N.L. VEGFR signaling during lymphatic vascular development: From progenitor cells to functional vessels. Dev. Dyn. 2015, 244, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The canonical notch signaling pathway: Structural and biochemical insights into shape, sugar, and force. Dev. Cell 2017, 41, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Koinuma, D.; Tsutsumi, S.; Vasilaki, E.; Kanki, Y.; Heldin, C.H.; Aburatani, H.; Miyazono, K. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef]
- Itoh, F.; Itoh, S.; Goumans, M.J.; Valdimarsdottir, G.; Iso, T.; Dotto, G.P.; Hamamori, Y.; Kedes, L.; Kato, M.; Ten Dijke, P. Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 2004, 23, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Rochon, E.R.; Wright, D.S.; Schubert, M.M.; Roman, B.L. Context-specific interactions between Notch and ALK1 cannot explain ALK1-associated arteriovenous malformations. Cardiovasc. Res. 2015, 107, 143–152. [Google Scholar] [CrossRef]
- Wöltje, K.; Jabs, M.; Fischer, A. Serum Induces transcription of Hey1 and Hey2 genes by Alk1 but not notch signaling in endothelial cells. PLoS ONE 2015, 10, e0120547. [Google Scholar] [CrossRef] [PubMed]
- Ricard, N.; Ciais, D.; Levet, S.; Subileau, M.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Bidart, M.; Feige, J.J.; Bailly, S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 2012, 119, 6162–6171. [Google Scholar] [CrossRef]
- Larrivée, B.; Prahst, C.; Gordon, E.; del Toro, R.; Mathivet, T.; Duarte, A.; Simons, M.; Eichmann, A. ALK1 signaling inhibits angiogenesis by cooperating with the notch pathway. Dev. Cell 2012, 22, 489–500. [Google Scholar] [CrossRef]
- Luna-Zurita, L.; Prados, B.; Grego-Bessa, J.; Luxán, G.; Del Monte, G.; Benguría, A.; Adams, R.H.; Pérez-Pomares, J.M.; De La Pompa, J.L. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J. Clin. Investig. 2010, 120, 3493–3507. [Google Scholar] [CrossRef]
- Garside, V.C.; Chang, A.C.; Karsan, A.; Hoodless, P.A. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell. Mol. Life Sci. 2013, 70, 2899–2917. [Google Scholar] [CrossRef]
- Beets, K.; Huylebroeck, D.; Moya, I.M.; Umans, L.; Zwijsen, A. Robustness in angiogenesis: Notch and BMP shaping waves. Trends Genet. 2013, 29, 140–149. [Google Scholar] [CrossRef]
- Bai, G.; Sheng, N.; Xie, Z.; Bian, W.; Yokota, Y.; Benezra, R.; Kageyama, R.; Guillemot, F.; Jing, N. Id sustains Hes1 expression to inhibit precocious neurogenesis by releasing negative autoregulation of Hes1. Dev. Cell 2007, 13, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tammela, T.; Yamamoto, M.; Anisimov, A.; Holopainen, T.; Kaijalainen, S.; Karpanen, T.; Lehti, K.; Ylä-Herttuala, S.; Alitalo, K. Notch restricts lymphatic vessel sprouting induced by vascular endothelial growth factor. Blood 2011, 118, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Murtomaki, A.; Uh, M.K.; Kitajewski, C.; Zhao, J.; Nagasaki, T.; Shawber, C.J.; Kitajewski, J. Notch signaling functions in lymphatic valve formation. Development 2014, 141, 2446–2451. [Google Scholar] [CrossRef] [PubMed]
- Michelini, S.; Ricci, M.; Serrani, R.; Barati, S.; Kenanoglu, S.; Veselenyiova, D.; Kurti, D.; Baglivo, M.; Basha, S.H.; Priya, S.; et al. NOTCH1: Review of its role in lymphatic development and study of seven families with rare pathogenic variants. Mol. Genet. Genomic Med. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Bernier-Latmani, J.; Cisarovsky, C.; Demir, C.S.; Bruand, M.; Jaquet, M.; Davanture, S.; Ragusa, S.; Siegert, S.; Dormond, O.; Benedito, R.; et al. DLL4 promotes continuous adult intestinal lacteal regeneration and dietary fat transport. J. Clin. Investig. 2015, 125, 4572–4586. [Google Scholar] [CrossRef]
- Franco, C.A.; Liebner, S.; Gerhardt, H. Vascular morphogenesis: A Wnt for every vessel? Curr. Opin. Genet. Dev. 2009, 19, 476–483. [Google Scholar] [CrossRef]
- Strutt, D.; Madder, D.; Chaudhary, V.; Artymiuk, P.J. Structure-function dissection of the Frizzled receptor in Drosophila melanogaster suggests different mechanisms of action in planar polarity and canonical Wnt signaling. Genetics 2012, 192, 1295–1313. [Google Scholar] [CrossRef] [PubMed]
- Gammons, M.V.; Renko, M.; Johnson, C.M.; Rutherford, T.J.; Bienz, M. Wnt signalosome assembly by DEP domain swapping of dishevelled. Mol. Cell 2016, 64, 92–104. [Google Scholar] [CrossRef]
- Brunt, L.; Scholpp, S. The function of endocytosis in Wnt signaling. Cell. Mol. Life Sci. 2018, 75, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Horsthuis, T.; Farin, H.F.; Grieskamp, T.; Norden, J.; Petry, M.; Wakker, V.; Moorman, A.F.M.; Christoffels, V.M.; Kispert, A. Tbx20 interacts with smads to confine tbx2 expression to the atrioventricular canal. Circ. Res. 2009, 105, 442–452. [Google Scholar] [CrossRef]
- Kashiwada, T.; Fukuhara, S.; Terai, K.; Tanaka, T.; Wakayama, Y.; Ando, K.; Nakajima, H.; Fukui, H.; Yuge, S.; Saito, Y.; et al. β-catenin-dependent transcription is central to bmp-mediated formation of venous vessels. Development 2015, 142, 497–509. [Google Scholar] [CrossRef]
- Cha, B.; Geng, X.; Mahamud, M.R.; Fu, J.; Mukherjee, A.; Kim, Y.; Jho, E.-H.; Kim, T.H.; Kahn, M.L.; Xia, L.; et al. Mechanotransduction activates canonical Wnt/β-catenin signaling to promote lymphatic vascular patterning and the development of lymphatic and lymphovenous valves. Genes Dev. 2016, 30, 1454–1469. [Google Scholar] [CrossRef]
- Cha, B.; Srinivasan, R.S. Mechanosensitive β-catenin signaling regulates lymphatic vascular development. BMB Rep. 2016, 49, 403–404. [Google Scholar] [CrossRef]
- Cha, B.; Geng, X.; Mahamud, M.R.; Zhang, J.Y.; Chen, L.; Kim, W.; Jho, E.H.; Kim, Y.; Choi, D.; Dixon, J.B.; et al. Complementary wnt sources regulate lymphatic vascular development via PROX1-Dependent Wnt/β-catenin signaling. Cell Rep. 2018, 25, 571–584.e5. [Google Scholar] [CrossRef]
- Nicenboim, J.; Malkinson, G.; Lupo, T.; Asaf, L.; Sela, Y.; Mayseless, O.; Gibbs-Bar, L.; Senderovich, N.; Hashimshony, T.; Shin, M.; et al. Lymphatic vessels arise from specialized angioblasts within a venous niche. Nature 2015, 522, 56–61. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Simeoni, I.; Downes, K.; Frazer, Z.C.; Megy, K.; Bernabeu-Herrero, M.E.; Shurr, A.; Brimley, J.; Patel, D.; Kell, L.; et al. Mutational and phenotypic characterization of hereditary hemorrhagic telangiectasia. Blood 2020, 136, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type. Nat. Genet. 1996, 13, 189–195. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrel, J.; et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, É.; Westermann, C.J.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fülöp, G.T.; Langa, C.; et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef]
- Hernandez, F.; Huether, R.; Carter, L.; Johnston, T.; Thompson, J.; Gossage, J.R.; Chao, E.; Elliott, A.M. Mutations in RASA1 and GDF2 identified in patients with clinical features of hereditary hemorrhagic telangiectasia. Hum. Genome Var. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Hashimoto, T.; Tihan, T.; Young, W.L.; Perry, V.; Lawton, M.T. Growth and regression of arteriovenous malformations in a patient with hereditary hemorrhagic telangiectasia. Case report. J. Neurosurg. 2007, 106, 470–477. [Google Scholar] [CrossRef]
- Peacock, H.M.; Caolo, V.; Jones, E.A.V. Arteriovenous malformations in hereditary haemorrhagic telangiectasia: Looking beyond ALK1-NOTCH interactions. Cardiovasc. Res. 2016, 109, 196–203. [Google Scholar] [CrossRef]
- Choi, E.J.; Walker, E.J.; Shen, F.; Paul Oh, S.; Arthur, H.M.; Young, W.L.; Su, H. Minimal homozygous endothelial deletion of eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc. Dis. 2012, 33, 540–547. [Google Scholar] [CrossRef]
- Bernabeu, C.; Bayrak-Toydemir, P.; McDonald, J.; Letarte, M. Potential second-hits in hereditary hemorrhagic telangiectasia. J. Clin. Med. 2020, 9, 3571. [Google Scholar] [CrossRef]
- Tielemans, B.; Delcroix, M.; Belge, C.; Quarck, R. TGFβ and BMPRII signalling pathways in the pathogenesis of pulmonary arterial hypertension. Drug Discov. Today 2019, 24, 703–716. [Google Scholar] [CrossRef]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial primary pulmonary hypertension (Gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C.; Aldred, M.; Brannon, C.A.; et al. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Gra¨f, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, J.; Lu, W. Bone morphogenetic protein signalling in pulmonary hypertension: Advances and therapeutic implications. Exp. Physiol. 2017, 102, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 2019, 17. [Google Scholar] [CrossRef]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Zafar, A.; Quadri, S.A.; Farooqui, M.; Ikram, A.; Robinson, M.; Hart, B.L.; Mabray, M.C.; Vigil, C.; Tang, A.T.; Kahn, M.L.; et al. Familial Cerebral Cavernous Malformations. Stroke 2019, 50, 1294–1301. [Google Scholar] [CrossRef]
- Maddaluno, L.; Rudini, N.; Cuttano, R.; Bravi, L.; Giampietro, C.; Corada, M.; Ferrarini, L.; Orsenigo, F.; Papa, E.; Boulday, G.; et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013, 498, 492–496. [Google Scholar] [CrossRef]
- Siu, S.C.; Silversides, C.K. Bicuspid Aortic Valve Disease. J. Am. Coll. Cardiol. 2010, 55, 2789–2800. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Crapanzano, F.; Schirone, L.; Allegra, A.; Pisano, C.; Ruvolo, G.; Forte, M.; Greco, E.; Cavarretta, E.; Marullo, A.G.M.; et al. Deregulation of Notch1 pathway and circulating endothelial progenitor cell (EPC) number in patients with bicuspid aortic valve with and without ascending aorta aneurysm. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Luyckx, I.; MacCarrick, G.; Kempers, M.; Meester, J.; Geryl, C.; Rombouts, O.; Peeters, N.; Claes, C.; Boeckx, N.; Sakalihasan, N.; et al. Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy. Eur. J. Hum. Genet. 2019, 27, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Derwall, M.; Malhotra, R.; Lai, C.S.; Beppu, Y.; Aikawa, E.; Seehra, J.S.; Zapol, W.M.; Bloch, K.D.; Yu, P.B. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Saeed, O.; Otsuka, F.; Polavarapu, R.; Karmali, V.; Weiss, D.; Davis, T.; Rostad, B.; Pachura, K.; Adams, L.; Elliott, J.; et al. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Vandersmissen, I.; Craps, S.; Depypere, M.; Coppiello, G.; van Gastel, N.; Maes, F.; Carmeliet, G.; Schrooten, J.; Jones, E.A.V.; Umans, L.; et al. Endothelial Msx1 transduces hemodynamic changes into an arteriogenic remodeling response. J. Cell Biol. 2015, 210, 1239–1256. [Google Scholar] [CrossRef]
- Pachori, A.S.; Custer, L.; Hansen, D.; Clapp, S.; Kemppa, E.; Klingensmith, J. Bone morphogenetic protein 4 mediates myocardial ischemic injury through JNK-dependent signaling pathway. J. Mol. Cell. Cardiol. 2010, 48, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Ikeda, K.; Akakabe, Y.; Koide, M.; Uraoka, M.; Yutaka, K.T.; Kurimoto-Nakano, R.; Takahashi, T.; Matoba, S.; Yamada, H.; et al. Paracrine osteogenic signals via bone morphogenetic protein-2 accelerate the atherosclerotic intimal calcification in vivo. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Boström, K.; Watson, K.E.; Horn, S.; Wortham, C.; Herman, I.M.; Demer, L.L. Bone morphogenetic protein expression in human atherosclerotic lesions. J. Clin. Investig. 1993, 91, 1800–1809. [Google Scholar] [CrossRef]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Van Der Veken, B.; De Meyer, G.; Martinet, W. Inhibition of VEGF receptor signaling attenuates intraplaque angiogenesis and plaque destabilization in a mouse model of advanced atherosclerosis. Atherosclerosis 2017, 263, e33–e34. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Brown, M.A.; Kaplan, F.S. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Cocks, M.; Mohan, A.; Meyers, C.A.; Ding, C.; Levi, B.; McCarthy, E.; James, A.W. Vascular patterning in human heterotopic ossification. Hum. Pathol. 2017, 63, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.; Kipnis, J.; Randolph, G.J.; Harvey, N.L. The Lymphatic Vasculature in the 21st Century: Novel Functional Roles in Homeostasis and Disease. Cell 2020, 182, 270–296. [Google Scholar] [CrossRef] [PubMed]
- Visser, J.; Van Geel, M.; Cornelissen, A.J.M.; Van Der Hulst, R.R.W.J.; Qiu, S.S. Breast cancer-related lymphedema and genetic predisposition: A systematic review of the literature. Lymphat. Res. Biol. 2019, 17, 288–293. [Google Scholar] [CrossRef]
- Kim, T.; Tafoya, E.; Chelliah, M.P.; Lekwuttikarn, R.; Li, J.; Sarin, K.Y.; Teng, J. Alterations of the MEK/ERK, BMP, and Wnt/β-catenin pathways detected in the blood of individuals with lymphatic malformations. PLoS ONE 2019, 14, e0213872. [Google Scholar] [CrossRef]
- Miaskowski, C.; Dodd, M.; Paul, S.M.; West, C.; Hamolsky, D.; Abrams, G.; Cooper, B.A.; Elboim, C.; Neuhaus, J.; Schmidt, B.L.; et al. Lymphatic and angiogenic candidate genes predict the development of secondary lymphedema following breast cancer surgery. PLoS ONE 2013, 8, e60164. [Google Scholar] [CrossRef]
- Liu, T.; Zou, X.Z.; Huang, N.; Ge, X.Y.; Yao, M.Z.; Liu, H.; Zhang, Z.; Hu, C.P. miR-27a promotes endothelial-mesenchymal transition in hypoxia-induced pulmonary arterial hypertension by suppressing BMP signaling. Life Sci. 2019, 227, 64–73. [Google Scholar] [CrossRef]
- Tian, F.; Zhou, A.X.; Smits, A.M.; Larsson, E.; Goumans, M.J.; Heldin, C.H.; Borén, J.; Akyürek, L.M. Endothelial cells are activated during hypoxia via endoglin/ALK-1/SMAD1/5 signaling in vivo and in vitro. Biochem. Biophys. Res. Commun. 2010, 392, 283–288. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| BMP Signaling Component | Animal Model | Function in LV | References | |
|---|---|---|---|---|
| BMP ligands | BMP2 | ZF, C, M | Anti-lymphangiogenic: restricts LEC specification | [32] |
| BMP9 | M, C | Anti-lymphangiogenic: Promotes LV maturation and valve maturation. Restricts LEC proliferation. Pro-lymph-vasculogenic: promotes LEC specification | [33,34,35] | |
| BMP type I receptors | ALK1 | M, C | Anti-lymphangiogenic: promotes LV maturation and remodeling | [33,34,36] |
| ALK3 | ZF | Pro-lymphangiogenic: promotes LEC numbers | [37] | |
| BMP type II Receptors | BMPRII | ZF M, C | Pro-lymphangiogenic: promotes LEC numbers Anti-lymphangiogenic: promotes LV maturation and remodeling | [37] [36] |
| ACTRIIB | M, C | Anti-lymphangiogenic: promotes LV maturation and remodeling | [36] | |
| Intracellular effectors | SMAD5 | ZF | Pro-lymphangiogenic: promotes LEC numbers | [37] |
| SMAD4 | C | Anti-lymphangiogenic: restricts LEC specification | [32] | |
| TMEM100 | M | Anti-lymphangiogenic: restricts LEC specification | [38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomarev, L.C.; Ksiazkiewicz, J.; Staring, M.W.; Luttun, A.; Zwijsen, A. The BMP Pathway in Blood Vessel and Lymphatic Vessel Biology. Int. J. Mol. Sci. 2021, 22, 6364. https://doi.org/10.3390/ijms22126364
Ponomarev LC, Ksiazkiewicz J, Staring MW, Luttun A, Zwijsen A. The BMP Pathway in Blood Vessel and Lymphatic Vessel Biology. International Journal of Molecular Sciences. 2021; 22(12):6364. https://doi.org/10.3390/ijms22126364
Chicago/Turabian StylePonomarev, Ljuba C., Jakub Ksiazkiewicz, Michael W. Staring, Aernout Luttun, and An Zwijsen. 2021. "The BMP Pathway in Blood Vessel and Lymphatic Vessel Biology" International Journal of Molecular Sciences 22, no. 12: 6364. https://doi.org/10.3390/ijms22126364
APA StylePonomarev, L. C., Ksiazkiewicz, J., Staring, M. W., Luttun, A., & Zwijsen, A. (2021). The BMP Pathway in Blood Vessel and Lymphatic Vessel Biology. International Journal of Molecular Sciences, 22(12), 6364. https://doi.org/10.3390/ijms22126364