
Novel Bacterial Production of Two Different Bioactive Forms of Human Stem-Cell Factor

 ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Design of Expression Plasmids
2.2. Expression and Solubility of the Recombinant Proteins
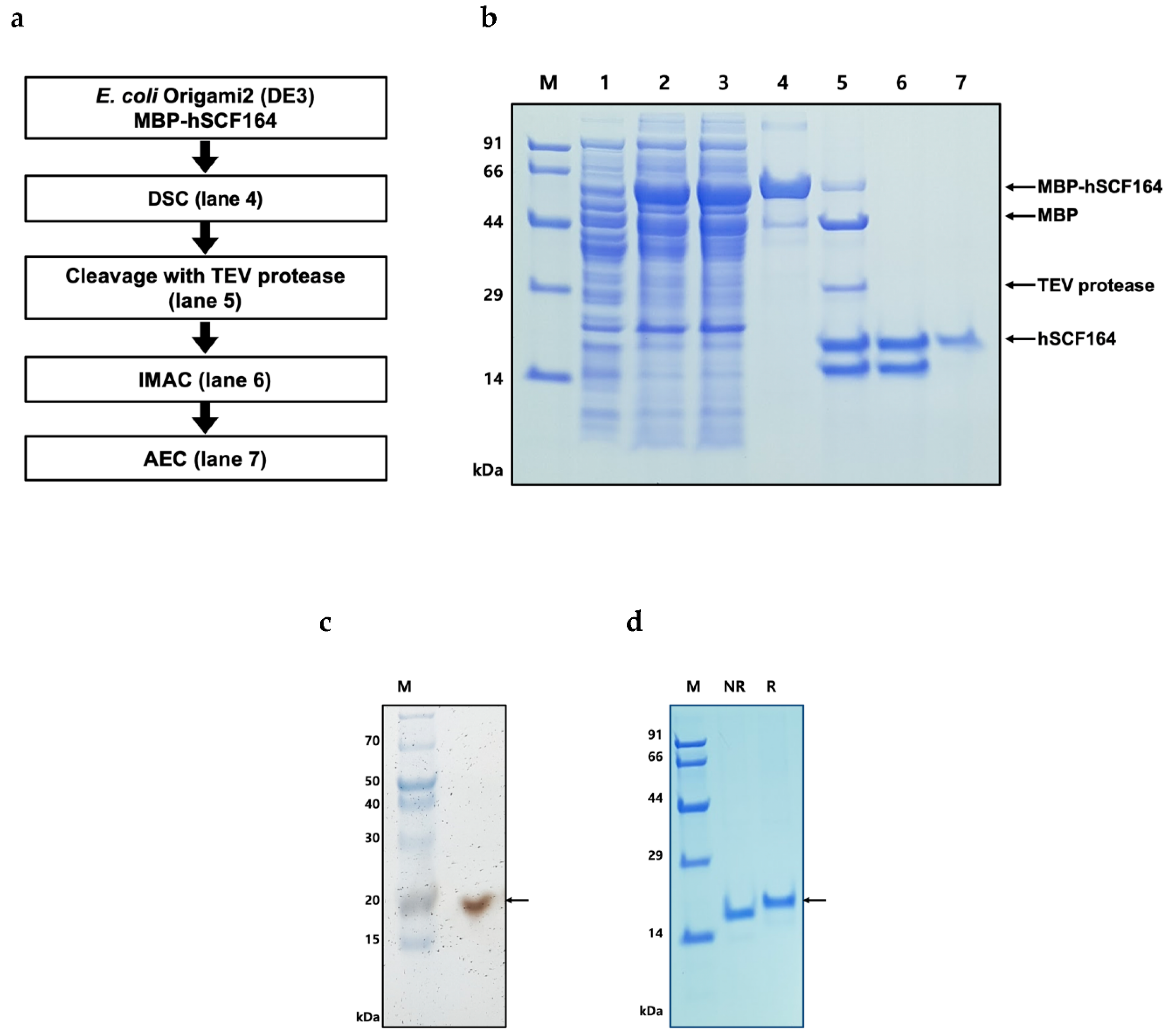
2.3. Purification of hSCF164
2.4. Purification of Full-Length His-hSCF248
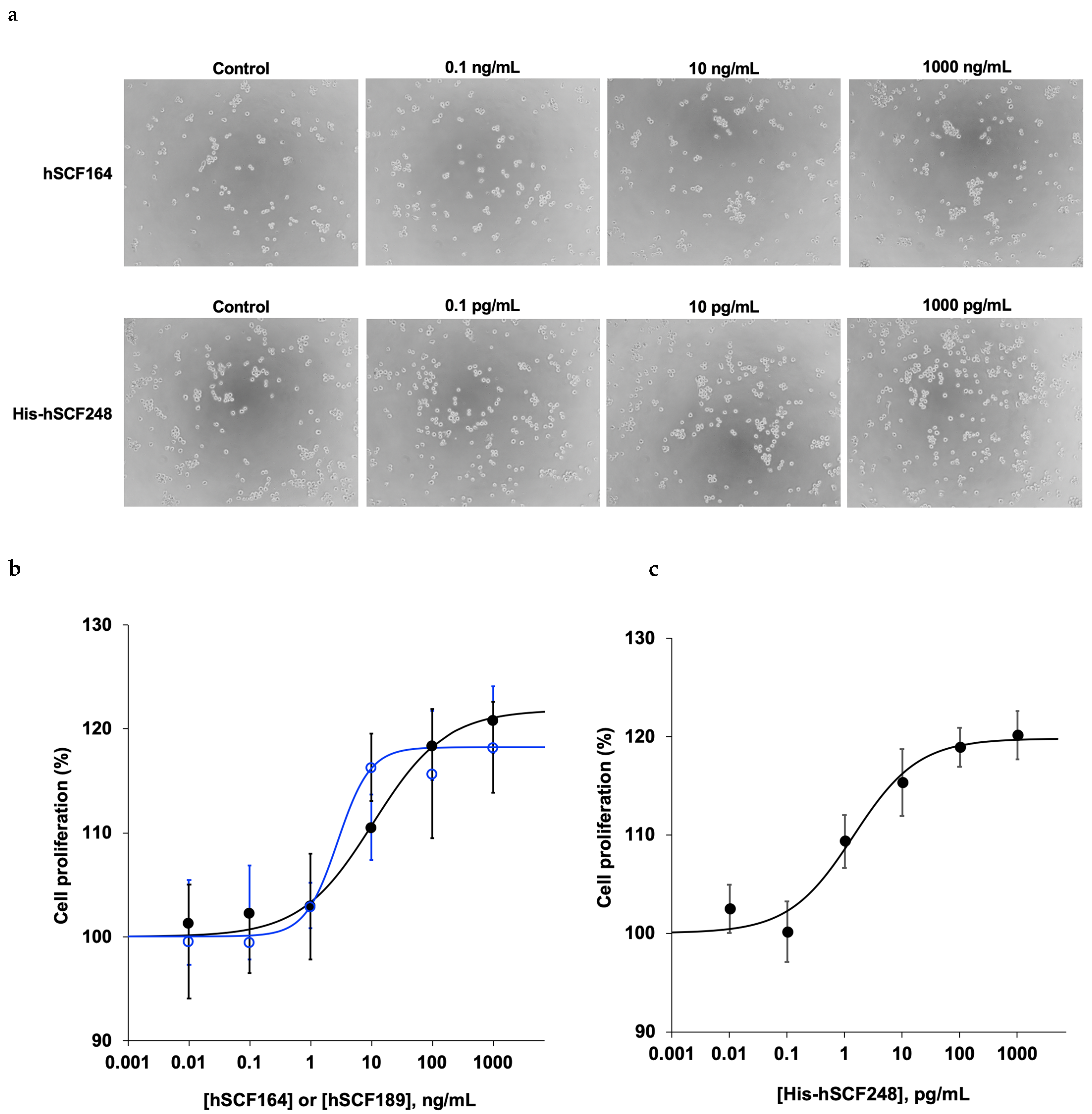
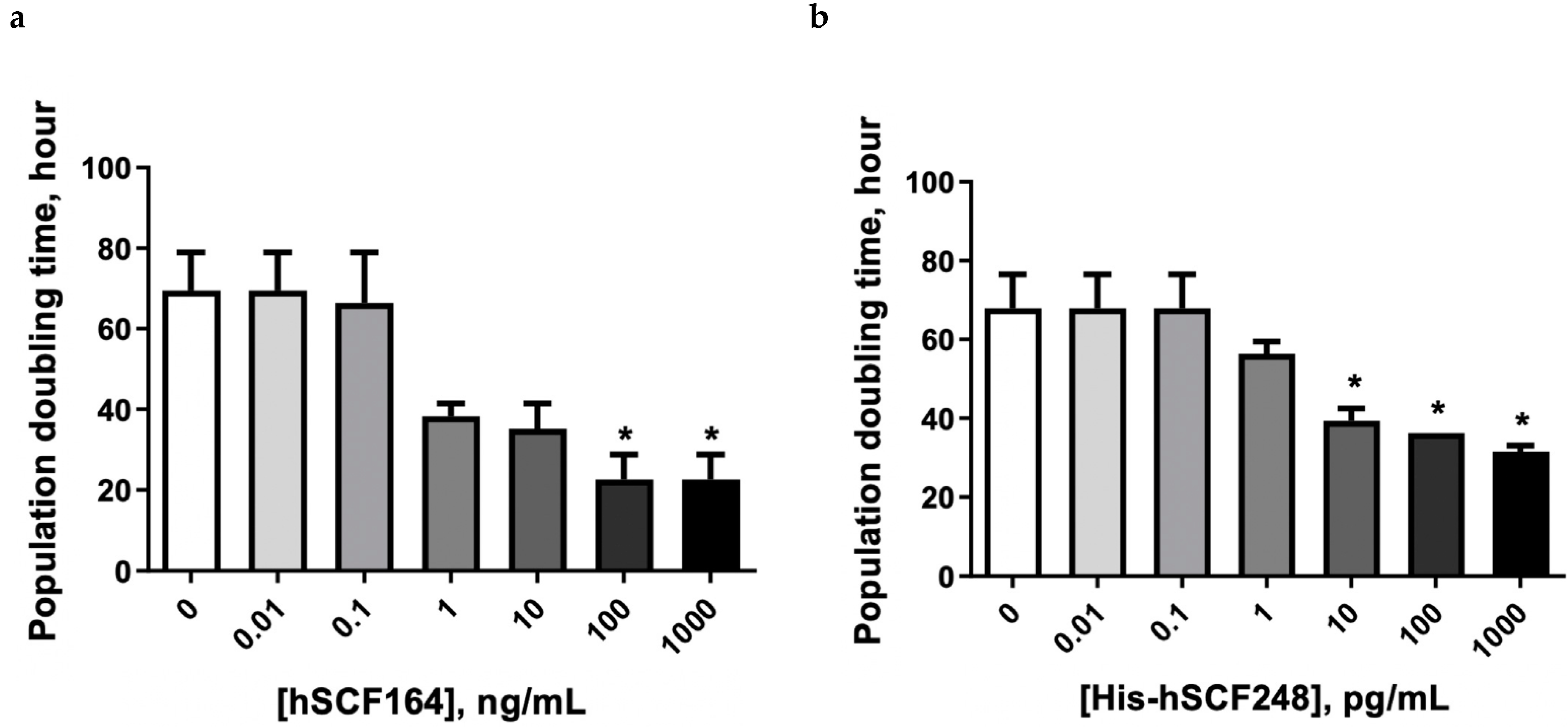
2.5. Effect of hSCF164 and His-hSCF248 on Cellular Proliferation
2.6. Effect of hSCF164 and His-hSCF248 on Cell Cycle
2.7. hSCF Regulates Gene Expression Involved in Cell Growth
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Construction of Plasmids
4.3. Expression and Solubility of Fusion Proteins
4.4. Purification and Tag Removal of Recombinant hSCF164 from E. coli
4.5. Purification of Recombinant His-hSCF248 from E. coli
4.6. Analysis in Electrophoresis and Silver Staining
4.7. Endotoxin Assay
4.8. In Vitro Activity Assay
4.9. RNA Preparation and Real-Time Quantitative PCR
4.10. Cell Cycle Assay
4.11. Protein Stability Assay
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ashman, L.K. The biology of stem cell factor and its receptor C-kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051. [Google Scholar] [CrossRef]
- Unni, S.K.; Modi, D.N.; Pathak, S.G.; Dhabalia, J.V.; Bhartiya, D. Stage-specific localization and expression of c-kit in the adult human testis. J. Histochem. Cytochem. 2009, 57, 861–869. [Google Scholar] [CrossRef]
- Erlandsson, A.; Larsson, J.; Forsberg-Nilsson, K. Stem cell factor is a chemoattractant and a survival factor for CNS stem cells. Exp. Cell Res. 2004, 301, 201–210. [Google Scholar] [CrossRef]
- Kapur, R.; Chandra, S.; Cooper, R.; McCarthy, J.; Williams, D.A. Role of p38 and ERK MAP kinase in proliferation of erythroid progenitors in response to stimulation by soluble and membrane isoforms of stem cell factor. Blood 2002, 100, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Verma, S.; Hsieh, I.C.; Hung, A.; Cheng, T.T.; Wang, S.Y.; Liu, Y.C.; Stanford, W.L.; Weisel, R.D.; Li, R.K.; et al. Stem cell factor attenuates vascular smooth muscle apoptosis and increases intimal hyperplasia after vascular injury. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Bashamboo, A.; Taylor, A.H.; Samuel, K.; Panthier, J.J.; Whetton, A.D.; Forrester, L.M. The survival of differentiating embryonic stem cells is dependent on the SCF-KIT pathway. J. Cell Sci. 2006, 119 Pt 15, 3039–3046. [Google Scholar] [CrossRef]
- Williams, L.A.; Davis-Dusenbery, B.N.; Eggan, K.C. SnapShot: Directed differentiation of pluripotent stem cells. Cell 2012, 149, 1174. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S. Humanized mice: A brief overview on their diverse applications in biomedical research. J. Cell Physiol. 2018, 233, 2889–2901. [Google Scholar] [CrossRef] [PubMed]
- Kanellakis, P.; Slater, N.J.; Du, X.J.; Bobik, A.; Curtis, D.J. Granulocyte colony-stimulating factor and stem cell factor improve endogenous repair after myocardial infarction. Cardiovasc. Res. 2006, 70, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.S.; Clogston, C.L.; Wypych, J.; Fausset, P.R.; Lauren, S.; Mendiaz, E.A.; Zsebo, K.M.; Langley, K.E. Amino acid sequence and post-translational modification of stem cell factor isolated from buffalo rat liver cell-conditioned medium. J. Biol. Chem. 1991, 266, 8102–8107. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, R.; Joachimiak, A.; Schlessinger, J.; Kong, X.P. Crystal structure of human stem cell factor: Implication for stem cell factor receptor dimerization and activation. Proc. Natl. Acad. Sci. USA 2000, 97, 7732–7737. [Google Scholar] [CrossRef]
- Lennartsson, J.; Ronnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.H.; Suggs, S.V.; Langley, K.E.; Lu, H.S.; Ting, J.; Okino, K.H.; Morris, C.F.; McNiece, I.K.; Jacobsen, F.W.; Mendiaz, E.A.; et al. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell 1990, 63, 203–211. [Google Scholar] [CrossRef]
- Majumdar, M.K.; Feng, L.; Medlock, E.; Toksoz, D.; Williams, D.A. Identification and mutation of primary and secondary proteolytic cleavage sites in murine stem cell factor cDNA yields biologically active, cell-associated protein. J. Biol. Chem. 1994, 269, 1237–1242. [Google Scholar] [CrossRef]
- Brannan, C.I.; Lyman, S.D.; Williams, D.E.; Eisenman, J.; Anderson, D.M.; Cosman, D.; Bedell, M.A.; Jenkins, N.A.; Copeland, N.G. Steel-Dickie mutation encodes a c-kit ligand lacking transmembrane and cytoplasmic domains. Proc. Natl. Acad. Sci. USA 1991, 88, 4671–4674. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.R.; Wu, G.M.; Mendiaz, E.A.; Syed, R.; Wypych, J.; Toso, R.; Mann, M.B.; Boone, T.C.; Narhi, L.O.; Lu, H.S.; et al. The majority of stem cell factor exists as monomer under physiological conditions. Implications for dimerization mediating biological activity. J. Biol. Chem. 1997, 272, 6406–6415. [Google Scholar] [CrossRef] [PubMed]
- Langley, K.E.; Mendiaz, E.A.; Liu, N.; Narhi, L.O.; Zeni, L.; Parseghian, C.M.; Clogston, C.L.; Leslie, I.; Pope, J.A.; Lu, H.S.; et al. Properties of variant forms of human stem cell factor recombinantly expressed in Escherichia coli. Arch. BioChem. Biophys. 1994, 311, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Pinchasi, D.; Zhou, M.; Lax, I.; Schlessinger, J. Kit receptor dimerization is driven by bivalent binding of stem cell factor. J. Biol. Chem. 1997, 272, 6311–6317. [Google Scholar] [CrossRef]
- Arakawa, T.; Yphantis, D.A.; Lary, J.W.; Narhi, L.O.; Lu, H.S.; Prestrelski, S.J.; Clogston, C.L.; Zsebo, K.M.; Mendiaz, E.A.; Wypych, J.; et al. Glycosylated and unglycosylated recombinant-derived human stem cell factors are dimeric and have extensive regular secondary structure. J. Biol. Chem. 1991, 266, 18942–18948. [Google Scholar] [CrossRef]
- Flanagan, J.G.; Leder, P. The kit ligand: A cell surface molecule altered in steel mutant fibroblasts. Cell 1990, 63, 185–194. [Google Scholar] [CrossRef]
- Zsebo, K.M.; Wypych, J.; McNiece, I.K.; Lu, H.S.; Smith, K.A.; Karkare, S.B.; Sachdev, R.K.; Yuschenkoff, V.N.; Birkett, N.C.; Williams, L.R.; et al. Identification, purification, and biological characterization of hematopoietic stem cell factor from buffalo rat liver--conditioned medium. Cell 1990, 63, 195–201. [Google Scholar] [CrossRef]
- Han, J.; Yan, X.; Zhu, J.; Zhi, X.; Zang, Y.; Shen, B.; Qin, J. Expression of a novel recombinant dual human stem cell factor in insect cells. Protein Expr. Purif. 2003, 31, 311–317. [Google Scholar] [CrossRef]
- Han, J.; Zang, Y.; Lu, H.; Zhu, J.; Qin, J. A novel recombinant dual human SCF expressed in and purified from silkworm, Bombyx mori, possesses higher bioactivity than recombinant monomeric human SCF. Eur. J. Haematol. 2004, 72, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zang, Y.; Zhu, J.; Lu, H.; Han, J.; Qin, J. Expression of a novel recombinant stem cell factor/macrophage colony-stimulating factor fusion protein in baculovirus-infected insect cells. Protein Expr. Purif. 2005, 41, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.D.; Narhi, L.O.; Chang, W.C.; Lu, H.S. Refolding and oxidation of recombinant human stem cell factor produced in Escherichia coli. J. Biol. Chem. 1996, 271, 11301–11308. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zang, Y.; Ze, Y.; Zhu, J.; Chen, T.; Han, J.; Qin, J. Expression, refolding, and characterization of a novel recombinant dual human stem cell factor. Protein Expr. Purif. 2005, 43, 126–132. [Google Scholar] [CrossRef]
- Lili, W.; Chaozhan, W.; Xindu, G. Expression, renaturation and simultaneous purification of recombinant human stem cell factor in Escherichia coli. Biotechnol. Lett. 2006, 28, 993–997. [Google Scholar] [CrossRef]
- Wang, C.; Liu, J.; Wang, L.; Geng, X. Solubilization and refolding with simultaneous purification of recombinant human stem cell factor. Appl. BioChem. Biotechnol. 2008, 144, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, C.; Geng, X. Fast preparation of recombinant human stem cell factor from inclusion bodies using different hydrophobic interaction chromatographic columns. Se Pu 2011, 29, 36–41. [Google Scholar] [CrossRef]
- Chen, L.H.; Cai, F.; Zhang, D.J.; Zhang, L.; Zhu, P.; Gao, S. Large-scale purification and characterization of recombinant human stem cell factor in Escherichia coli. Biotechnol. Appl. BioChem. 2017, 64, 509–518. [Google Scholar] [CrossRef]
- Bals, C.; Schambach, A.; Meyer, J.; Scheper, T.; Rinas, U. Expression and purification of bioactive soluble murine stem cell factor from recombinant Escherichia coli using thioredoxin as fusion partner. J. Biotechnol. 2011, 152, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Akuta, T.; Kikuchi-Ueda, T.; Imaizumi, K.; Oshikane, H.; Nakaki, T.; Okada, Y.; Sultana, S.; Kobayashi, K.; Kiyokawa, N.; Ono, Y. Expression of bioactive soluble human stem cell factor (SCF) from Escherichia coli by coproduction of thioredoxin and efficient purification using arginine in affinity chromatography. Protein Expr. Purif. 2015, 105, 1–7. [Google Scholar] [CrossRef]
- Ueda, T.; Akuta, T.; Kikuchi-Ueda, T.; Imaizumi, K.; Ono, Y. Improving the soluble expression and purification of recombinant human stem cell factor (SCF) in endotoxin-free Escherichia coli by disulfide shuffling with persulfide. Protein Expr. Purif. 2016, 120, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, H.; Tao, X.; Luo, Y.; Wang, H.; He, J.; Fang, Q.; Guo, S.; Song, C. SCF promotes the production of IL-13 via the MEK-ERK-CREB signaling pathway in mast cells. Exp. Ther. Med. 2019, 18, 2491–2496. [Google Scholar] [CrossRef]
- Abbaspour Babaei, M.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Devel. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lim, J.; Woo, K.C.; Kim, K.T. Piperonylic acid stimulates keratinocyte growth and survival by activating epidermal growth factor receptor (EGFR). Sci. Rep. 2018, 8, 162. [Google Scholar] [CrossRef]
- Jeong, H.; Kim, H.J.; Lee, S.J. Complete Genome Sequence of Escherichia coli Strain BL21. Genome Announc. 2015, 3, e00134. [Google Scholar] [CrossRef] [PubMed]
- Hadj Sassi, A.; Trigui-Lahiani, H.; Abdeljalil, S.; Gargouri, A. Enhancement of solubility, purification and inclusion-bodies-refolding of an active pectin lyase from Penicillium occitanis expressed in Escherichia coli. Int. J. Biol. Macromol. 2017, 95, 256–262. [Google Scholar] [CrossRef]
- Wang, L.L.; Geng, X.D.; Han, H. Cloning, expression, renaturation and purification of soluble hSCF. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2004, 20, 402–405. [Google Scholar] [PubMed]
- Nguyen, M.T.; Prima, M.J.; Song, J.A.; Kim, J.; Do, B.H.; Yoo, J.; Park, S.; Jang, J.; Lee, S.; Lee, E.; et al. Prokaryotic soluble overexpression and purification of oncostatin M using a fusion approach and genetically engineered E. coli strains. Sci. Rep. 2019, 9, 13706. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Heo, Y.; Do, B.H.; Baek, S.; Kim, C.J.; Jang, Y.J.; Lee, W.; Choe, H. Bacterial overexpression and purification of soluble recombinant human serum albumin using maltose-binding protein and protein disulphide isomerase. Protein Expr. Purif. 2020, 167, 105530. [Google Scholar] [CrossRef] [PubMed]
- Shirano, Y.; Shibata, D. Low temperature cultivation of Escherichia coli carrying a rice lipoxygenase L-2 cDNA produces a soluble and active enzyme at a high level. FEBS Lett. 1990, 271, 128–130. [Google Scholar] [CrossRef]
- Ferrer, M.; Chernikova, T.N.; Timmis, K.N.; Golyshin, P.N. Expression of a temperature-sensitive esterase in a novel chaperone-based Escherichia coli strain. Appl. Environ. MicroBiol. 2004, 70, 4499–4504. [Google Scholar] [CrossRef]
- Vera, A.; Gonzalez-Montalban, N.; Aris, A.; Villaverde, A. The conformational quality of insoluble recombinant proteins is enhanced at low growth temperatures. Biotechnol. Bioeng. 2007, 96, 1101–1106. [Google Scholar] [CrossRef]
- Yi, A.R.; Lee, S.R.; Jang, M.U.; Park, J.M.; Eom, H.J.; Han, N.S.; Kim, T.J. Cloning of dextransucrase gene from Leuconostoc citreum HJ-P4 and its high-level expression in E. coli by low temperature induction. J. MicroBiol. Biotechnol. 2009, 19, 829–835. [Google Scholar]
- Kim, E.K.; Moon, J.C.; Lee, J.M.; Jeong, M.S.; Oh, C.; Ahn, S.M.; Yoo, Y.J.; Jang, H.H. Large-scale production of soluble recombinant amyloid-beta peptide 1-42 using cold-inducible expression system. Protein Expr. Purif. 2012, 86, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Song, J.A.; Koo, B.K.; Chong, S.H.; Kwak, J.; Ryu, H.B.; Nguyen, M.T.; Vu, T.T.; Jeong, B.; Kim, S.W.; Choe, H. Expression and purification of biologically active human FGF2 containing the b'a' domains of human PDI in Escherichia coli. Appl. BioChem. Biotechnol. 2013, 170, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Koo, B.K.; Thi Vu, T.T.; Song, J.A.; Chong, S.H.; Jeong, B.; Ryu, H.B.; Moh, S.H.; Choe, H. Prokaryotic soluble overexpression and purification of bioactive human growth hormone by fusion to thioredoxin, maltose binding protein, and protein disulfide isomerase. PLoS ONE 2014, 9, e89038. [Google Scholar] [CrossRef] [PubMed]
- Nallamsetty, S.; Kapust, R.B.; Tozser, J.; Cherry, S.; Tropea, J.E.; Copeland, T.D.; Waugh, D.S. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr. Purif. 2004, 38, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.K.; Simmons, C.R.; Wiener, M.C. Inhibition of tobacco etch virus protease activity by detergents. Protein Expr. Purif. 2003, 27, 109–114. [Google Scholar] [CrossRef]
- Butt, T.R.; Edavettal, S.C.; Hall, J.P.; Mattern, M.R. SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 2005, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.S. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 2011, 80, 283–293. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expression Level (%) | Solubility (%) | ||||
|---|---|---|---|---|---|
| Tag | 18 °C | 30 °C | 18 °C | 30 °C | |
| His6 | 16 ± 0.23 | 38.2 ± 0.5 | 59.4 ± 8 | 12.1 ± 5.2 | |
| hSCF164 | PDIb’a’ | 34.2 ± 1.1 | 22.1 ± 3.8 | 95.6 ± 2.9 | 4.9 ± 1.6 |
| MBP | 35 ± 3 | 48.2 ± 5.2 | 96.8 ± 2.3 | 84.7 ± 11.2 | |
| Expression Level (%) | Solubility (%) | ||||
|---|---|---|---|---|---|
| Strain | 18 °C | 30 °C | 18 °C | 30 °C | |
| His-hSCF248 | Origami 2(DE3) | 29 ± 10.9 | 36.7 ± 5.9 | 35.1 ± 14.4 | 6.2 ± 3.2 |
| BL21(DE3) | 56.3 ± 5.2 | 62.5 ± 4.1 | 61.2 ± 17.9 | 9.7 ± 3.5 | |
| Purification Step | Total Protein (mg) | Purity (%) | hSCF164 (mg) | Yield (%) |
| Supernatant | 61.44 | 35.5 | 21.8 | 100 |
| Dextrin | 9.90 | 86.3 | 8.54 | 39.2 |
| IMAC | 3.66 | 54.6 | 1.99 | 23.3 |
| Anion exchange | 0.24 | 99.6 | 0.24 | 12.0 |
| Purification Step | Total Protein (mg) | Purity (%) | His-hSCF248 (mg) | Yield (%) |
| Supernatant | 60.8 | 27.4 | 16.65 | 100 |
| Anion exchange | 21.9 | 48.5 | 10.02 | 60.2 |
| Gel filtration | 2.5 | 96.3 | 2.40 | 23.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.; de Paula, M.N.; Baek, S.; Ta, H.K.K.; Nguyen, M.T.; Jeong, T.-H.; Kim, C.J.; Jang, Y.J.; Choe, H. Novel Bacterial Production of Two Different Bioactive Forms of Human Stem-Cell Factor. Int. J. Mol. Sci. 2021, 22, 6361. https://doi.org/10.3390/ijms22126361
Lee E, de Paula MN, Baek S, Ta HKK, Nguyen MT, Jeong T-H, Kim CJ, Jang YJ, Choe H. Novel Bacterial Production of Two Different Bioactive Forms of Human Stem-Cell Factor. International Journal of Molecular Sciences. 2021; 22(12):6361. https://doi.org/10.3390/ijms22126361
Chicago/Turabian StyleLee, Eunyoung, Michelle Novais de Paula, Sangki Baek, Huynh Kim Khanh Ta, Minh Tan Nguyen, Taeck-Hyun Jeong, Chong Jai Kim, Yeon Jin Jang, and Han Choe. 2021. "Novel Bacterial Production of Two Different Bioactive Forms of Human Stem-Cell Factor" International Journal of Molecular Sciences 22, no. 12: 6361. https://doi.org/10.3390/ijms22126361
APA StyleLee, E., de Paula, M. N., Baek, S., Ta, H. K. K., Nguyen, M. T., Jeong, T.-H., Kim, C. J., Jang, Y. J., & Choe, H. (2021). Novel Bacterial Production of Two Different Bioactive Forms of Human Stem-Cell Factor. International Journal of Molecular Sciences, 22(12), 6361. https://doi.org/10.3390/ijms22126361