
Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression


Abstract
1. Introduction
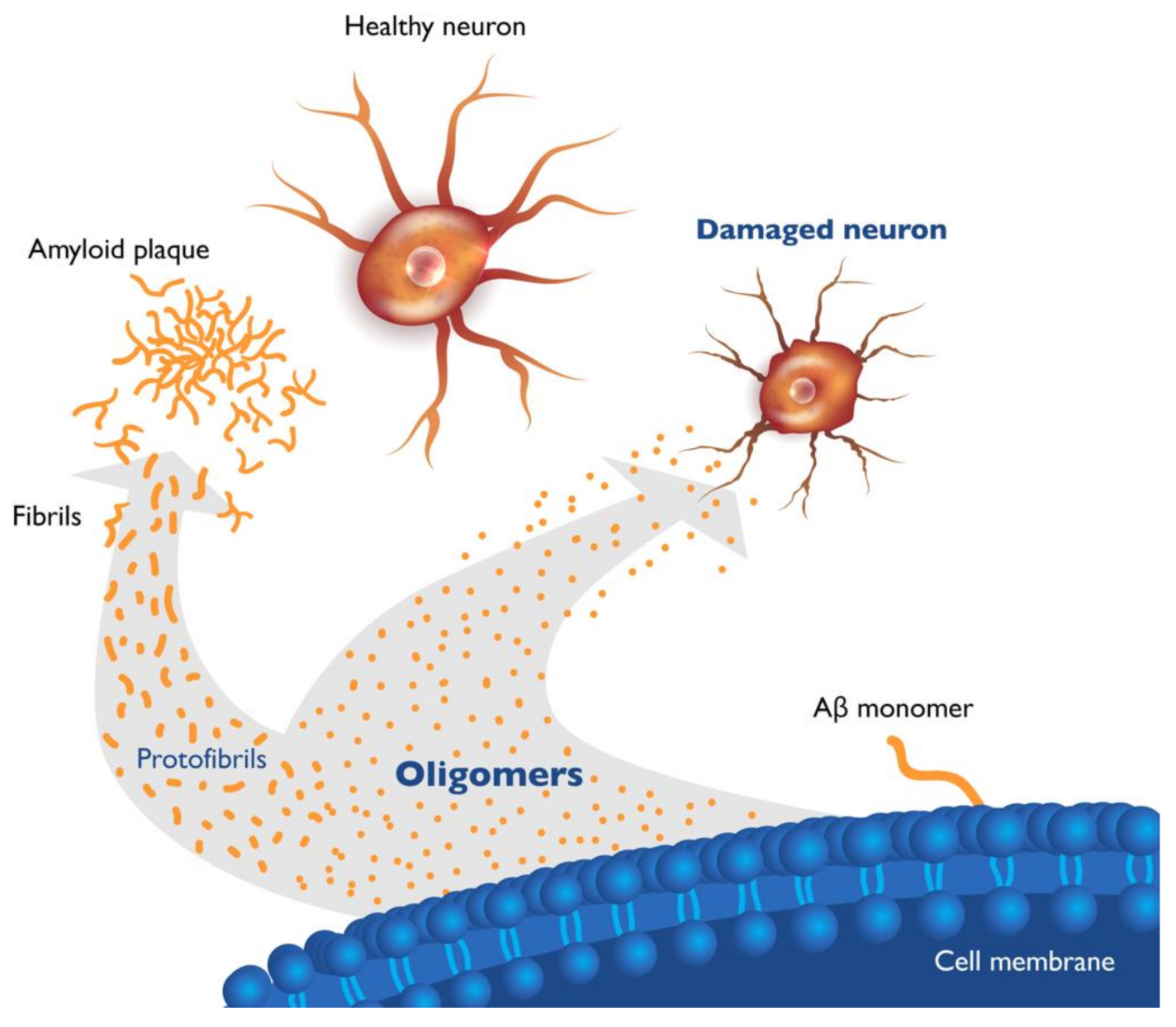
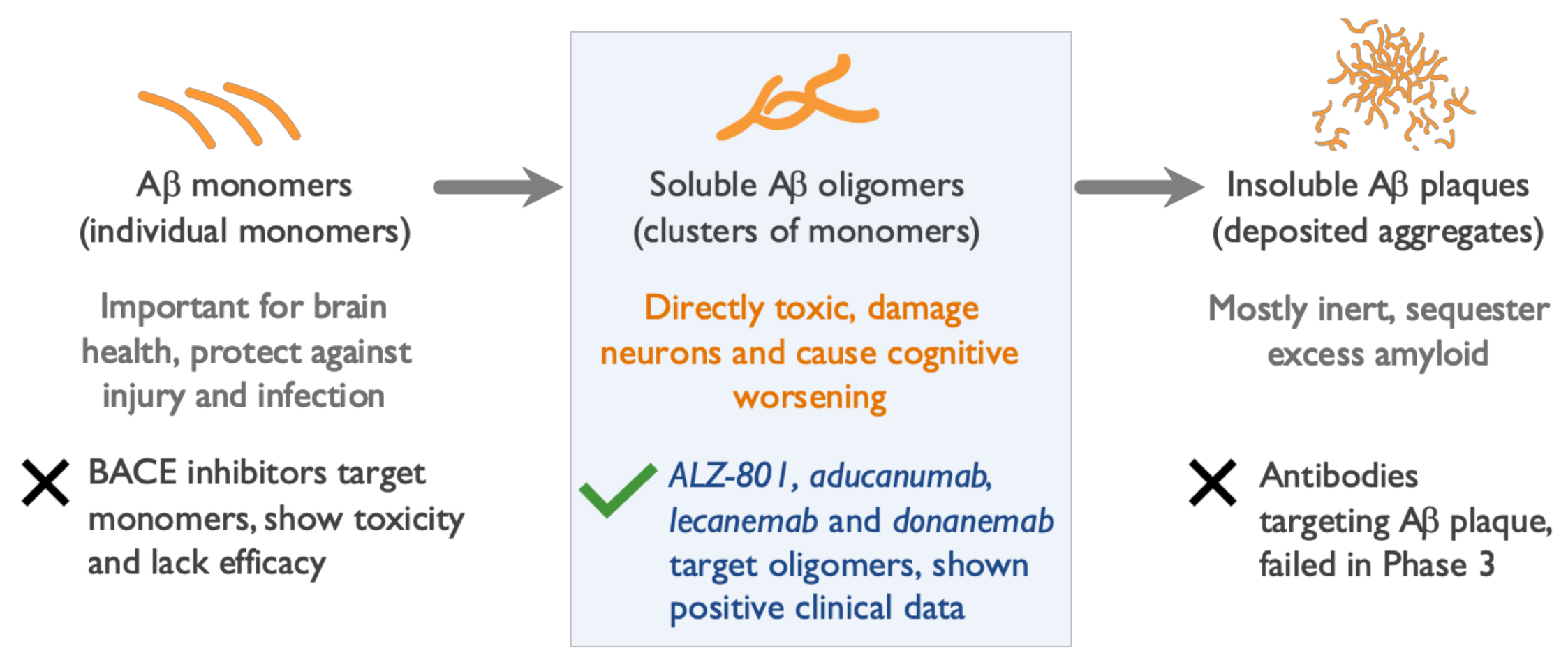
2. Soluble Beta Amyloid (Aβ) Oligomers Play an Early Role in Alzheimer ’s Disease (AD) Pathogenesis and Drive Disease Progression
3. Genetic Evidence Points to a Central Role of Aβ Oligomers in AD Pathogenesis
4. The ε4 Allele of Apolipoprotein E (APOE4) Genotype Increases Brain Oligomer Concentration and Accelerates Course of AD
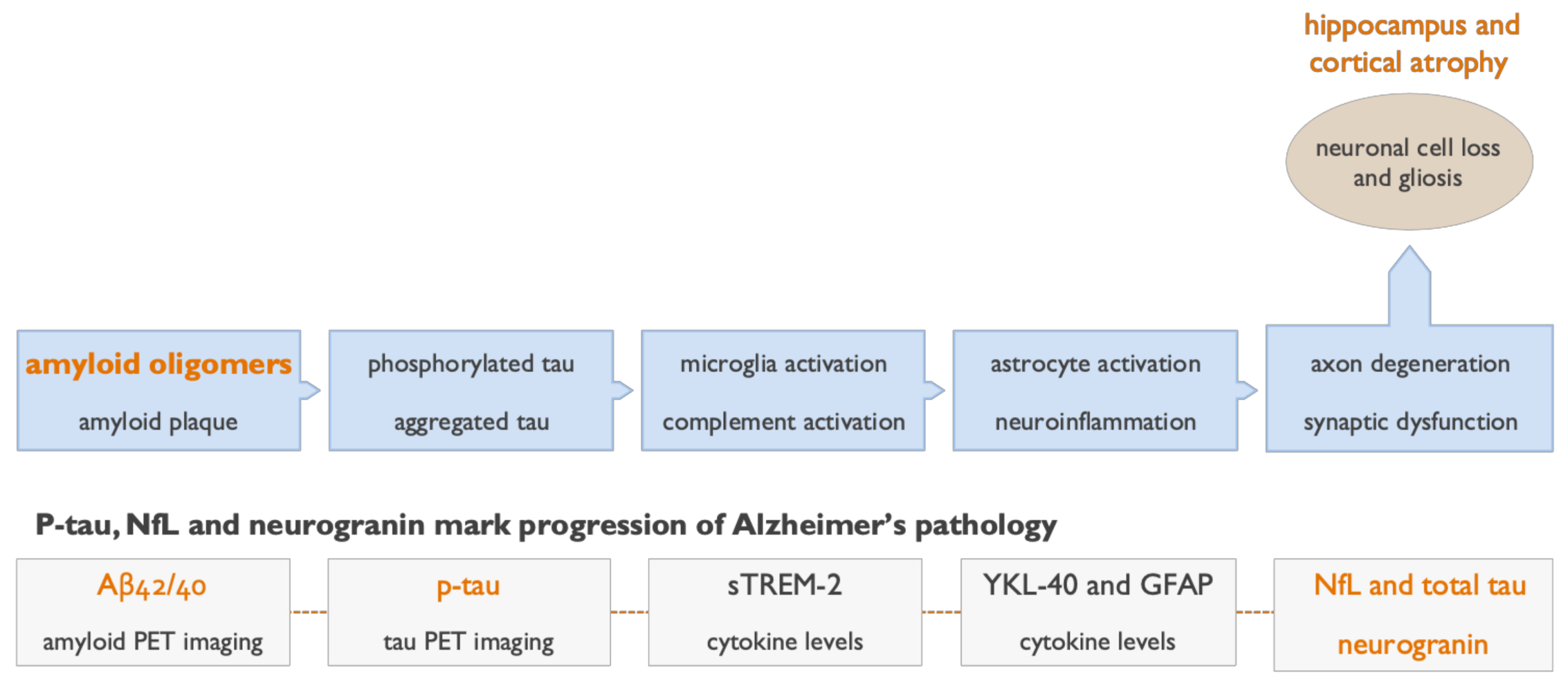
5. Clinical and Biomarker Data Show Aβ Oligomers to Be Effective Therapeutic Targets in AD
6. Amyloid Plaque Clearance Does Not Correlate with Clinical Efficacy of Anti-Amyloid Agents
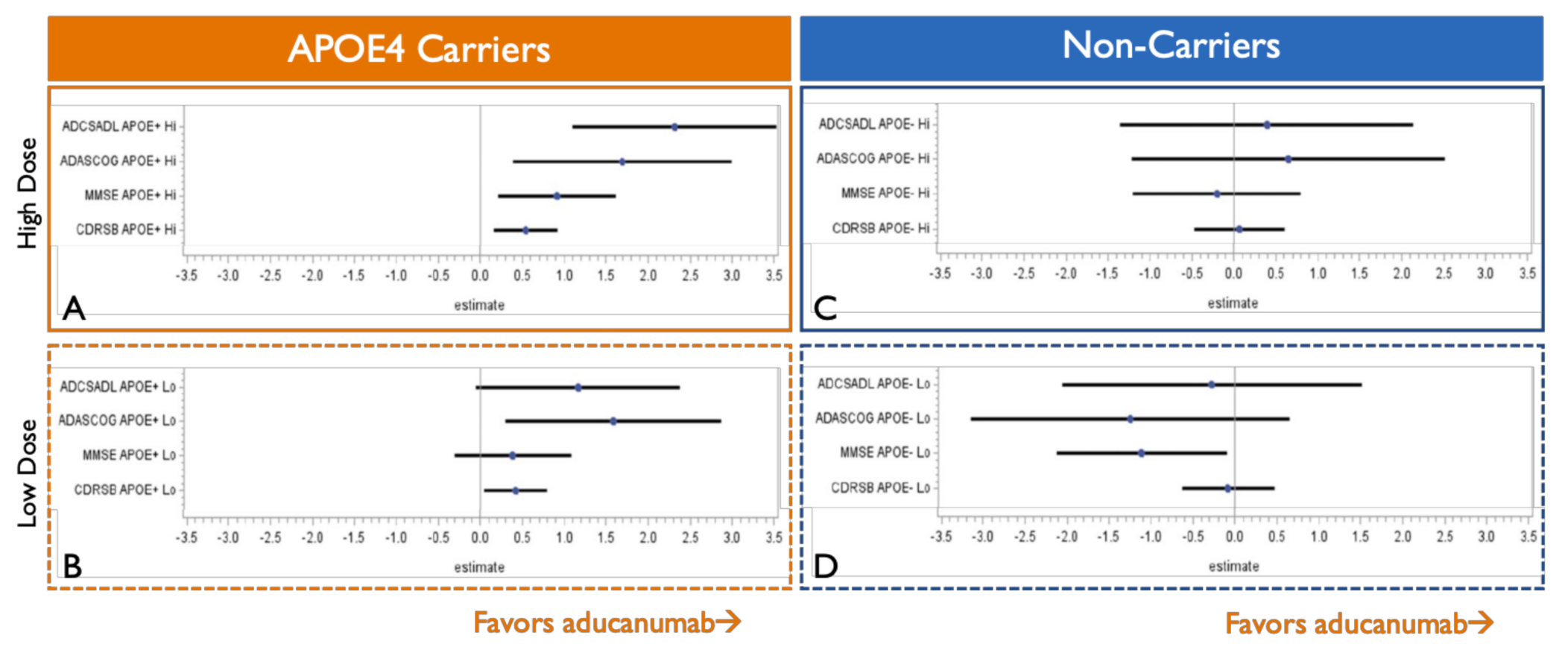
7. APOE4 Carriers Show Stronger and Less Variable Clinical Efficacy than Non-Carriers
8. Oral ALZ-801 Selectively and Fully Inhibits Formation of Aβ Oligomers in Human Brain
9. Conclusions
Funding
Conflicts of Interest
References
- Dickson, D.W. Neuropathological diagnosis of Alzheimer’s disease: A perspective from longitudinal clinicopathological studies. Neurobiol. Aging 1997, 18 (Suppl.4), S21–S26. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carillo, M.C.; Dunn, B.; Budd Haeberlein, S.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. A Is for Amyloid. J. Prev. Alzheimers Dis. 2020, 7, 140–141. [Google Scholar] [CrossRef]
- Ashe, K.H. The biogenesis and biology of amyloid β oligomers in the brain. Alzheimer’s Dement. 2020, 16, 1561–1567. [Google Scholar] [CrossRef]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimers Dis. 2018, 64 (Suppl. 1), S567–S610. [Google Scholar] [CrossRef]
- Anand, K.; Sabbagh, M. Amyloid Imaging: Poised for Integration into Medical Practice. Neurotherapeutics 2017, 14, 54–61. [Google Scholar] [CrossRef]
- Johnson, K.A.; Minoshima, S.; Bohnen, N.I.; Donohoe, K.J.; Foster, N.L.; Hersovitch, P.; Karlawish, J.H.; Rowe, C.C.; Carrillo, M.C.; Hartley, D.M.; et al. Appropriate use criteria for amyloid PET: A report of the Amyloid Imaging Task Force (AIT), the Society of Nuclear Medicine and Molecular Imaging (SNMMI) and the Alzheimer Association (AA). Alzheimers Dement. 2013, 9, E1–E16. [Google Scholar] [CrossRef]
- Lois, C.; Gonzalez, I.; Johnson, K.A.; Price, J.C. PET imaging of tau protein targets: A methodology perspective. Brain Imaging Behav. 2019, 13, 333–344. [Google Scholar] [CrossRef]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: A longitudinal study. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzo, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Sutphena, C.L.; McCue, L.; Herriese, E.M.; Xiong, B.; Ladensone, J.H.; Holtzman, D.M.; Fagan, A.M.; ADNI. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer’s disease. Alzheimers Dement. 2018, 14, 869–879. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Fluid biomarker-based molecular phenotyping of Alzheimer’s disease patients in research and clinical settings. Prog. Mol. Biol. Transl. Sci. 2019, 168, 3–23. [Google Scholar] [CrossRef]
- Leuzy, A.; Ashton, N.J.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordverg, A.; Ossenkoppele, R.; Zetterberg, H.; et al. 2020 update on the clinical validity of cerebrospinal fluid amyloid, tau, and phospho-tau as biomarkers for Alzheimer’s disease in the context of a structured 5-phase development framework. Nucl. Med. Mol. Imaging 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; von Hehn, C.; Tian, Y.; Chalkias, S.; Muralidharan, K.K.; Chen, T.; Wu, S.; Skordos, L.; Nisenbaum, L.; Rajagovindan, R.; et al. EMERGE and ENGAGE Topline Results: Two Phase 3 Studies to Evaluate Aducanumab in Patients with Early Alzheimer’s Disease; CTAD: San Diego, CA, USA, 2019. [Google Scholar]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Koyama, A.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alz. Res. Therapy 2021, 13, 8. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Evans, C.D.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.D.; Sims, J.R.; Brys, M.; et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 10, 90. [Google Scholar] [CrossRef]
- Abushakra, S.; Porsteinsson, A.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Hendrix, S.; Wang, P.; Shen, L.; et al. Clinical benefits of tramiprosate in Alzheimer’s disease are associated with higher number of APOE4 alleles: The “APOE4 gene-dose effect”. J. Prev. Alz. Dis. 2016, 3, 219–228. [Google Scholar] [CrossRef]
- Abushakra, S.; Porsteinsson, A.; Scheltens, P.; Sadowsky, C.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Wang, P.; et al. Clinical effects of tramiprosate in APOE4/4 homozygous patients with mild Alzheimer’s disease suggest disease modification potential. J. Prev. Alz. Dis. 2017, 4, 149–156. [Google Scholar] [CrossRef]
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical pharmacokinetics and safety of ALZ-801, a novel prodrug of tramiprosate in development for the treatment of Alzheimer’s disease. Clin. Pharmacokinet. 2018, 57, 315–333. [Google Scholar] [CrossRef]
- Lannfelt, L.; Söderberg, L.; Laudon, H.; Sahlin, C.; Johannesson, M.; Nygren, P.; Möller, C. BAN2401 shows stronger binding to soluble aggregated amyloid-beta species than aducanumab. BioArctic poster presentation. In Proceedings of the Alzheimer’s Association International Conference, Los Angeles, CA, USA, 14–18 July 2019. [Google Scholar]
- Linse, S.; Scheidt, T.; Bernfur, K.; Vendruscolo, M.; Dobson, C.M.; Cohen, S.I.A.; Sileikis, E.; Lundqvist, M.; Qian, F.; O’Malley, T.; et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat. Struct. Mol. Biol. 2020, 27, 1125–1133. [Google Scholar] [CrossRef]
- Uhlmann, R.E.; Rother, C.; Rasmussen, J.; Schelle, J.; Bergmann, C.; Ullrich Gavilanes, E.M.; Fritschi, S.K.; Buehler, A.; Baumann, F.; Skodras, A.; et al. Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nat. Neurosci. 2020, 23, 1580–1588. [Google Scholar] [CrossRef]
- Kocis, P.; Tolar, M.; Yu, J.; Sinko, W.; Ray, S.; Blennow, K.; Fillit, H.; Hey, J.A. Elucidating the Aβ42 anti-aggregation mechanism of action of tramiprosate in Alzheimer’s disease: Integrating molecular analytical methods, pharmacokinetic and clinical data. CNS Drugs 2017, 31, 495–509. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.U. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Iljina, M.; Garcia, G.A.; Dear, A.J.; Flink, J.; Narayan, P.; Michaels, T.C.; Dobson, C.M.; Frenkel, D.; Knowles, T.P.J.; Klenerman, D. Quantitative analysis of co-oligomer formation by amyloid-beta peptide isoforms. Sci. Rep. 2016, 6, 28658. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Gaspar, R.C.; Villarreal, S.A.; Bowles, N.; Hepler, R.W.; Joyce, J.G.; Shugrue, P. Oligomers of β-amyloid are sequestered into and seed new plaque s in the brains of an AD mouse model. Exp. Neurol. 2010, 223, 394–400. [Google Scholar] [CrossRef]
- Janelidze, S.; Zetterberg, H.; Mattsson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; van Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: Better diagnostic markers of Alzheimer disease. Ann. Clin. Transl. Neurol. 2016, 3, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.R.; Liu, R.T. The toxicity and polymorphism of β-amyloid oligomers. Int. J. Mol. Sci. 2020, 21, 4477. [Google Scholar] [CrossRef]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion channel formation by amyloid-β42 oligomers but not amyloid-β40 in cellular membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef]
- Canevari, L.; Abramov, A.Y.; Duchen, M.R. Toxicity of amyloid beta peptide: Tales of calcium, mitochondria, and oxidative stress. Neurochem. Res. 2004, 29, 637–650. [Google Scholar] [CrossRef]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 2019, 365, 559–565. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular receptors of amyloid β oligomers (AβOs) in Alzheimer’s disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Park, S.A.; Han, S.M.; Kim, C.E. New fluid biomarkers tracking non-amyloid-β and non-tau pathology in Alzheimer’s disease. Exp. Mol. Med. 2020, 52, 556–568. [Google Scholar] [CrossRef]
- Bateman, R.J.; Aisen, P.S.; DeStrooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.A.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef]
- Tomiyama, T.; Shimada, H. APP Osaka mutation in familial Alzheimer’s disease—Its discovery, phenotypes, and mechanism of recessive inheritance. Int. J. Mol. Sci. 2020, 21, 1413. [Google Scholar] [CrossRef]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef]
- Schöll, M.; Wall, A.; Thordardottir, S.; Ferreira, D.; Bogdanovic, N.; Långström, B.; Almkvist, O.; Graff, C.; Nordberg, A. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology 2012, 79, 229–236. [Google Scholar] [CrossRef]
- Limegrover, C.S.; LeVine, H.; Izzo, N.J., 3rd; Yurko, R.; Mozzoni, K.; Rehak, C.; Sadlek, K.; Safferstein, H.; Catalano, S.A. Alzheimer’s protection effect of A673T mutation may be driven by lower Aβ oligomer binding affinity. J. Neurochem. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Strittmater, W.J.; Roses, A.D. Apolipoprotein E and Alzheimer’s disease. Annu. Rev. Neurosci. 1996, 19, 53–77. [Google Scholar] [CrossRef]
- Sando, S.B.; Melquist, S.; Cannon, A.; Hutton, M.L.; Sletvold, O.; Saltvedt, I.; Saltvedt, I.; White, L.R.; Lydersen, S.; Aasly, J.O. APOE ε4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol. 2008, 8, 9. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Koffie, R.M.; Hashimoto, T.; Tai, H.C.; Kay, K.R.; Serrano-Pozo, A.; Joyner, D.; Hou, S.; Kopeikina, K.J.; Frosch, M.P.; Lee, V.M.; et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain. 2012, 135 Pt 7, 2155–2168. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Tokuda, T.; Kasai, T.; Ishigami, N.; Hidaka, H.; Kondo, M.; Allsop, D.; Nakagawa, M. High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J. 2010, 24, 2716–2726. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012, 32, 15181–15192. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Bilousova, T.; Jungbauer, L.; Roeske, S.K.; Youmans, K.L.; Yu, C.; Poon, W.W.; Cornwell, L.B.; Miller, C.A.; Vinters, H.V.; et al. Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 2013, 288, 5914–5926. [Google Scholar] [CrossRef]
- Degenhardt, E.K.; Witte, M.M.; Case, M.G.; Yu, P.; Henley, D.B.; Hochstetlet, H.M.; D’Souza, D.N.; Trzepacz, P.T. Florbetapir F18 PET amyloid neuroimaging and characteristics in patients with mild and moderate Alzheimer dementia. Psychosomatics 2016, 57, 208–216. [Google Scholar] [CrossRef]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef]
- Klein, G.; Delmar, P.; Voyle, N.; Rehal, S.; Hofmann, C.; Abi-Saab, D.; Andjelkovic, M.; Ristic, S.; Wang, G.; Bateman, R.; et al. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: A PET substudy interim analysis. Alzheimers Res. Ther. 2019, 11, 101. [Google Scholar] [CrossRef]
- Blennow, K.; Shaw, L.M.; Stomrud, E.; Mattsson, N.; Toledo, J.B.; Buck, K.; Wahl, S.; Eichenlaub, U.; Lifke, V.; Simon, M.; et al. Predicting clinical decline and conversion to Alzheimer’s disease or dementia using novel elecsys Aβ(1-42), p-Tau and t-Tau CSF immunoassays. Sci. Rep. 2019, 9, 19024. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma p-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217, e20200861. [Google Scholar] [CrossRef]
- Gauthier, S.; Aisen, P.S.; Ferris, S.H.; Saumier, D.; Duong, A.; Haine, D.; Garceau, D.; Suhy, J.; Oh, J.; Lau, W.; et al. Effect of tramiprosate in patients with mild-to-moderate Alzheimer’s disease: Exploratory analyses of the MRI sub-group of the Alphase study. J. Nutr. Health Aging 2009, 13, 550–557. [Google Scholar] [CrossRef]
- Abushakra, S.; Porsteinsson, A.; Sabbagh, M.; Bracoud, L.; Schaerer, J.; Power, A.; Hey, J.A.; Scott, D.; Suhy, J.; Tolar, M. APOE ε4/ε4 homozygotes with early Alzheimer’s disease show accelerated hippocampal atrophy and cortical thinning that correlates with cognitive decline. Alzheimer’s Dement. 2020, 6, e12117. [Google Scholar] [CrossRef]
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and identification of an endogenous metabolite of tramiprosate and its prodrug ALZ-801 that inhibits beta amyloid oligomer formation in the human brain. CNS Drugs 2018, 32, 849–861. [Google Scholar] [CrossRef]
- Scheltens, P.; Hallikainen, M.; Grimmer, T.; Duning, T.; Gouw, A.A.; Teunissen, C.E.; Wink, A.M.; Maruff, P.; Harrison, J.; van Baal, C.M.; et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: Results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res. Ther. 2018, 10, 107. [Google Scholar] [CrossRef]
- Combined FDA and Biogen PCNS Drugs Advisory Committee Briefing Document (NDA/BLA# 761178). Available online: https://www.fda.gov/advisory-committees/advisory-committee-calendar/november-6-2020-meeting-peripheral-and-central-nervous-system-drugs-advisory-committee-meeting#event-materials (accessed on 6 May 2021).
- Swanson, C.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Bradley, H.; Rabe, M.; Totsuka, K.; Lai, R.; Gordon, R.; et al. The GAP study of BAN2401 study 201 in Early AD. Persistence of BAN2401-mediated amyloid reductions post-treatment: A preliminary comparison of amyloid status between the core phase of BAN2401-G000-201 and baseline of the open-label ex-tension phase in subjects with early Alzheimer’s disease. In Proceedings of the Clinical Trials on Alzheimer’s Disease Conference, San Diego, CA, USA, 4–7 December 2019. [Google Scholar]
- Gervais, F.; Paquette, J.; Morissette, C.; Krywkowski, P.; Yu, M.; Azzi, M.; Lacombe, D.; Kong, X.; Aman, A.; Laurin, J.; et al. Targeting soluble Abeta peptide with tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 2007, 28, 537–547. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical & Biomarker Profile | Biogen Aducanumab IV Infusion 10 mg/kg Monthly | Eli Lilly Donanemab IV Infusion 1400 mg Monthly | Eisai Lecanemab IV Infusion 10 mg/kg Twice per Month | Alzheon ALZ-801/Tramiprosate Oral Tablet 265 mg Twice Daily | |
|---|---|---|---|---|---|
| Selectivity for Oligomers * | + | + | ++ | +++ Blocks oligomer formation | |
| Study Population | Early AD All genotypes | Early AD All genotypes | Early AD All genotypes | Early AD APOE4 carriers | Mild AD APOE4/4 homozygotes |
| Cognition ADAS-cog (% benefit vs. placebo) | 27% p = 0.0097 | 32% p = 0.04 | 47% p = 0.017 | 84% Not reported | 125% p = 0.0001 |
| Function CDR-SB (% benefit vs. placebo) | 22% p = 0.012 | 23% p = NS | 26% p = NS | 60% Not reported | 81% p = 0.0197 |
| CSF p-tau181 (% benefit vs. placebo) | 15% | Not reported | 13% | Ongoing biomarker study | |
| Imaging Biomarkers | Significant decrease in tau PET signal | Significant decrease in tau PET signal | Increase in hippocampal atrophy (7.6%, not significant) | Significant decrease of hippocampal atrophy | |
| Brain Edema (% vs. placebo) | 35% (42% in APOE4) | 27% | 10% | 15% | 0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. https://doi.org/10.3390/ijms22126355
Tolar M, Hey J, Power A, Abushakra S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. International Journal of Molecular Sciences. 2021; 22(12):6355. https://doi.org/10.3390/ijms22126355
Chicago/Turabian StyleTolar, Martin, John Hey, Aidan Power, and Susan Abushakra. 2021. "Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression" International Journal of Molecular Sciences 22, no. 12: 6355. https://doi.org/10.3390/ijms22126355
APA StyleTolar, M., Hey, J., Power, A., & Abushakra, S. (2021). Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. International Journal of Molecular Sciences, 22(12), 6355. https://doi.org/10.3390/ijms22126355