
Chimeric Stimuli-Responsive Liposomes as Nanocarriers for the Delivery of the Anti-Glioma Agent TRAM-34

 ,
,  ,
, 
 , ,
, ,  ,
,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
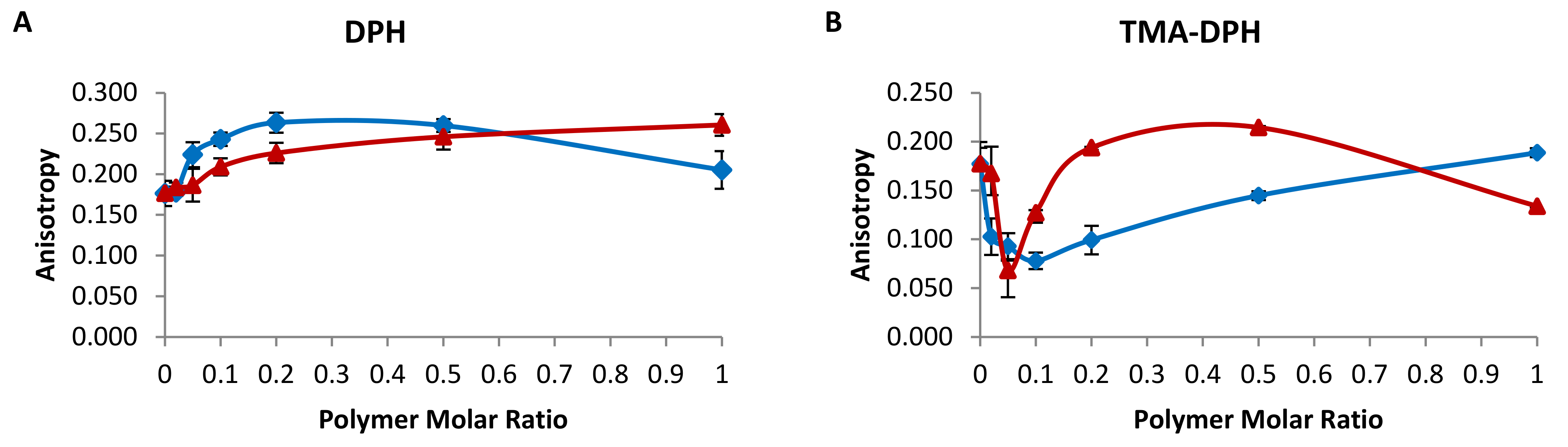
2.1. Membrane Fluidity of Chimeric Bilayers
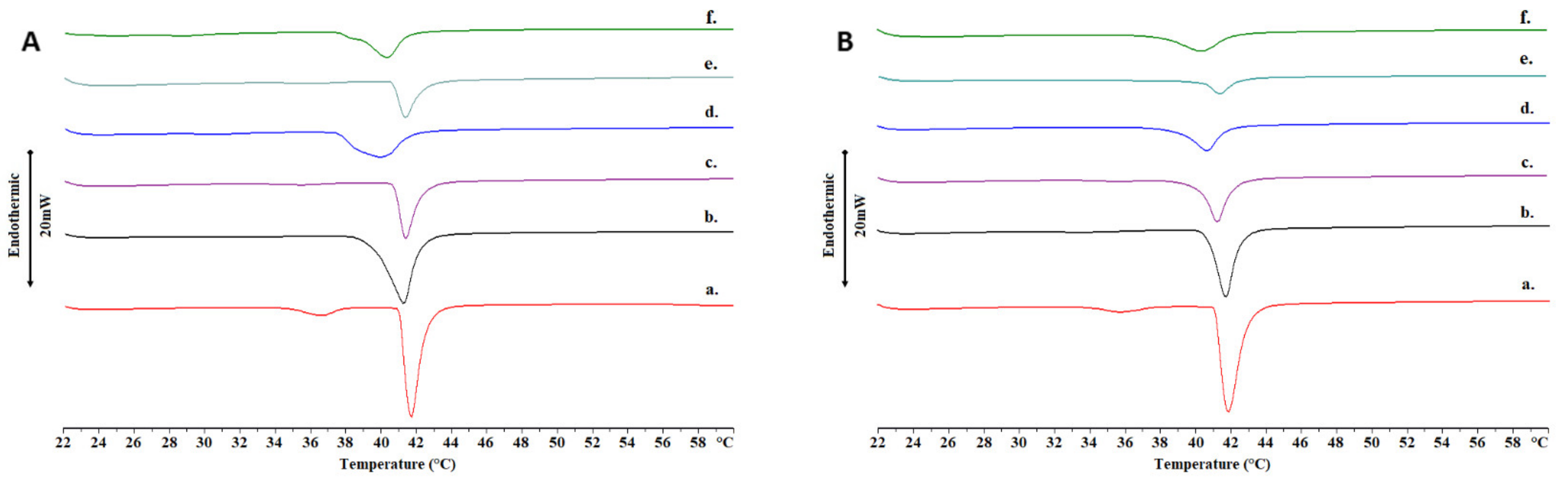
2.2. Thermotropic Behavior of Chimeric Bilayers with TRAM-34
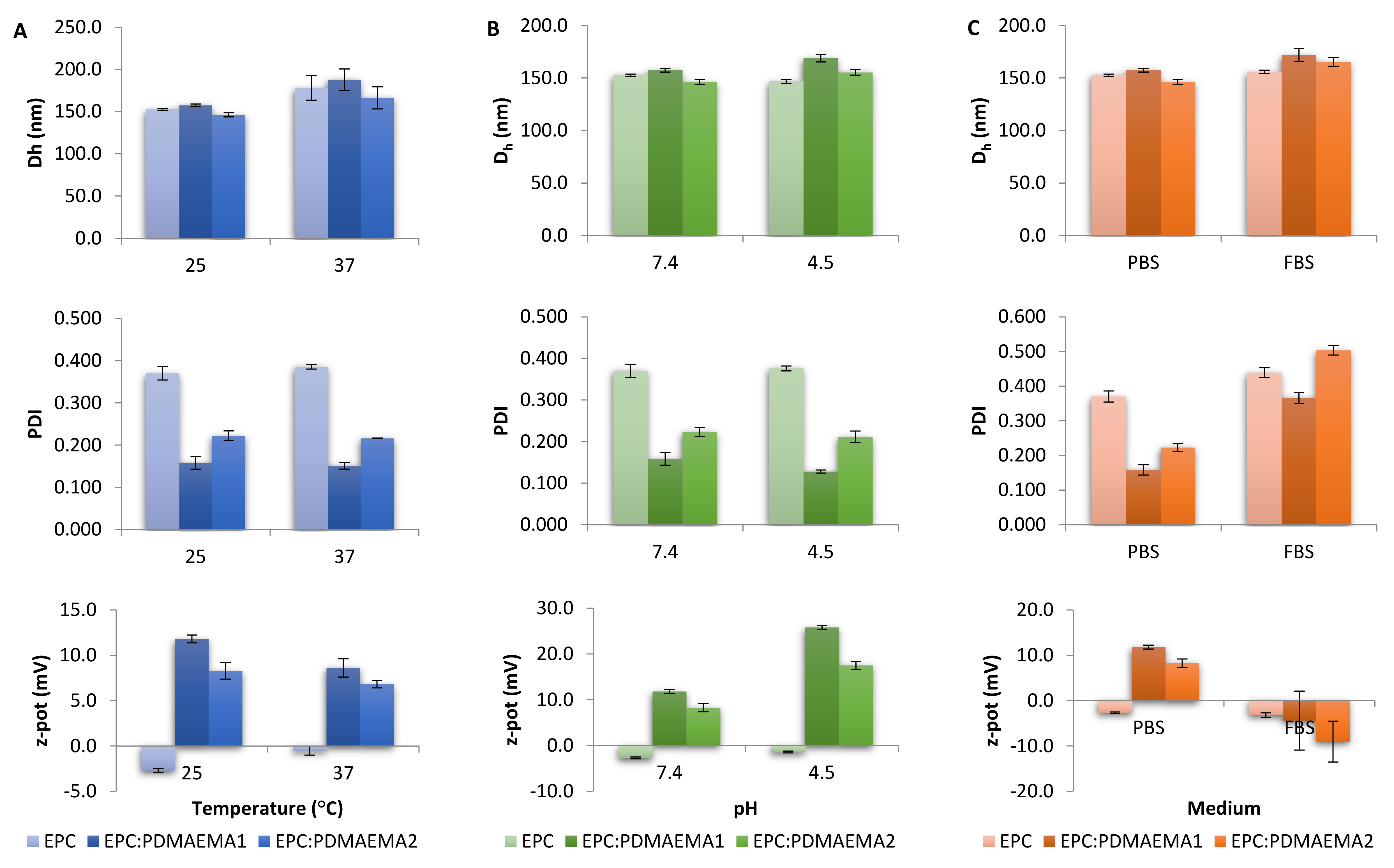
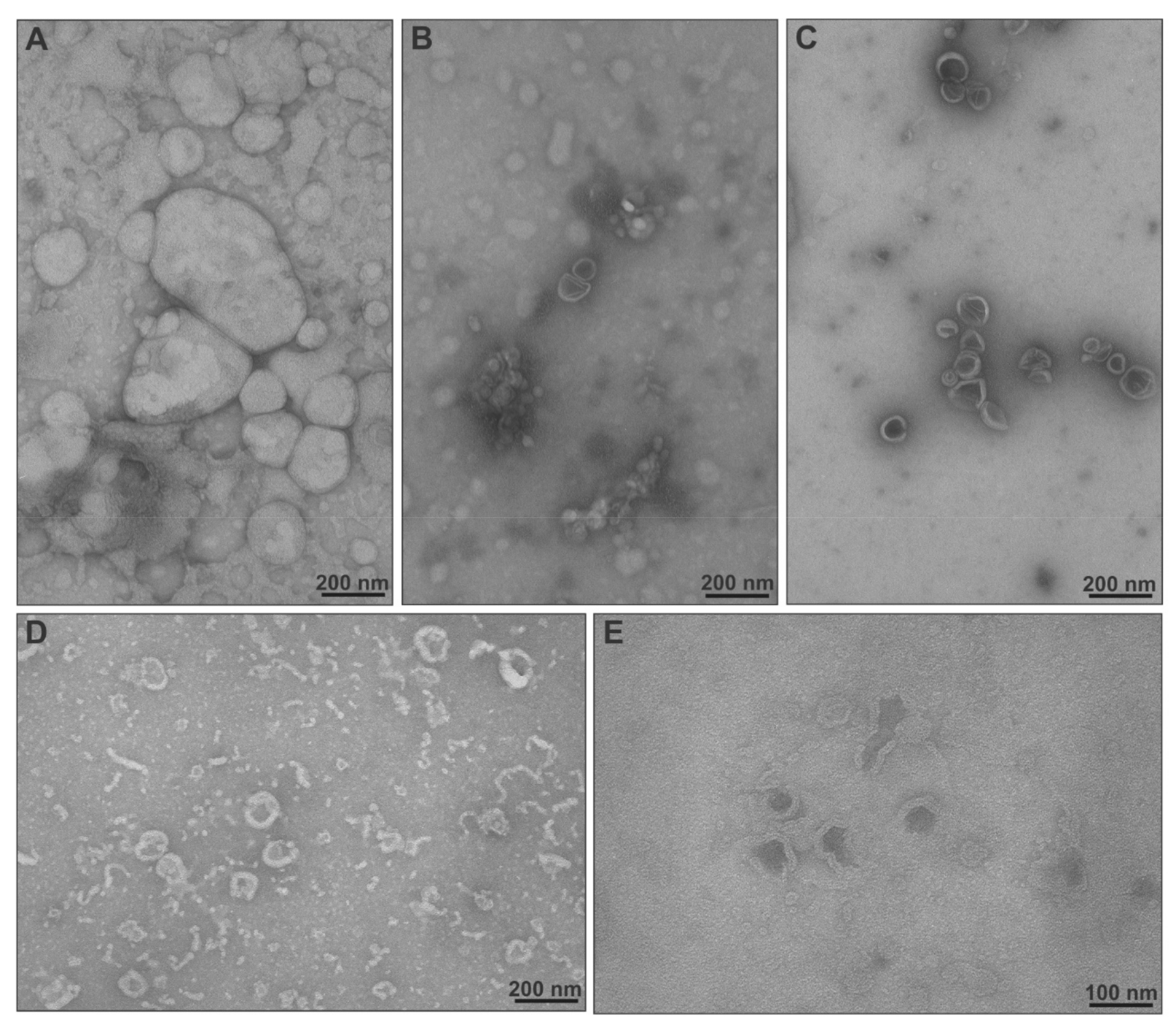
2.3. Physicochemical Characteristics, Stimuli-Responsiveness and Protein Interactions of Chimeric Nanocarriers
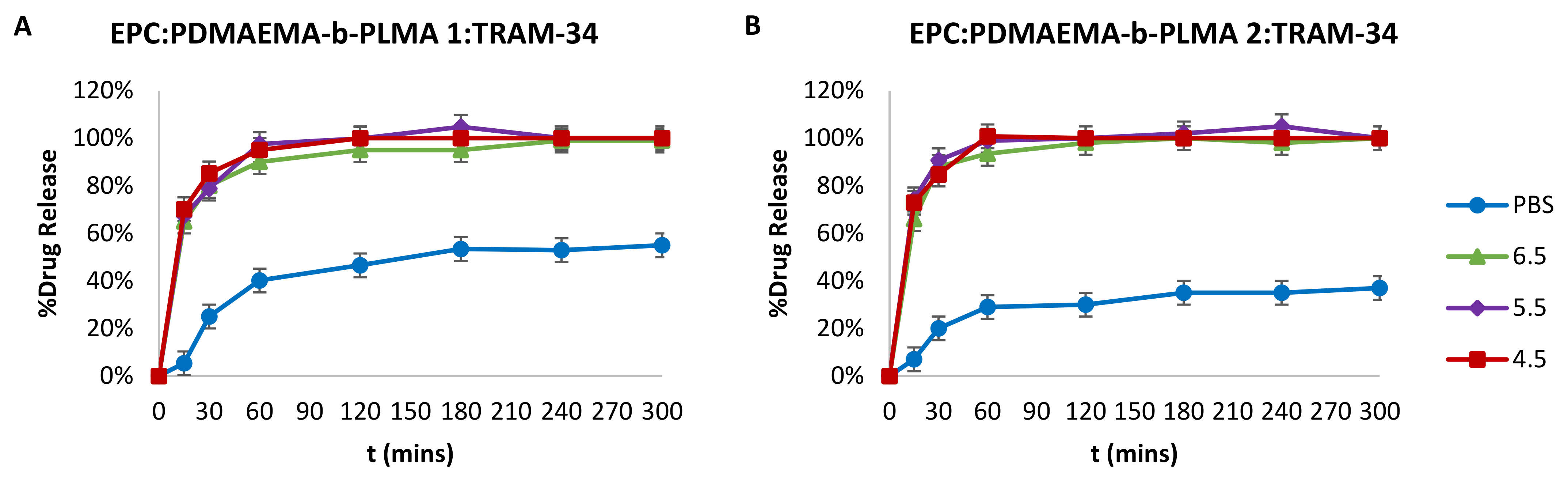
2.4. Drug Entrapment Efficiency % (EE%) and Release in Acidic Conditions
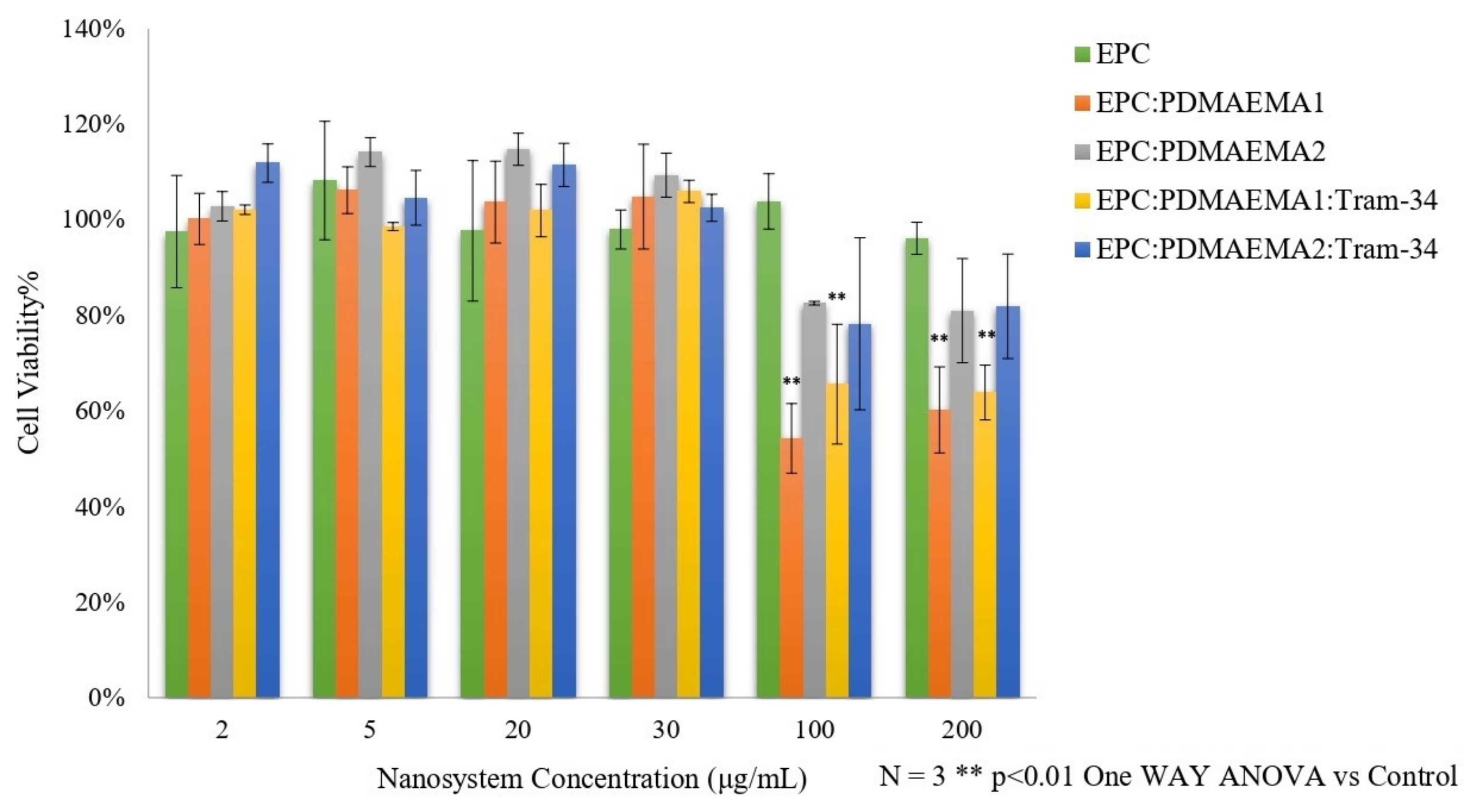
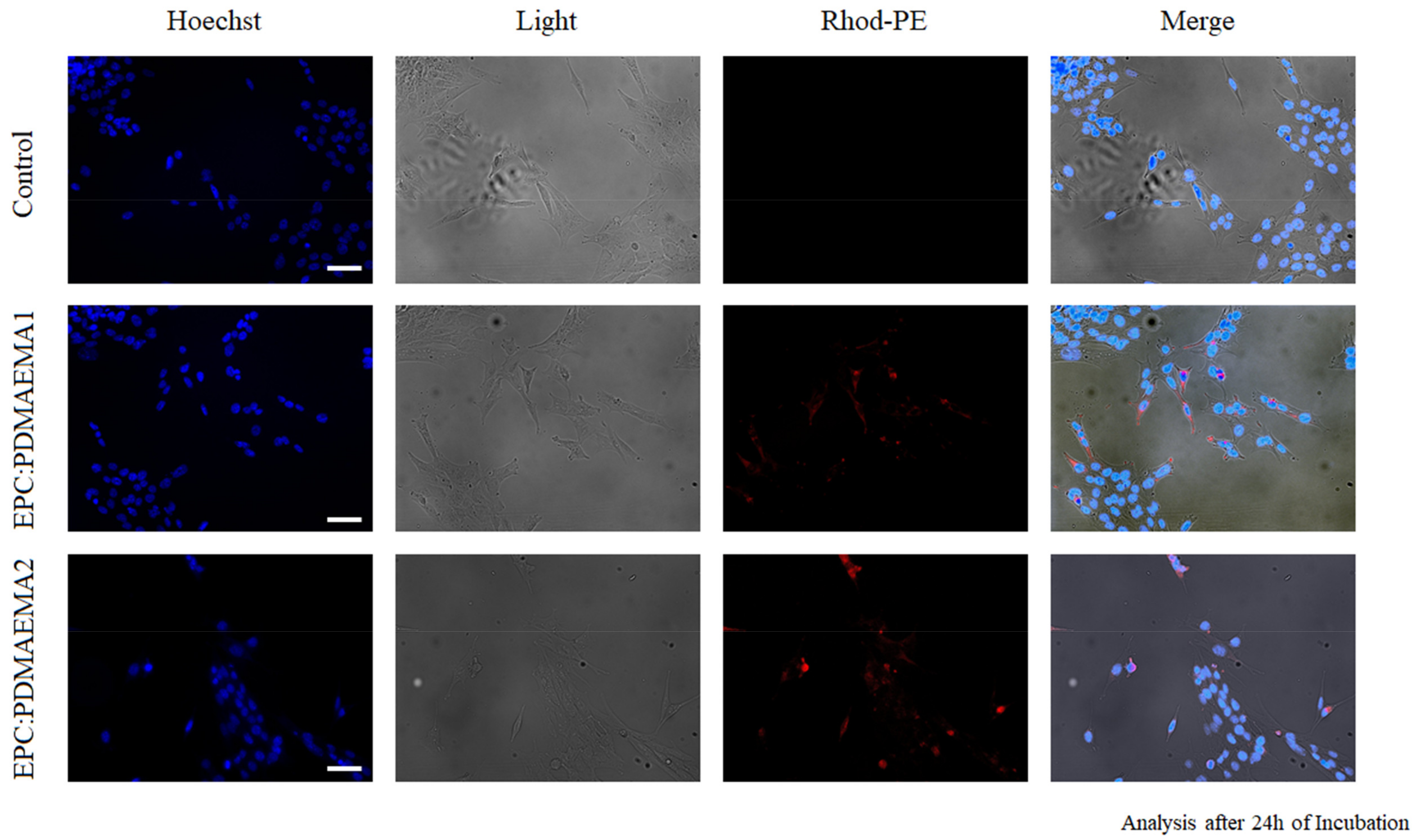
2.5. In Vitro Toxicity and Uptake of Chimeric Nanocarriers by HEK-293 Cells
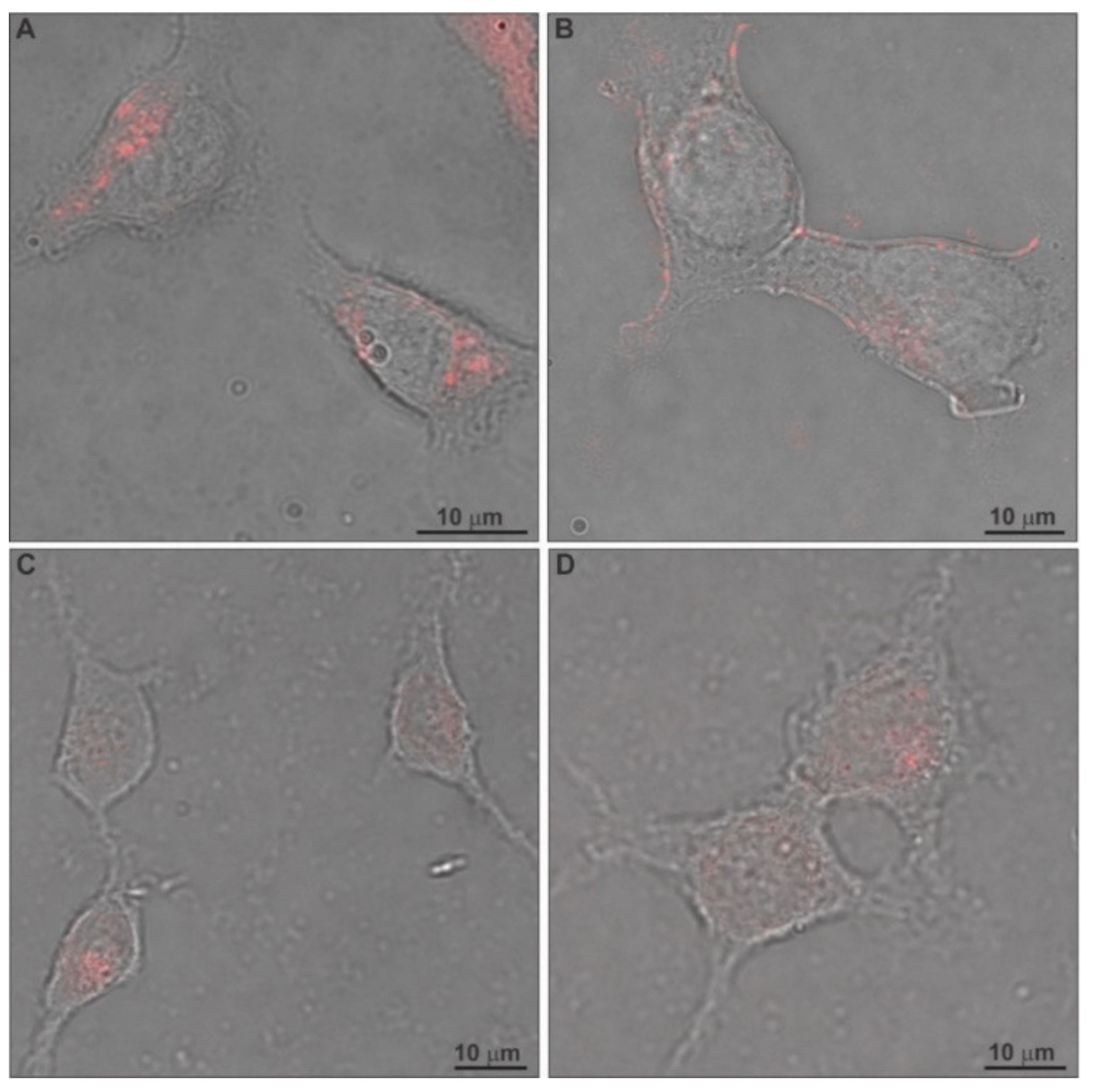
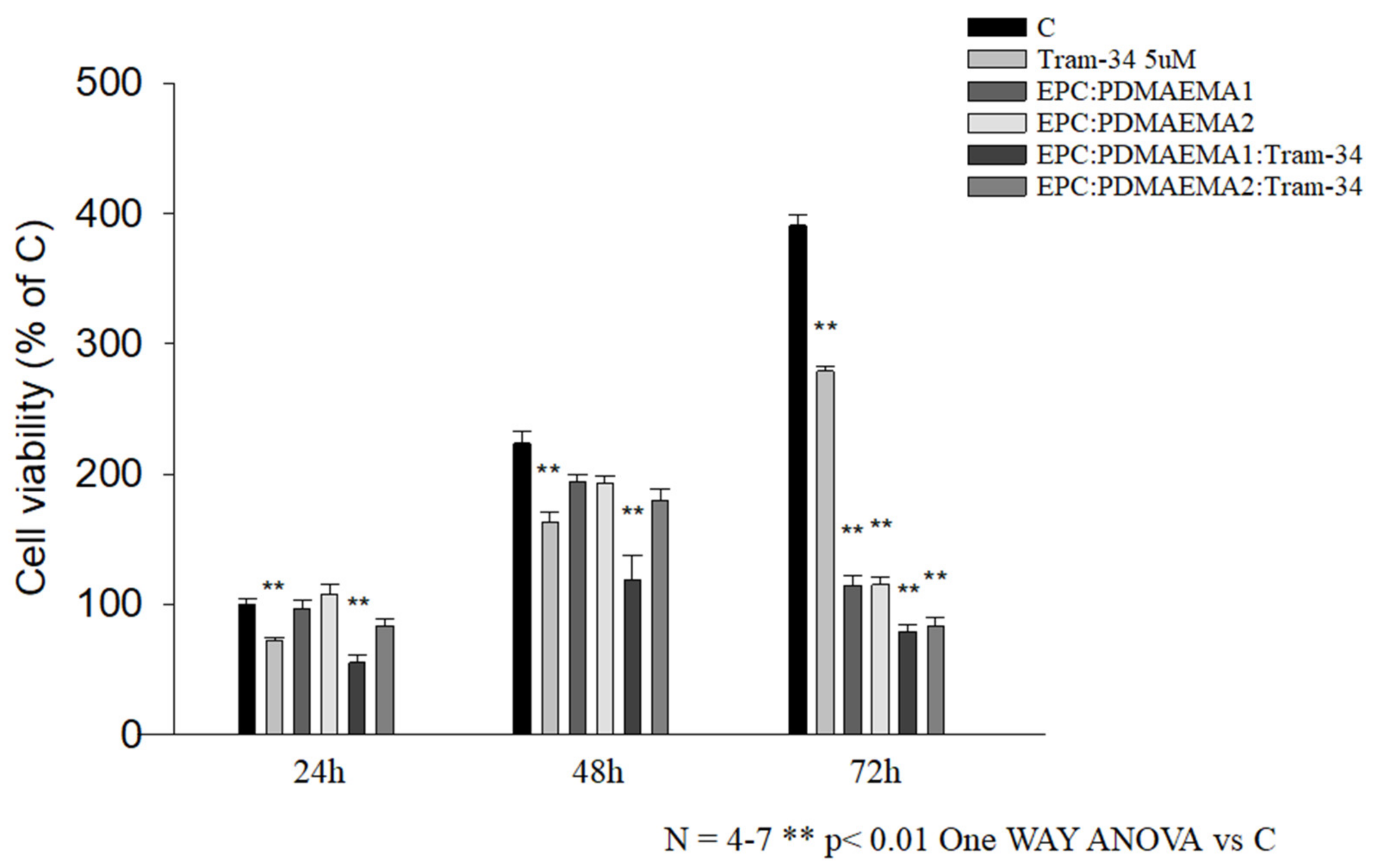
2.6. In Vitro Antiproliferative Effect and Uptake of Drug-Loaded Chimeric Nanocarriers by GL261 Cells
2.7. In Vivo Toxicity of Chimeric Nanocarriers
3. Materials and Methods
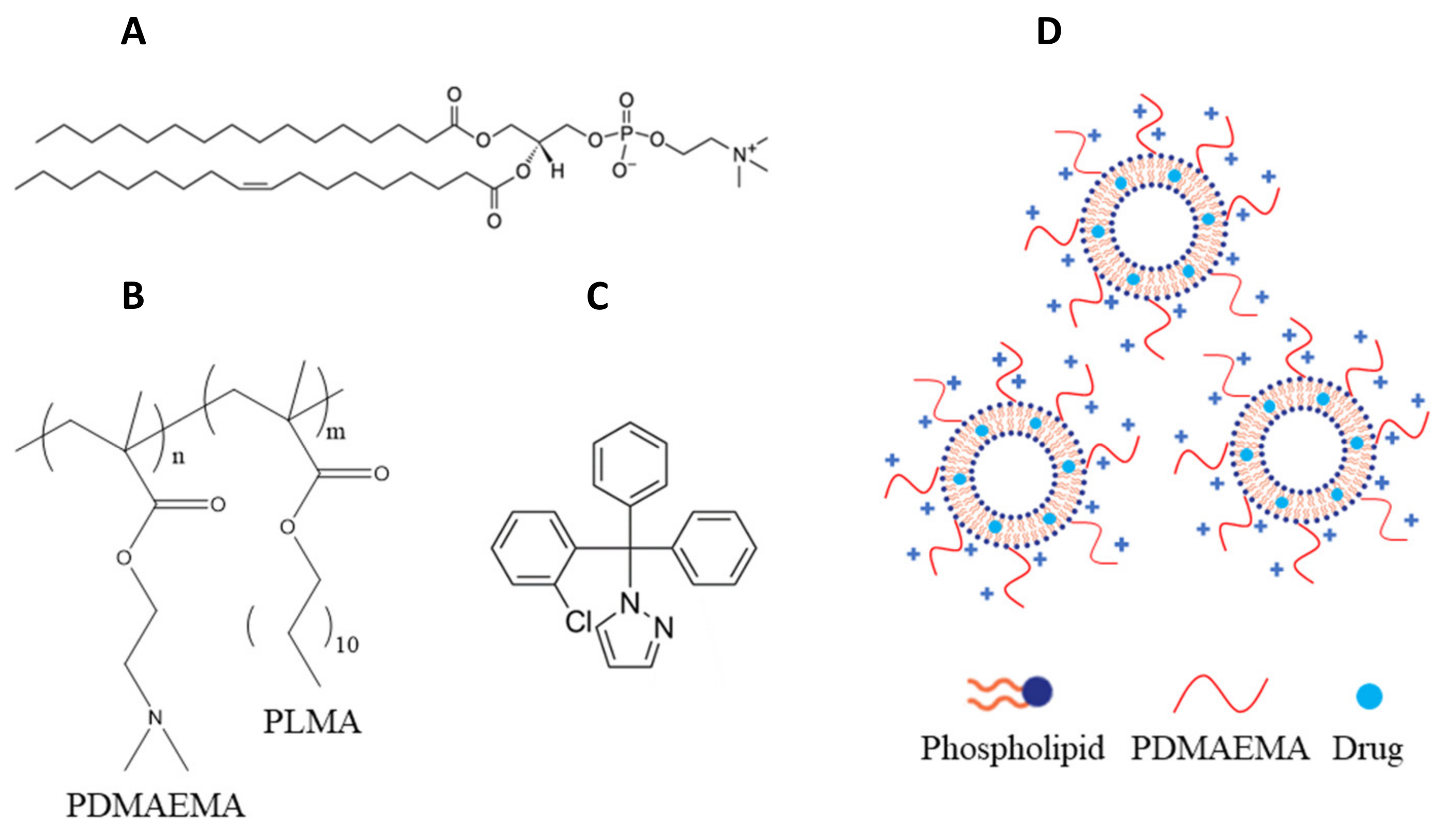
3.1. Materials
3.2. Preparation of Chimeric Bilayers
3.3. Fluorescence Anisotropy/Polarization (FA)
3.4. Differential Scanning Calorimetry (DSC)
3.5. Preparation of Chimeric Nanocarriers
3.6. Light Scattering Techniques
3.7. Transmission Electron Microscopy (TEM)
3.8. Drug Entrapment Efficiency % (EE%) and Release Studies
3.9. Normal Cell Culture
3.10. Normal Cell Viability In Vitro
3.11. Confocal Laser Scanning Microscopy (CLSM)
3.12. Glioma Cell Culture
3.13. Glioma Cell Viability In Vitro
3.14. Fluorescence Microscopy (FM)
3.15. In Vivo Toxicity
3.16. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- El-Sawy, H.S.; Al-Abd, A.M.; Ahmed, T.A.; El-Say, K.M.; Torchilin, V.P. Stimuli-responsive nano-architecture drug-delivery systems to solid tumor micromilieu: Past, present, and future perspectives. ACS Nano 2018, 12, 10636–10664. [Google Scholar] [CrossRef] [PubMed]
- Naziris, N.; Pippa, N.; Pispas, S.; Demetzos, C. Stimuli-responsive drug delivery nanosystems: From bench to clinic. Curr. Nanomed. 2016, 6, 1–20. [Google Scholar] [CrossRef]
- Demetzos, C.; Pippa, N. Advanced drug delivery nanosystems (aDDnSs): A mini-review. Drug Deliv. 2014, 21, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Luo, L.; Wang, Y.; Wu, Q.; Dai, H.B.; Li, J.S.; Durkan, C.; Wang, N.; Wang, G.X. Endogenous pH-responsive nanoparticles with programmable size changes for targeted tumor therapy and imaging applications. Theranostics 2018, 8, 3038–3058. [Google Scholar] [CrossRef] [PubMed]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Allhenn, D.; Boushehri, M.A.; Lamprecht, A. Drug delivery strategies for the treatment of malignant gliomas. Int. J. Pharm. 2012, 436, 299–310. [Google Scholar] [CrossRef]
- Ohya, S.; Kito, H. Ca2+-activated K+ channel KCa3.1 as a therapeutic target for immune disorders. Biol. Pharm. Bull. 2018, 41, 1158–1163. [Google Scholar] [CrossRef]
- Catacuzzeno, L.; Fioretti, B.; Franciolini, F. Expression and role of the intermediate-conductance calcium-activated potassium channel KCa3.1 in glioblastoma. J. Signal Transduct. 2012, 2012, 421564. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.F.; Zhou, Y.; Soboloff, J. WT1/EGR1-mediated control of STIM1 expression and function in cancer cells. Front. Biosci. 2011, 16, 2402–2415. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, G.; Catalano, M.; Sciaccaluga, M.; Chece, G.; Cipriani, R.; Rosito, M.; Grimaldi, A.; Lauro, C.; Cantore, G.; Santoro, A.; et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis. 2013, 4, e773. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, G.; Grimaldi, A.; Chece, G.; Porzia, A.; Esposito, V.; Santoro, A.; Salvati, M.; Mainiero, F.; Ragozzino, D.; Di Angelantonio, S.; et al. KCa3.1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget 2016, 7, 30781–30796. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.; D’Alessandro, G.; Golia, M.T.; Grössinger, E.M.; Di Angelantonio, S.; Ragozzino, D.; Santoro, A.; Esposito, V.; Wulff, H.; Catalano, M.; et al. KCa3.1 inhibition switches the phenotype of glioma-infiltrating microglia/macrophages. Cell Death Dis. 2016, 7, e2174. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, P.; Mangino, G.; Fioretti, B.; Catacuzzeno, L.; Puca, R.; Ponti, D.; Miscusi, M.; Franciolini, F.; Ragona, G.; Calogero, A. The inhibition of KCa3.1 channels activity reduces cell motility in glioblastoma derived cancer stem cells. PLoS ONE 2012, 7, e47825. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.J.; Zhu, Y.; Zhang, Q.Y.; Mongin, A.A.; Hough, L.B. TRAM-34, a putatively selective blocker of intermediate-conductance, calcium-activated potassium channels, inhibits cytochrome P450 activity. PLoS ONE 2013, 8, e63028. [Google Scholar] [CrossRef]
- Wulff, H.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Delineation of the clotrimazole/TRAM-34 binding site on the intermediate conductance calcium-activated potassium channel, IKCa1. J. Biol. Chem. 2001, 276, 32040–32045. [Google Scholar] [CrossRef]
- Wulff, H.; Miller, M.J.; Hansel, W.; Grissmer, S.; Cahalan, M.D.; Chandy, K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA 2000, 97, 8151–8156. [Google Scholar] [CrossRef]
- Xu, H.; Paxton, J.W.; Wu, Z. Enhanced pH-responsiveness, cellular trafficking, cytotoxicity and long-circulation of PEGylated liposomes with post-insertion technique using gemcitabine as a model drug. Pharm. Res. 2015, 32, 2428–2438. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Chang, H.I.; Yeh, M.K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar]
- Bruggemann, E.P.; Melchior, D.L. Alterations in the organization of phosphatidylcholine/cholesterol bilayers by tetrahydrocannabinol. J. Biol. Chem. 1983, 258, 8298–8303. [Google Scholar] [CrossRef]
- Naziris, N.; Pippa, N.; Chrysostomou, V.; Pispas, S.; Demetzos, C.; Libera, M.; Trzebicka, B. Morphological diversity of block copolymer/lipid chimeric nanostructures. J. Nanopart. Res. 2017, 19, 347–357. [Google Scholar] [CrossRef]
- Naziris, N.; Pippa, N.; Stellas, D.; Chrysostomou, V.; Pispas, S.; Demetzos, C.; Libera, M.; Trzebicka, B. Development and evaluation of stimuli-responsive chimeric nanostructures. AAPS PharmSciTech 2018, 19, 2971–2989. [Google Scholar] [CrossRef]
- Zhang, Y.; Maji, S.; Greiner, A. PDMAEMA based gene delivery materials. Mater. Today 2012, 15, 388–393. [Google Scholar]
- Samsonova, O.; Pfeiffer, C.; Hellmund, M.; Merkel, O.M.; Kissel, T. Low molecular weight pDMAEMA-block-pHEMA blockcopolymers synthesized via RAFT-polymerization: Potential non-viral gene delivery agents? Polymer 2011, 3, 693–718. [Google Scholar] [CrossRef]
- Zengin, A.; Karakose, G.; Caykara, T. Poly(2-(dimethylamino)ethyl methacrylate) brushes fabricated by surface-mediated RAFT polymerization and their response to pH. Eur. Polym. J. 2013, 49, 3350–3358. [Google Scholar] [CrossRef]
- Ionov, M.; Klajnert, B.; Gardikis, K.; Hatziantoniou, S.; Palecz, B.; Salakhutdinov, B.; Cladera, J.; Zamaraeva, M.; Demetzos, C.; Bryszewska, M. Effect of amyloid beta peptides Aβ1-28 and Aβ25-40 on model lipid membranes. J. Therm. Anal. Calorim. 2010, 99, 741–747. [Google Scholar] [CrossRef]
- Wei, X.; Cohen, R.; Barenholz, Y. Insights into composition/structure/function relationships of Doxil® gained from “high-sensitivity” differential scanning calorimetry. Eur. J. Pharm. Biopharm. 2016, 104, 260–270. [Google Scholar] [CrossRef]
- Naziris, N.; Skandalis, A.; Forys, A.; Trzebicka, B.; Fessas, D.; Pispas, S.; Demetzos, C. A thermal analysis and physicochemical study on thermoresponsive chimeric liposomal nanosystems. J. Therm. Anal. Calorim. 2020, 141, 751–766. [Google Scholar] [CrossRef]
- Zhang, B.; Song, Y.; Wang, T.; Yang, S.; Zhang, J.; Liu, Y.; Zhang, N.; Garg, S. Efficient co-delivery of immiscible hydrophilic/hydrophobic chemotherapeutics by lipid emulsions for improved treatment of cancer. Int. J. Nanomed. 2017, 12, 2871–2886. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Demetzos, C. Differential Scanning Calorimetry (DSC): A tool to study the thermal behavior of lipid bilayers and liposomal stability. J. Liposome Res. 2008, 18, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidi, A.; Naziris, N.; Chountoulesi, M.; Kiriakidi, S.; Sartori, B.; Kolokouris, D.; Amentisch, H.; Mali, G.; Ntountaniotis, D.; Demetzos, C.; et al. Comparative perturbation effects exerted by the influenza A M2 WT protein inhibitors amantadine and the spiro[pyrrolidine-2,2′-adamantane] variant AK13 to membrane bilayers studied using biophysical experiments and molecular dynamics simulations. J. Phys. Chem. B 2018, 122, 9877–9895. [Google Scholar] [CrossRef]
- Tribet, C.; Vial, F. Flexible macromolecules attached to lipid bilayers: Impact on fluidity, curvature, permeability and stability of the membranes. Soft Matter 2008, 4, 68–81. [Google Scholar] [CrossRef]
- Roy, B.; Guha, P.; Bhattarai, R.; Nahak, P.; Karmakar, G.; Chettri, P.; Panda, A.K. Influence of lipid composition, pH, and temperature on physicochemical properties of liposomes with curcumin as model drug. J. Oleo Sci. 2016, 65, 399–411. [Google Scholar] [CrossRef]
- Saptarshi, S.R.; Duschl, A.; Lopata, A.L. Interaction of nanoparticles with proteins: Relationto bio-reactivity of the nanoparticle. J. Nanobiotechnol. 2013, 11, 26–37. [Google Scholar] [CrossRef]
- Böhme, U.; Scheler, U. Effective charge of bovine serum albumin determined by electrophoresis NMR. Chem. Phys. Letters 2007, 435, 342–345. [Google Scholar] [CrossRef]
- Chou, M.J.; Yu, H.Y.; Hsia, J.C.; Chen, Y.H.; Hung, T.T.; Chao, H.M.; Chern, E.; Huang, Y.Y. Highly efficient intracellular protein delivery by cationic polyethyleneimine-modified gelatin nanoparticles. Materials 2018, 11, 301. [Google Scholar] [CrossRef] [PubMed]
- Ruozi, B.; Belletti, D.; Tombesi, A.; Tosi, G.; Bondioli, L.; Forni, F.; Vandelli, M.A. AFM, ESEM, TEM, and CLSM in liposomal characterization: A comparative study. Int. J. Nanomed. 2011, 6, 557–563. [Google Scholar] [CrossRef]
- Franken, L.E.; Boekema, E.J.; Stuart, M.C.A. Transmission electron microscopy as a tool for the characterization of soft materials: Application and interpretation. Adv. Sci. 2017, 4, 1600476. [Google Scholar] [CrossRef] [PubMed]
- Popplewell, J.F.; Swann, M.J.; Freeman, N.J.; McDonnell, C.; Ford, R.C. Quantifying the effects of melittin on liposomes. Biochim. Biophys. Acta. 2007, 1768, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.S.; Choi, J.Y.; Kim, J.O.; Lee, M.K.; Kim, S.H.; Lim, S.J. Development of paclitaxel-loaded liposomal nanocarrier stabilized by triglyceride incorporation. Int. J. Nanomed. 2016, 11, 4465–4477. [Google Scholar]
- Ruysschaert, T.; Marque, A.; Duteyrat, J.L.; Lesieur, S.; Winterhalter, M.; Fournier, D. Liposome retention in size exclusion chromatography. BMC Biotechnol. 2005, 5, 11. [Google Scholar] [CrossRef]
- Fahr, A.; van Hoogevest, P.; Kuntsche, J.; Leigh, M.L. Lipophilic drug transfer between liposomal and biological membranes: What does it mean for parenteral and oral drug delivery? J. Liposome Res. 2006, 16, 281–301. [Google Scholar] [CrossRef]
- Pippa, N.; Meristoudi, A.; Pispas, S.; Demetzos, C. Temperature-dependent drug release from DPPC:C12H25-PNIPAM-COOH liposomes: Control of the drug loading/release by modulation of the nanocarriers’ components. Int. J. Pharm. 2015, 485, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Naziris, N.; Pippa, N.; Meristoudi, A.; Pispas, S.; Demetzos, C. Design and development of pH-responsive HSPC:C12H25-PAA chimeric liposomes. J. Liposome Res. 2017, 27, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Bahadar, H.; Maqbool, F.; Niaz, K.; Abdollahi, M. Toxicity of nanoparticles and an overview of current experimental models. Iran. Biomed. J. 2016, 20, 1–11. [Google Scholar] [PubMed]
- Angius, F.; Floris, A. Liposomes and MTT cell viability assay: An incompatible affair. Toxicol. In Vitro 2015, 29, 314–319. [Google Scholar] [CrossRef]
- Roursgaard, M.; Knudsen, K.B.; Northeved, H.; Persson, M.; Christensen, T.; Kumar, P.E.K.; Permin, A.; Andresen, T.L.; Gjetting, T.; Lykkesfeldt, J.; et al. In vitro toxicity of cationic micelles and liposomes in cultured human hepatocyte (HepG2) and lung epithelial (A549) cell lines. Toxicol. In Vitro 2016, 36, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Bitoque, D.B.; Simão, S.; Oliveira, A.V.; Machado, S.; Duran, M.R.; Lopes, E.; da Costa, A.M.; Silva, G.A. Efficiency of RAFT-synthesized PDMAEMA in gene transfer to the retina. J. Tissue Eng. Regen. Med. 2017, 11, 265–275. [Google Scholar] [CrossRef]
- Kang, J.H.; Jang, W.Y.; Ko, Y.T. The effect of surface charges on the cellular uptake of liposomes investigated by live cell imaging. Pharm. Res. 2017, 34, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Gan, L.S. Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int. J. Cell Biol. 2013, 2013, 703545. [Google Scholar] [CrossRef]
- Goren, D.; Horowitz, A.T.; Tzemach, D.; Tarshish, M.; Zalipsky, S.; Gabizon, A. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin. Cancer Res. 2000, 6, 1949–1957. [Google Scholar]
- Liu, X.; Chen, Y.; Li, H.; Huang, N.; Jin, Q.; Ren, K.; Ji, J. Enhanced retention and cellular uptake of nanoparticles in tumors by controlling their aggregation behavior. ACS Nano 2013, 7, 6244–6257. [Google Scholar] [CrossRef] [PubMed]
- Mohr, C.J.; Steudel, F.A.; Gross, D.; Ruth, P.; Lo, W.Y.; Hoppe, R.; Schroth, W.; Brauch, H.; Huber, S.M.; Lukowski, R. Cancer-associated intermediate conductance Ca2+-activated K⁺ channel KCa3.1. Cancers 2019, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Iatrou, H.; Dimas, K.; Gkikas, M.; Tsimblouli, C.; Sofianopoulou, S. Polymersomes from polypeptide containing triblock Co- and terpolymers for drug delivery against pancreatic cancer: Asymmetry of the external hydrophilic blocks. Macromol. Biosci. 2014, 14, 1222–1238. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Willson, J.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. 2015. Available online: https://dtp.cancer.gov/organization/btb/acute_toxicity.htm (accessed on 10 January 2021).
- Naziris, N.; Saitta, F.; Chrysostomou, V.; Libera, M.; Trzebicka, B.; Fessas, D.; Pispas, S.; Demetzos, C. pH-responsive chimeric liposomes: From nanotechnology to biological assessment. Int. J. Pharm. 2020, 574, 118849. [Google Scholar] [CrossRef]
- Bilalis, P.; Skoulas, D.; Karatzas, A.; Marakis, J.; Stamogiannos, A.; Tsimblouli, C.; Sereti, E.; Stratikos, E.; Dimas, K.; Vlassopoulos, D.; et al. Self-healing pH- and enzyme stimuli-responsive hydrogels for targeted delivery of gemcitabine to treat pancreatic cancer. Biomacromolecules 2018, 19, 3840–3852. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanosystem | Dh 1 (nm) | SD 2 | PDI 3 | SD | z-pot 4 (mV) | SD |
|---|---|---|---|---|---|---|
| ΕPC | 152.7 | 1.0 | 0.370 | 0.016 | −2.7 | 0.2 |
| ΕPC:PDMAEMA1 | 157.3 | 1.7 | 0.158 | 0.015 | 11.8 | 0.4 |
| ΕPC:PDMAEMA2 | 146.3 | 2.5 | 0.223 | 0.011 | 8.3 | 0.9 |
| ΕPC:TRAM-34 | 167.7 | 3.9 | 0.335 | 0.022 | −1.2 | 0.1 |
| ΕPC:PDMAEMA1:TRAM-34 | 164.4 | 8.0 | 0.236 | 0.052 | 14.0 | 1.2 |
| ΕPC:PDMAEMA 2:TRAM-34 | 139.2 | 0.8 | 0.193 | 0.006 | 12.4 | 0.8 |
| Nanosystem | Initial Concentration (mg/mL) | Concentration before Extrusion (mg/mL) | SD 1 | IE% 2 | Concentration after Extrusion (mg/mL) | SD | IE% |
|---|---|---|---|---|---|---|---|
| EPC:TRAM-34 | 1.20 | 0.18 | 0.03 | 15 | 0.00 | 0.00 | 0 |
| EPC:PDMAEMA 1: TRAM-34 | 1.20 | 0.96 | 0.06 | 80 | 0.88 | 0.05 | 73 |
| EPC:PDMAEMA 2: TRAM-34 | 1.20 | 0.82 | 0.05 | 68 | 0.74 | 0.04 | 62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naziris, N.; Pippa, N.; Sereti, E.; Chrysostomou, V.; Kędzierska, M.; Kajdanek, J.; Ionov, M.; Miłowska, K.; Balcerzak, Ł.; Garofalo, S.; et al. Chimeric Stimuli-Responsive Liposomes as Nanocarriers for the Delivery of the Anti-Glioma Agent TRAM-34. Int. J. Mol. Sci. 2021, 22, 6271. https://doi.org/10.3390/ijms22126271
Naziris N, Pippa N, Sereti E, Chrysostomou V, Kędzierska M, Kajdanek J, Ionov M, Miłowska K, Balcerzak Ł, Garofalo S, et al. Chimeric Stimuli-Responsive Liposomes as Nanocarriers for the Delivery of the Anti-Glioma Agent TRAM-34. International Journal of Molecular Sciences. 2021; 22(12):6271. https://doi.org/10.3390/ijms22126271
Chicago/Turabian StyleNaziris, Nikolaos, Natassa Pippa, Evangelia Sereti, Varvara Chrysostomou, Marta Kędzierska, Jakub Kajdanek, Maksim Ionov, Katarzyna Miłowska, Łucja Balcerzak, Stefano Garofalo, and et al. 2021. "Chimeric Stimuli-Responsive Liposomes as Nanocarriers for the Delivery of the Anti-Glioma Agent TRAM-34" International Journal of Molecular Sciences 22, no. 12: 6271. https://doi.org/10.3390/ijms22126271
APA StyleNaziris, N., Pippa, N., Sereti, E., Chrysostomou, V., Kędzierska, M., Kajdanek, J., Ionov, M., Miłowska, K., Balcerzak, Ł., Garofalo, S., Limatola, C., Pispas, S., Dimas, K., Bryszewska, M., & Demetzos, C. (2021). Chimeric Stimuli-Responsive Liposomes as Nanocarriers for the Delivery of the Anti-Glioma Agent TRAM-34. International Journal of Molecular Sciences, 22(12), 6271. https://doi.org/10.3390/ijms22126271