
Uremic Toxins and Frailty in Patients with Chronic Kidney Disease: A Molecular Insight


Abstract
1. Uremic Toxins in Chronic Kidney Disease (CKD): An Introduction to Their Sources and Adverse Influences
2. Uremic Toxins and Their Associations with Cellular Degeneration: A Potential Origin of Frailty
2.1. Frailty: What Is It and What Causes It?
2.1.1. Senescence
2.1.2. Mitochondrial Dysfunction
2.1.3. Stem Cell Exhaustion and Telomere Attrition
2.1.4. Oxidative Stress and Inflammation
2.2. Cellular Transport of Uremic Toxins Varies between Cell Types
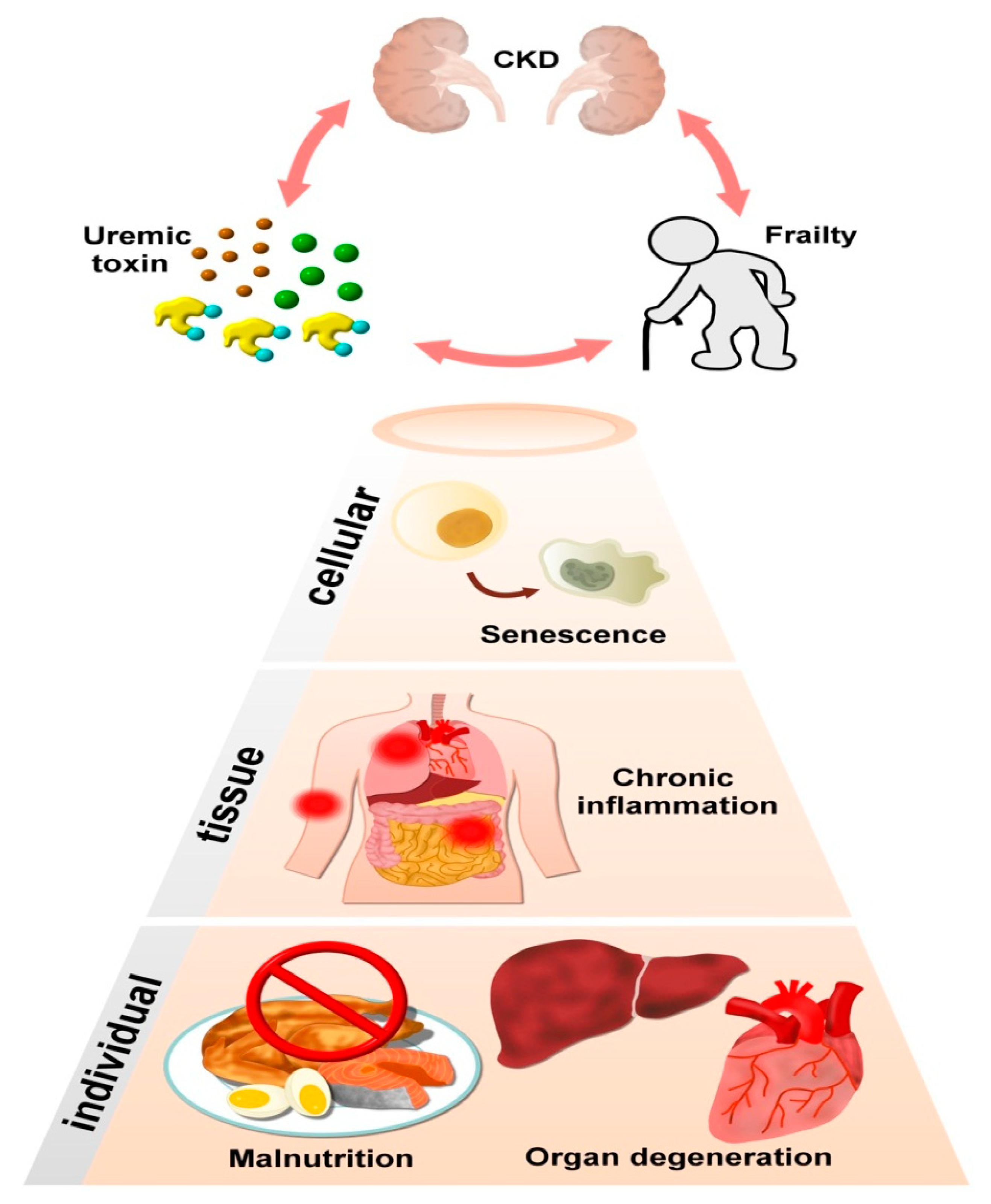
3. CKD, UTs, and Frailty Are Situated within a Vicious Circle
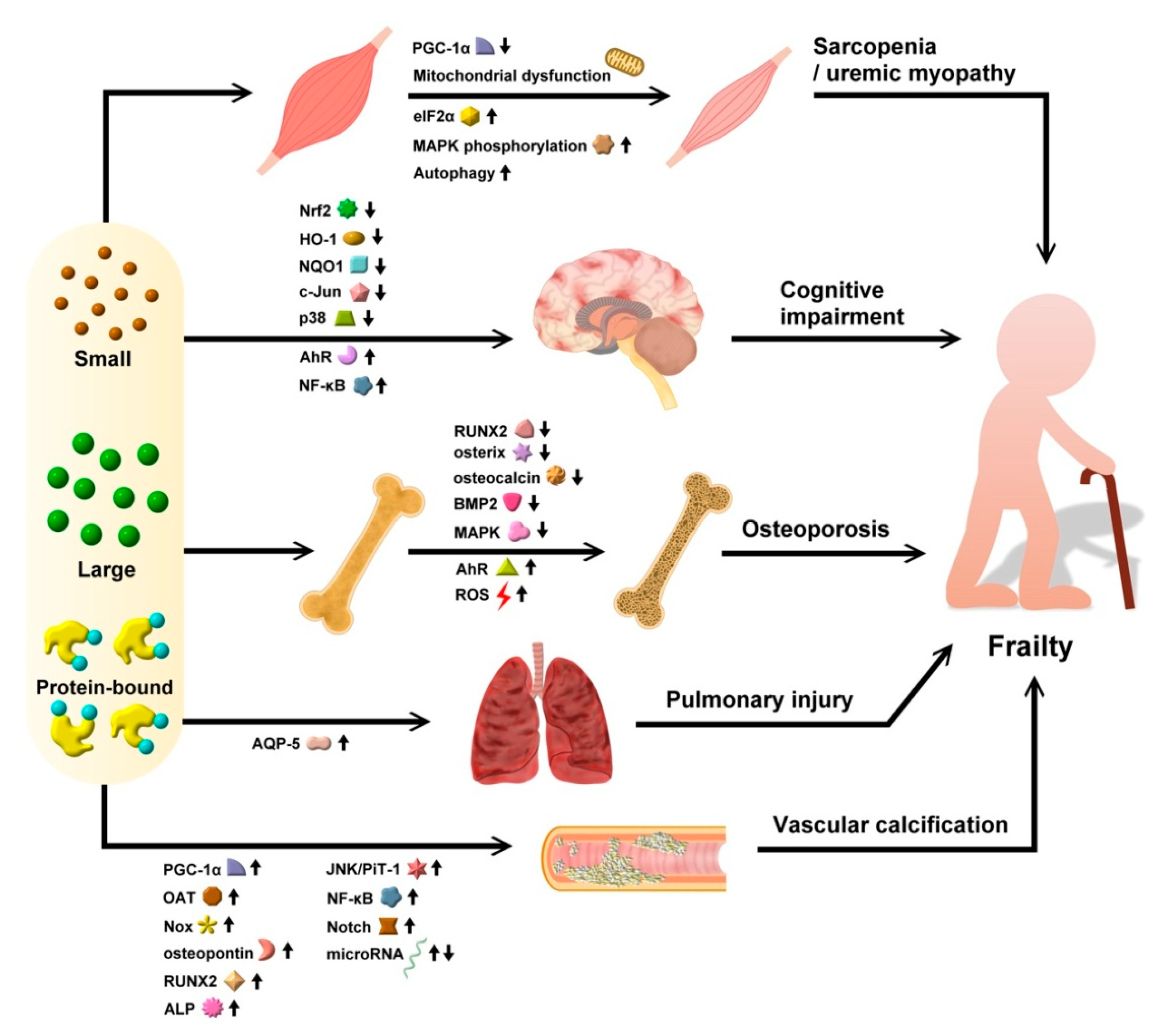
4. Uremic Toxins and Frailty: From Specific Molecular Linkage to Tissue Relevance and Clinical Evidence
4.1. Evidence for Uremic Toxins in Precipitating Sarcopenia
4.1.1. Protein-Bound UTs
4.1.2. Other UTs
4.2. Evidence for Uremic Toxins in Predisposing Patients to Cognitive Impairment/Frailty
4.2.1. Protein-Bound UTs
4.2.2. Other UTs
4.3. Evidence for Uremic Toxins in Inducing Osteoporosis
4.3.1. Protein-Bound UTs
4.3.2. Other UTs
4.4. Evidence for Uremic Toxins in Causing Cardiopulmonary Deconditioning
4.4.1. Cardiovascular System
Protein-Bound UTs
Other UTs
4.4.2. Pulmonary System
4.5. Frailty Accelerates Renal Progression and Possibly Increased UT Levels
5. Mediators of Organ Degeneration and Frailty as Emerging Diagnostics and Therapeutic Targets
6. Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; on behalf of the European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Chao, C.T.; Lin, S.H. Uremic Vascular Calcification: The Pathogenic Roles and Gastrointestinal Decontamination of Uremic Toxins. Toxins 2020, 12, 812. [Google Scholar] [CrossRef]
- Hu, J.R.; Coresh, J.; Inker, L.A.; Levey, A.S.; Zheng, Z.; Rebholz, C.M.; Adrienne, T.; Appel, L.J.; Chen, J.; Sarnak, M.J.; et al. Serum metabolites are associated with all-cause mortality in chronic kidney disease. Kidney Int. 2018, 94, 381–389. [Google Scholar] [CrossRef]
- Rysz, J.; Franczyk, B.; Ławiński, J.; Olszewski, R.; Ciałkowska-Rysz, A.; Gluba-Brzózka, A. The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins 2021, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Santana Machado, T.; Poitevin, S.; Paul, P.; McKay, N.; Jourde-Chiche, N.; Legris, T.; Mouly-Bandini, A.; Dignat-George, F.; Brunet, P.; Masereeuw, R.; et al. Indoxyl Sulfate Upregulates Liver P-Glycoprotein Expression and Activity through Aryl Hydrocarbon Receptor Signaling. J. Am. Soc. Nephrol. 2018, 29, 906–918. [Google Scholar] [PubMed]
- André, C.; Choukroun, G.; Bennis, Y.; Kamel, S.; Lemaire-Hurtel, A.S.; Masmoudi, K.; Bodeau, S.; Liabeuf, S. Potential interactions between uremic toxins and drugs: An application in kidney transplant recipients treated with calcineurin inhibitors. Nephrol. Dial. Transplant. 2021. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, M.; Wang, J.; Yang, K.; Shi, Y.; Zhang, J.; Zhang, B.; Zhang, L.; Zhao, J.; C-STRIDE. Geriatric nutrition risk index is associated with renal progression, cardiovascular events and all-cause mortality in chronic kidney disease. J. Nephrol. 2020, 33, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Steinmeyer, Z.; Delpierre, C.; Soriano, G.; Steinmeyer, A.; Ysebaert, L.; Balardy, L.; Sourdet, S. Hemoglobin concentration; a pathway to frailty. BMC Geriatr. 2020, 20, 202. [Google Scholar] [CrossRef]
- Chao, C.T.; Wang, J.; Huang, J.W.; Chan, D.C.; Hung, K.Y.; Chien, K.L.; COGENT Study Group. Chronic kidney disease–related osteoporosis is associated with incident frailty among patients with diabetic kidney disease: A propensity score–matched cohort study. Osteoporos. Int. 2020, 31, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Nobili, A.; Vitale, G. Frailty and sarcopenia: From theory to clinical implementation and public health relevance. Eur. J. Intern. Med. 2016, 35, 1–9. [Google Scholar] [CrossRef]
- Theou, O.; Walston, J.; Rockwood, K. Operationalizing Frailty Using the Frailty Phenotype and Deficit Accumulation Approaches. Interdiscip. Top. Gerontol. Geriatr. 2015, 41, 66–73. [Google Scholar]
- Cunha, A.I.L.; Veronese, N.; de Melo Borges, S.; Ricci, N.A. Frailty as a predictor of adverse outcomes in hospitalized older adults: A systematic review and meta-analysis. Ageing Res. Rev. 2019, 56, 100960. [Google Scholar] [CrossRef]
- Chao, C.T.; Hsu, Y.H.; Chang, P.Y.; He, Y.T.; Ueng, R.S.; Lai, C.F.; Chiang, C.K.; Huang, J.W.; Huang, S.J. Simple self-report FRAIL scale might be more closely associated with dialysis complications than other frailty screening instruments in rural chronic dialysis patients. Nephrology 2015, 20, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Wang, J.; Huang, J.W.; Chan, D.C.; Chien, K.L. Frailty Predicts an Increased Risk of End-Stage Renal Disease with Risk Competition by Mortality among 165,461 Diabetic Kidney Disease Patients. Aging Dis. 2019, 10, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Lee, Y.H.; Li, C.M.; Han, D.S.; Huang, J.W.; Huang, K.C. Advanced Age and Chronic Kidney Disease Modify the Association Between Metabolic Syndrome and Frailty Among Community-Dwelling Elderly. Rejuvenation Res. 2019, 23, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Chao, C.T.; Huang, J.W.; Huang, K.C. Vascular Calcification as an Underrecognized Risk Factor for Frailty in 1783 Community-Dwelling Elderly Individuals. J. Am. Heart Assoc. 2020, 9, e017308-e. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.I.; Chiang, C.L.; Chao, C.T.; Chiang, C.K.; Huang, J.W. Gustatory Dysfunction Is Closely Associated With Frailty in Patients With Chronic Kidney Disease. J. Ren. Nutr. 2021, 31, 49–56. [Google Scholar] [CrossRef] [PubMed]
- White, W.E.; Yaqoob, M.M.; Harwood, S.M. Aging and uremia: Is there cellular and molecular crossover? World J. Nephrol. 2015, 4, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Schroth, J.; Thiemermann, C.; Henson, S.M. Senescence and the Aging Immune System as Major Drivers of Chronic Kidney Disease. Front. Cell. Dev. Biol. 2020, 8, 564461. [Google Scholar] [CrossRef]
- Adijiang, A.; Higuchi, Y.; Nishijima, F.; Shimizu, H.; Niwa, T. Indoxyl sulfate, a uremic toxin, promotes cell senescence in aorta of hypertensive rats. Biochem. Biophys. Res. Commun. 2010, 399, 637–641. [Google Scholar] [CrossRef]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am. J. Physiol. Cell Physiol. 2012, 303, C126–C134. [Google Scholar] [CrossRef]
- Koizumi, M.; Tatebe, J.; Watanabe, I.; Yamazaki, J.; Ikeda, T.; Morita, T. Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Cellular Senescence in Human Umbilical Vein Endothelial Cells. J. Atheroscler. Thromb. 2014, 21, 904–916. [Google Scholar] [CrossRef]
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729. [Google Scholar] [CrossRef]
- Lee, J.H.; Yun, C.W.; Hur, J.; Lee, S.H. Fucoidan Rescues p-Cresol-Induced Cellular Senescence in Mesenchymal Stem Cells via FAK-Akt-TWIST Axis. Mar. Drugs 2018, 16, 121. [Google Scholar] [CrossRef]
- Tsai, Y.T.; Yeh, H.Y.; Chao, C.T.; Chiang, C.K. Superoxide Dismutase 2 (SOD2) in Vascular Calcification: A Focus on Vascular Smooth Muscle Cells, Calcification Pathogenesis, and Therapeutic Strategies. Oxid. Med. Cell. Longev. 2021, 2021, 6675548. [Google Scholar] [CrossRef] [PubMed]
- Chalupsky, M.; Goodson, D.A.; Gamboa, J.L.; Roshanravan, B. New insights into muscle function in chronic kidney disease and metabolic acidosis. Curr. Opin. Nephrol. Hypertens. 2021, 30, 369–376. [Google Scholar] [CrossRef]
- Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Stat3 Contributes to Indoxyl Sulfate-Induced Inflammatory and Fibrotic Gene Expression and Cellular Senescence. Am. J. Nephrol. 2012, 36, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Mafra, D.; Borges, N.A.; Lindholm, B.; Shiels, P.G.; Evenepoel, P.; Stenvinkel, P. Food as medicine: Targeting the uraemic phenotype in chronic kidney disease. Nat. Rev. Nephrol. 2021, 17, 153–171. [Google Scholar] [CrossRef]
- Romanello, V. The interplay between mitochonodrial morphology and myomitokines in aging sarcopenia. Int. J. Mol. Sci. 2021, 22, 91. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of e3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Cheng, M.L.; Pan, H.C.; Lee, J.H.; Lee, C.C. Protein-bound uremic toxins impaired mitochondrial dynamics and functions. Oncotarget 2017, 8, 77722–77733. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Kramann, R.; Mallau, M.; Makowska, A.; van Roeyen, C.R.; Rong, S.; Buecher, E.B.; Boor, P.; Kovacova, K.; Zok, S.; et al. Mesenchymal stem cells from rats with chronic kidney disease exhibit premature senescence and loss of regenerative potential. PLoS ONE 2014, 9, e92115. [Google Scholar] [CrossRef]
- Yoon, Y.Y.; Lee, J.H.; Yun, C.W.; Lee, S.H. Pioglitazone improves the function of human mesenchymal stem cells in chronic kidney disease patients. Int. J. Mol. Sci. 2019, 20, 2314. [Google Scholar] [CrossRef]
- Han, Y.S.; Kim, S.M.; Lee, J.H.; Jung, S.K.; Noh, H.; Lee, S.H. Melatonin protects chronic kidney disease mesenchymal stem cells against senescence via PrPc-dependent enhancement of the mitochondrial function. J. Pineal Res. 2019, 66, e12535. [Google Scholar] [CrossRef]
- Perlman, A.S.; Chevalier, J.M.; Wilkinson, P.; Liu, H.; Parker, T.; Levine, D.M.; Sloan, B.J.; Gong, A.; Sherman, R.; Farrel, F.X. Serum Inflammatory and Immune Mediators Are Elevated in Early Stage Diabetic Nephropathy. Ann. Clin. Lab. Sci. 2015, 45, 256–263. [Google Scholar]
- Fazzini, F.; Lamina, C.; Raschenberger, J.; Schultheiss, U.T.; Kotsis, F.; Schonherr, S.; Weissensteiner, H.; Forer, L.; Steinbrenner, I.; Meiselbach, H.; et al. GCKD investigators. Results from the German Chronic Kidney Disease (GCKD) study support association of relative telomere length with mortality in a large cohort of patients with moderate chronic kidney disease. Kidney Int. 2020, 98, 488–497. [Google Scholar] [CrossRef]
- Tsirpanlis, G.; Chatzipanagiotou, S.; Boufidou, F.; Kordinas, V.; Alevyzaki, F.; Zoga, M.; Kyritsis, I.; Stamatelou, K.; Triantafyllis, G.; Nicolaou, C. Telomerase Activity Is Decreased in Peripheral Blood Mononuclear Cells of Hemodialysis Patients. Am. J. Nephrol. 2006, 26, 91–96. [Google Scholar] [CrossRef]
- Zhang, R.; Saredy, J.; Shao, Y.; Yao, T.; Liu, L.; Saaoud, F.; Yang, W.Y.; Sun, Y.; Johnson, C.; Drummer, C.; et al. End-stage renal disease is different from chronic kidney disease in upregulating ROS-modulated proinflammatory secretome in PBMCs—a novel multiple-hit model for disease progression. Redx Biol. 2020, 34, 101460. [Google Scholar] [CrossRef]
- Zhang, J.; Rane, G.; Dai, X.; Shanmugam, M.K.; Arfuso, F.; Samy, R.P.; Lai, M.K.P.; Kappei, D.; Kumar, A.P.; Sethi, G. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res. Rev. 2016, 25, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Kooman, J.P.; Kotanko, P.; Schols, A.M.W.J.; Shiels, P.G.; Stenvinkel, P. Chronic kidney disease and premature ageing. Nat. Rev. Nephrol. 2014, 10, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Graboski, A.L.; Redinbo, M.R. Gut-derived protein-bound uremic toxins. Toxins 2020, 12, 590. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Otagiri, M.; Maruyama, T. Update on the pharmacokinetics and redox properties of protein-bound uremic toxins. J. Pharm. Sci. 2011, 100, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Peel, N.M.; Krosch, M.; Hubbard, R.E. Frailty and chronic kidney disease: A systematic review. Arch. Gerontol. Geriatr. 2017, 68, 135–142. [Google Scholar] [CrossRef]
- Lee, S.Y.; Wang, J.; Chao, C.T.; Chien, K.L.; Huang, J.W. Frailty modifies the association between opioid use and mortality in chronic kidney disease patients with diabetes: A population-based cohort study. Aging 2020, 12, 21730–21746. [Google Scholar] [CrossRef]
- Ebert, T.; Pawelzik, S.C.; Witasp, A.; Arefin, S.; Hobson, S.; Kublickiene, K.; Shiels, P.G.; Back, M.; Stenvinkel, P. Inflammation and Premature Ageing in Chronic Kidney Disease. Toxins 2020, 12, 227. [Google Scholar] [CrossRef]
- Wu, P.Y.; Chao, C.T.; Chan, D.C.; Huang, J.W.; Hung, K.Y. Contributors, risk associates, and complications of frailty in patients with chronic kidney disease: A scoping review. Ther. Adv. Chronic Dis. 2019, 10, 2040622319880382. [Google Scholar] [CrossRef] [PubMed]
- Fahal, I.H. Uraemic sarcopenia: Aetiology and implications. Nephrol. Dial. Transplant. 2014, 29, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Tang, C.H.; Cheng, R.W.Y.; Wang, M.Y.H.; Hung, K.Y. Protein-energy wasting significantly increases healthcare utilization and costs among patients with chronic kidney disease: A propensity-score matched cohort study. Curr. Med. Res. Opin. 2017, 33, 1705–1713. [Google Scholar] [CrossRef]
- Rodrigues, G.G.C.; Dellê, H.; Brito, R.B.O.; Cardoso, V.O.; Fernandes, K.P.S.; Mesquita-Ferrari, R.A.; Cunha, R.S.; Stinghen, A.E.M.; Dalboni, M.A.; Barreto, F.C. Indoxyl Sulfate Contributes to Uremic Sarcopenia by Inducing Apoptosis in Myoblasts. Arch. Med. Res. 2020, 51, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Enoki, Y.; Watanabe, H.; Arake, R.; Fujimura, R.; Ishiodori, K.; Imafuku, T.; Nishida, K.; Sugimoto, R.; Nagao, S.; Miyamura, S.; et al. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J. Cachexia Sarcopenia Muscle 2017, 8, 735–747. [Google Scholar] [CrossRef]
- Jheng, J.R.; Chen, Y.S.; Ao, U.I.; Chan, D.C.; Huang, J.W.; Hung, K.Y.; Tarng, D.C.; Chiang, C.K. The double-edged sword of endoplasmic reticulum stress in uremic sarcopenia through myogenesis perturbation. J. Cachexia Sarcopenia Muscle 2018, 9, 570–584. [Google Scholar] [CrossRef]
- Changchien, C.Y.; Lin, Y.H.; Cheng, Y.C.; Chang, H.H.; Peng, Y.S.; Chen, Y. Indoxyl sulfate induces myotube atrophy by ROS-ERK and JNK-MAFbx cascades. Chem. Biol. Interact. 2019, 304, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Saigusa, D.; Mishima, E.; Uchida, T.; Miura, D.; Morikawa-Ichinose, T.; Kisu, K.; Sekimoto, A.; Saito, R.; Oe, Y.; et al. Impact of the Oral Adsorbent AST-120 on Organ-Specific Accumulation of Uremic Toxins: LC-MS/MS and MS Imaging Techniques. Toxins 2017, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, J.; Ueda, S.; Yamagishi, S.I.; Nohara, N.; Nagasawa, H.; Wakabayashi, K.; Matsui, T.; Yuichiro, H.; Kadoguchi, T.; Otsuka, T.; et al. Association of advanced glycation end products with sarcopenia and frailty in chronic kidney disease. Sci. Rep. 2020, 10, 17647. [Google Scholar] [CrossRef] [PubMed]
- Pajek, M.; Jerman, A.; Osredkar, J.; Ponikvar, J.B.; Pajek, J. Association of Uremic Toxins and Inflammatory Markers with Physical Performance in Dialysis Patients. Toxins 2018, 10, 403. [Google Scholar] [CrossRef] [PubMed]
- Saoi, M.; Li, A.; McGlory, C.; Stokes, T.; von Allmen, M.T.; Phillips, S.M.; Britz-McKibbin, P. Metabolic Perturbations from Step Reduction in Older Persons at Risk for Sarcopenia: Plasma Biomarkers of Abrupt Changes in Physical Activity. Metabolites 2019, 9, 134. [Google Scholar] [CrossRef]
- Caldiroli, L.; Armelloni, S.; Eskander, A.; Messa, P.; Rizzo, V.; Margiotta, E.; Cesari, M.; Vettoretti, S. Association between the uremic toxins indoxyl-sulfate and p-cresyl-sulfate with sarcopenia and malnutrition in elderly patients with advanced chronic kidney disease. Exp. Gerontol. 2021, 147, 111266. [Google Scholar] [CrossRef]
- Kelaiditi, E.; Cesari, M.; Canevelli, M.; Abellan van Kan, G.; Ousset, P.J.; Gillette-Guyonnet, S.; Ritz, P.; Duveau, F.; Soto, M.E.; Provencher, V.; et al. Cognitive frailty: Rational and definition from an (I.A.N.A./I.A.G.G.) International Consensus Group. J. Nutr. Health Aging 2013, 17, 726–734. [Google Scholar] [CrossRef]
- Bu, Z.; Huang, A.; Xue, M.; Li, Q.; Bai, Y.; Xu, G. Cognitive frailty as a predictor of adverse outcomes among older adults: A systematic review and meta-analysis. Brain Behav. 2021, 11, e01926. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Wu, P.H.; Tsai, Y.C.; Hsu, Y.L.; Wang, H.Y.; Kuo, M.C.; Kuo, P.L.; Hwang, S.J. Indoxyl Sulfate Induces Apoptosis Through Oxidative Stress and Mitogen-Activated Protein Kinase Signaling Pathway Inhibition in Human Astrocytes. J. Clin. Med. 2019, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Karbowska, M.; Hermanowicz, J.M.; Tankiewicz-Kwedlo, A.; Kalaska, B.; Kaminski, T.W.; Nosek, K.; Wisniewska, R.J.; Pawlak, D. Neurobehavioral effects of uremic toxin-indoxyl sulfate in the rat model. Sci. Rep. 2020, 10, 9483. [Google Scholar] [CrossRef]
- Sun, C.Y.; Li, J.R.; Wang, Y.Y.; Lin, S.Y.; Ou, Y.C.; Lin, C.J.; Wang, J.D.; Liao, S.L.; Chen, C.J. Indoxyl sulfate caused behavioral abnormality and neurodegeneration in mie with unilateral nephrectomy. Aging 2021, 13, 6681–6701. [Google Scholar] [CrossRef]
- Sun, C.Y.; Li, J.R.; Wang, Y.Y.; Lin, S.Y.; Ou, Y.C.; Lin, C.J.; Wang, J.D.; Liao, S.L.; Chen, C.J. p-Cresol Sulfate Caused Behavior Disorders and Neurodegeneration in Mice with Unilateral Nephrectomy Involving Oxidative Stress and Neuroinflammation. Int. J. Mol. Sci. 2020, 21, 6687. [Google Scholar] [CrossRef]
- Bobot, M.; Thomas, L.; Moyon, A.; Fernandez, S.; McKay, N.; Balasse, L.; Garrigue, P.; Brige, P.; Chopinet, S.; Poitevin, S.; et al. Uremic Toxic Blood-Brain Barrier Disruption Mediated by AhR Activation Leads to Cognitive Impairment during Experimental Renal Dysfunction. J. Am. Soc. Nephrol. 2020, 31, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Huang, M.F.; Liang, S.S.; Hwang, S.J.; Tsai, J.C.; Liu, T.L.; Wu, P.H.; Yang, Y.H.; Kuo, K.C.; Kuo, M.C.; et al. Indoxyl sulfate, not p-cresyl sulfate, is associated with cognitive impairment in early-stage chronic kidney disease. Neurotoxicology 2016, 53, 148–152. [Google Scholar] [CrossRef]
- Lin, Y.T.; Wu, P.H.; Lee, H.H.; Mubanga, M.; Chen, C.S.; Kuo, M.C.; Chiu, Y.W.; Kuo, P.L.; Hwang, S.J. Indole-3 acetic acid increased risk of impaired cognitive function in patients receiving hemodialysis. Neurotoxicology 2019, 73, 85–91. [Google Scholar] [CrossRef]
- Lee, S.Y.; Wang, J.; Chao, C.T.; Chien, K.L.; Huang, J.W. Frailty is associated with a higher risk of developing delirium and cognitive impairment among patients with diabetic kidney disease: A longitudinal population-based cohort study. Diabetic Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mair, R.D.; Nguyen, H.; Huang, T.T.; Plummer, N.S.; Sirich, T.L.; Meyer, T.W. Accumulation of uremic solutes in the cerebrospinal fluid in experimental acute renal failure. Am. J. Physiol. Renal Physiol. 2019, 317, F296–F302. [Google Scholar] [CrossRef]
- Watanabe, K.; Sato, E.; Mishima, E.; Watanabe, M.; Abe, T.; Takahashi, N.; Nakayama, M. Effect of uremic toxins on hippocampal cell damage: Analysis in vitro and in rat model of chronic kidney disease. Heliyon 2021, 7, e06221. [Google Scholar] [CrossRef]
- Assem, M.; Lando, M.; Grissi, M.; Kamel, S.; Massy, Z.A.; Chillon, J.M.; Henaut, L. The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders. Toxins 2018, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Hypercoagulability is independently associated with kynurenine pathway activation in dialysed uraemic patients. Thromb. Haemost. 2009, 102, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.B.; Chang, C.C.; Li, L.C.; Lee, W.C.; Lin, C.N.; Li, S.C.; Moi, S.H.; Yang, C.H. Mutual Interaction of Clinical Factors and Specific microRNAs to Predict Mild Cognitive Impairment in Patients Receiving Hemodialysis. Cells 2020, 9, 2303. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Watanabe, T.; Nakayama, M. Cerebro-renal interactions: Impact of uremic toxins on cognitive function. Neurotoxicology 2014, 44, 184–193. [Google Scholar] [CrossRef]
- Li, Y.; Pi, H.C.; Yang, Z.K.; Dong, J. Associations between small and middle molecules clearance and the change of cognitive function in peritoneal dialysis. J. Nephrol. 2020, 33, 839–848. [Google Scholar] [CrossRef]
- Evenepoel, P.; Cunningham, J.; Ferrari, S.; Haarhaus, M.; Javaid, M.K.; Lafage-Proust, M.H.; Prieto-Alhambra, D.; Torres, P.U.; Cannata-Andia, J.; EUROD Workgroup. European Consensus Statement on the diagnosis and management of osteoporosis in chronic kidney disease stages G4–G5D. Nephrol. Dial. Transplant. 2021, 36, 42–59. [Google Scholar] [CrossRef]
- Kazama, J.J.; Iwasaki, Y.; Fukagawa, M. Uremic osteoporosis. Kidney Int. Suppl. 2013, 3, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Iwasaki, Y.; Yamato, H.; Mori, Y.; Komaba, H.; Watanabe, H.; Maruyama, T.; Fukagawa, M. p-Cresyl sulfate induces osteoblast dysfunction through activating JNK and p38 MAPK pathways. Bone 2013, 56, 347–354. [Google Scholar] [CrossRef]
- Liu, W.C.; Shyu, J.F.; Lin, Y.F.; Chiu, H.W.; Lim, P.S.; Lu, C.L.; Zheng, C.M.; Hou, Y.C.; Chen, P.H.; Lu, K.C. Resveratrol Rescue Indoxyl Sulfate-Induced Deterioration of Osteoblastogenesis via the Aryl Hydrocarbon Receptor /MAPK Pathway. Int. J. Mol. Sci. 2020, 21, 7483. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tominari, T.; Hirata, M.; Matsumoto, C.; Hirata, J.; Murphy, G.; Nagase, H.; Miyaura, C.; Inada, M. Indoxyl sulfate, a uremic toxin in chronic kidney disease, suppresses both bone formation and bone resorption. FEBS Open Bio. 2017, 7, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kwak, K.A.; Gil, H.W.; Song, H.Y.; Hong, S.Y. Indoxyl sulfate promotes apoptosis in cultured osteoblast cells. BMC Pharmacol. Toxicol. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Canziani, M.E.F.; Tomiyama, C.; Higa, A.; Mozar, A.; Glorieux, G.; Vanholder, R.; Massy, Z.; de Carvalho, A.B. Association between indoxyl sulfate and bone histomorphometry in pre-dialysis chronic kidney disease patients. J. Bras. Nefrol. 2014, 36, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Lanza, D.; Perna, A.F.; Oliva, A.; Vanholder, R.; Pletinck, A.; Guastafierro, S.; Di Nunzio, A.; Vigorito, C.; Capasso, G.; Jankowski, V.; et al. Impact of the uremic milieu on the osteogenic potential of mesenchymal stem cells. PLoS ONE 2015, 10, e0116468. [Google Scholar]
- Yano, S.; Yamaguchi, T.; Kanazawa, I.; Ogawa, N.; Hayashi, K.; Yamauchi, M.; Sugimoto, T. The uraemic toxin phenylacetic acid inhibits osteoblastic proliferation and differentiation: An implication for the pathogenesis of low turnover bone in chronic renal failure. Nephrol. Dial. Transplant. 2007, 22, 3160–3165. [Google Scholar] [CrossRef]
- Peng, Y.S.; Ding, H.C.; Lin, Y.T.; Syu, J.P.; Chen, Y.; Wang, S.M. Uremic toxin p-cresol induces disassembly of gap junctions of cardiomyocytes. Toxicology 2012, 302, 11–17. [Google Scholar] [CrossRef]
- Niwa, T. Role of Indoxyl Sulfate in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Experimental and Clinical Effects of Oral Sorbent AST-120. Ther. Apher. Dial. 2011, 15, 120–124. [Google Scholar] [CrossRef]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef]
- Chen, J.; Gu, Y.; Zhang, H.; Ning, Y.; Song, N.; Hu, J.; Cai, J.; Shi, Y.; Ding, X.; Zhang, X. Amelioration of Uremic Toxin Indoxyl Sulfate-Induced Osteoblastic Calcification by SET Domain Containing Lysine Methyltransferase 7/9 Protein. Nephron 2019, 141, 287–294. [Google Scholar] [CrossRef]
- He, X.; Jiang, H.; Gao, F.; Liang, S.; Wei, M.; Chen, L. Indoxyl sulfate-induced calcification of vascular smooth muscle cells via the PI3K/Akt/NF-κB signaling pathway. Microsc. Res. Tech. 2019, 82, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yisireyili, M.; Goto, S.; Kato, K.; Cheng, X.W.; Nakayama, T.; Matsushita, T.; Niwa, T.; Murohara, T.; Takeshita, K. Indoxyl Sulfate-induced Vascular Calcification is mediated through Altered Notch Signaling Pathway in Vascular Smooth Muscle Cells. Int. J. Med. Sci. 2020, 17, 2703–2717. [Google Scholar] [CrossRef]
- Hou, Y.C.; Lu, C.L.; Yuan, T.H.; Liao, M.T.; Chao, C.T.; Lu, K.C. The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective. Int. J. Mol. Sci. 2020, 21, 980. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Yeh, H.Y.; Yuan, T.H.; Chiang, C.K.; Chen, H.W. MicroRNA-125b in vascular diseases: An updated systematic review of pathogenetic implications and clinical applications. J. Cell. Mol. Med. 2019, 23, 5884–5894. [Google Scholar] [CrossRef]
- Chao, C.T.; Liu, Y.P.; Su, S.F.; Yeh, H.Y.; Chen, H.Y.; Lee, P.J.; Chen, W.J.; Lee, Y.M.; Huang, J.W.; Chiang, C.K.; et al. Circulating MicroRNA-125b Predicts the Presence and Progression of Uremic Vascular Calcification. Arterioscler Thromb. Vasc. Biol. 2017, 37, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Yeh, H.Y.; Tsai, Y.T.; Chiang, C.K.; Chen, H.W. A combined microRNA and target protein-based panel for predicting the probability and severity of uraemic vascular calcification: A translational study. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Chao, C.T.; Yeh, H.Y.; Tsai, Y.T.; Chuang, P.H.; Yuan, T.H.; Huang, J.W.; Chen, H.W. Natural and non-natural antioxidative compounds: Potential candidates for treatment of vascular calcification. Cell Death Discov. 2019, 5, 145. [Google Scholar] [CrossRef]
- Obokata, M.; Kurosawa, K.; Ishida, H.; Ito, K.; Ogawa, T.; Ando, Y.; Kurabayashi, M.; Negishi, K. Echocardiography-based pressure–volume loop assessment in the evaluation for the effects of indoxyl sulfate on cardiovascular function. J. Echocardiogr. 2019, 17, 35–43. [Google Scholar] [CrossRef]
- Chao, C.T.; Yeh, H.Y.; Tsai, Y.T.; Yuan, T.H.; Liao, M.T.; Huang, J.W.; Chen, H.W. Astaxanthin Counteracts Vascular Calcification In Vitro Through an Early Up-Regulation of SOD2 Based on a Transcriptomic Approach. Int. J. Mol. Sci. 2020, 21, 8530. [Google Scholar] [CrossRef]
- Zhou, C.; Shi, Z.; Ouyang, N.; Ruan, X. Hyperphosphatemia and Cardiovascular Disease. Front. Cell. Dev. Biol. 2021, 9, 644363. [Google Scholar] [CrossRef]
- Belmokhtar, K.; Ortillon, J.; Jaisson, S.; Massy, Z.A.; Boulagnon Rombi, C.; Doué, M.; Maurice, P.; Fritz, G.; Gillery, P.; Schmidt, A.M.; et al. Receptor for advanced glycation end products: A key molecule in the genesis of chronic kidney disease vascular calcification and a potential modulator of sodium phosphate co-transporter PIT-1 expression. Nephrol. Dial. Transplant. 2019, 34, 2018–2030. [Google Scholar] [CrossRef] [PubMed]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, J.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol. Dial. Transplant. 2018, 33, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Kuczera, P.; Adamczak, M.; Wiecek, A. Fibroblast Growth Factor-23-A Potential Uremic Toxin. Toxins 2016, 8, 369. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, N.; Sagata, M.; Saigo, C.; Yoneda, G.; Yamamoto, Y.; Nomura, Y.; Nishi, K.; Fujino, R.; Jono, H.; Saito, H. Indoxyl Sulfate as a Mediator Involved in Dysregulation of Pulmonary Aquaporin-5 in Acute Lung Injury Caused by Acute Kidney Injury. Int. J. Mol. Sci. 2016, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Gunta, S.S.; Mak, R.H. Ghrelin and leptin pathophysiology in chronic kidney disease. Pediatr. Nephrol. 2013, 28, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Yeh, H.Y.; Han, D.S.; Huang, J.W.; Huang, K.C. Determinants of circulating microRNA-125b, a risk factor of vascular calcification, among community-dwelling older adults. Clin. Transl. Med. 2020, 10, e145. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Han, D.S.; Huang, J.W. Circulating microRNA-125b levels are associated with the risk of vascular calcification in healthy community-dwelling older adults. Front. Cardiovasc. Med. 2021, 8, 624313. [Google Scholar] [CrossRef]
- Mahmassani, Z.S.; McKenzie, A.I.; Petrocelli, J.J.; de Hart, N.M.; Reidy, P.T.; Fix, D.K.; Ferrara, P.J.; Funai, K.; Drummond, M.J. Short-term metformin ingestion by healthy older adults improves myoblast function. Am. J. Physiol. Cell. Physiol. 2021, 320, C566–C576. [Google Scholar] [CrossRef]
- Chao, C.T.; Wang, J.; Chien, K.L.; Cohort of GEriatric Nephrology in NTUH (COGENT) Study Group. Both pre-frailty and frailty increase healthcare utilization and adverse health outcomes in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2018, 17, 130. [Google Scholar] [CrossRef]
- Chao, C.T.; Huang, J.W.; Chiang, C.K.; Hung, K.Y.; Cohort of GEriatric Nephrology in NTUH (COGENT) Study Group. Applicability of laboratory deficit-based frailty index in predominantly older patients with end-stage renal disease under chronic dialysis: A pilot test of its correlation with survival and self-reported instruments. Nephrology 2020, 25, 73–81. [Google Scholar] [CrossRef]
- Walston, J.; Buta, B.; Xue, Q.L. Frailty screening and interventions: Considerations for clinical practice. Clin. Geriatr. Med. 2018, 34, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Madero, M.; Cano, K.B.; Campos, I.; Tao, X.; Maheshwari, V.; Brown, J.; Cornejo, B.; Handelman, G.; Thijssen, S.; Kotanko, P. Removal of Protein-Bound Uremic Toxins during Hemodialysis Using a Binding Competitor. Clin. J. Am. Soc. Nephrol. 2019, 14, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Lobel, L.; Cao, Y.G.; Fenn, K.; Glickman, J.N.; Garrett, W.S. Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science 2020, 369, 1518. [Google Scholar] [CrossRef]
- Sirich, T.L.; Fong, K.; Larive, B.; Beck, G.J.; Chertow, G.M.; Levin, N.W.; Kliger, A.S.; Plummer, N.S.; Meyer, T.W.; Frequent Hemodialysis Network (FHN) Rrial Group. Limited reduction in uremic solute concentrations with increased dialysis frequency and time in the Frequent Hemdialysis Network Daily Trial. Kidney Int. 2017, 91, 1186–1192. [Google Scholar] [CrossRef] [PubMed]
- Reis, T.; Martino, F.; Dias, P.; de Freitas, G.R.R.; da Silva Filho, E.R.; de Azevedo, M.L.C.; Reis, F.; Cozzolino, M.; Rizo-Topete, L.; Ronco, C. Removal of middle molecules with medium cutoff dialyzer in patients on short frequent hemodialysis. Hemodial. Int. 2021, 25, 180–187. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Category | Subtype | Species | Adverse Effects | ||||
|---|---|---|---|---|---|---|---|
| Senescence | Inflammation | OS | Mt Dysfunction | SC Damages | |||
| Free, water soluble, LMW molecules | Reactive carbonyl group | 2-Hexenal | (+) | (+) | (+) | ||
| Hexanal | (+) | (+) | (+) | ||||
| 2-Nonenal | (+) | (+) | (+) | (+) | |||
| Nonanal | (+) | (+) | |||||
| 2-Octenal | (+) | (+) | (+) | ||||
| 4-OH-Hexenal | (+) | (+) | |||||
| 4-OH-Nonenal | (+) | (+) | (+) | ||||
| Malondialdehyde | (+) | (+) | (+) | ||||
| Nicotinamide | 4-Pyridone-3-carboxamide-1β-ribonucleoside-triphosphate (4PYTP) | (+) | (+) | ||||
| 4-Pyridone-3-carboxamide-1β-ribonucleoside-monophosphate (4PYMP) | (+) | (+) | |||||
| Purine | 8-hydroxy-2’-deoxyguanosine | (+) | (+) | (+) | (+) | (+) | |
| Hypoxanthine | (+) | (+) | (+) | ||||
| Neopterin | (+) | (+) | (+) | (+) | |||
| Uric acid | (+) | (+) | (+) | (+) | (+) | ||
| Guanidine | α-Keto-δ-guanidinovaleric acid | (+) | |||||
| Guanidinoacetic acid | (+) | (+) | (+) | ||||
| Guanidinosuccinic acid | (+) | (+) | |||||
| Methylguanidine | (+) | (+) | (+) | (+) | |||
| Amine | ADMA | (+) | (+) | (+) | (+) | (+) | |
| Dimethylamine | (+) | (+) | (+) | ||||
| Monomethylamine | (+) | (+) | (+) | ||||
| Middle molecules | Protein | α1-acid glycoprotein | (+) | ||||
| α1-microglobulin | (+) | (+) | |||||
| β2-microglobulin | (+) | (+) | (+) | ||||
| Adiponectin | (+) | (+) | (+) | (+) | |||
| Complement factor D | (+) | (+) | (+) | ||||
| FGF-23 | (+) | (+) | (+) | (+) | |||
| Leptin | (+) | (+) | (+) | (+) | |||
| Parathyroid hormone | (+) | (+) | (+) | ||||
| Retinol binding protein | (+) | (+) | (+) | ||||
| Soluble intracellular adhesion molecule-1 | (+ | (+) | (+) | ||||
| Cytokine | Interleukin-6 | (+) | (+) | (+) | (+) | (+) | |
| Interleukin-8 | (+) | (+) | (+) | (+) | (+) | ||
| Resistin | (+) | (+) | (+) | (+) | (+) | ||
| Tumor necrosis factor-α | (+) | (+) | (+) | (+) | (+) | ||
| Protein-bound molecules | Reactive carbonyl group | Acrolein | (+) | (+) | (+) | (+) | (+) |
| AGE | Carboxymethyllysine | (+) | (+) | (+) | (+) | (+) | |
| Pentosidine | (+) | (+) | (+) | ||||
| Hippurate | Hippuric acid | (+) | (+) | ||||
| Amino acid | Homocysteine | (+) | (+) | (+) | (+) | (+) | |
| Indole | Indoxyl sulfate | (+) | (+) | (+) | (+) | (+) | |
| Indole-3-acetic acid | (+) | (+) | (+) | (+) | |||
| Kynurenic acid | (+) | (+) | (+) | (+) | |||
| Phenol | p-Cresylsulfate | (+) | (+) | (+) | (+) | (+) | |
| Polyamine | Spermidine | (+) | (+) | ||||
| Toxin Types | UT Species | Cell Type Involved | Molecular Mediators |
|---|---|---|---|
| Protein-bound | Indoxyl sulfate | Muscle/myoblasts | PGC-1α downregulation |
| Muscle/myoblasts | myoD, myoG, MHC downregulation | ||
| Muscle/myoblasts | MAPK phosphorylation increase, atrogin-1 upregulation | ||
| Brain/astrocytes, glial cells | Nrf2, HO-1, NADPH dehydrogenase quinone 1 down-regulation | ||
| Brain/astrocytes, glial cells | AhR, NF-κB upregulation | ||
| Brain/astrocytes | MAPK, c-Jun, p38 downregulation | ||
| Brain/neural stem cells | BDNF down regulation | ||
| Bone/MSCs | MAPK downregulation but AhR activation | ||
| Bone/osteoblasts | Osterix, osteocalcin, BMP2 downregulation | ||
| Bone/osteoclasts | RANKL downregulation | ||
| Vessel/VSMCs | Prelamin A, JNK, PiT-1, SET7/9, PI3K/Akt/NF-κB, Notch upregulation | ||
| Lung/pneumocytes | AQP5 upregulation | ||
| p-cresol | Bone/osteoblasts | JNK/p38 activation | |
| Heart/cardiomyocytes | PKCα activation, Cx43-related gap junction disintegration | ||
| Small molecular | Methylglyoxal | Brain/neurons | ROS activation |
| Phenylacetic acid | Bone/osteoblasts | PTH response impairment | |
| Phosphate | Vessel/VSMCs | PiT-1, PiT-2, NF-κB, Wnt/β-catenin | |
| Large molecular | TNF-α | Vessel/VSMCs | ERK/AP1/c-Fos upregulation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, C.-T.; Lin, S.-H. Uremic Toxins and Frailty in Patients with Chronic Kidney Disease: A Molecular Insight. Int. J. Mol. Sci. 2021, 22, 6270. https://doi.org/10.3390/ijms22126270
Chao C-T, Lin S-H. Uremic Toxins and Frailty in Patients with Chronic Kidney Disease: A Molecular Insight. International Journal of Molecular Sciences. 2021; 22(12):6270. https://doi.org/10.3390/ijms22126270
Chicago/Turabian StyleChao, Chia-Ter, and Shih-Hua Lin. 2021. "Uremic Toxins and Frailty in Patients with Chronic Kidney Disease: A Molecular Insight" International Journal of Molecular Sciences 22, no. 12: 6270. https://doi.org/10.3390/ijms22126270
APA StyleChao, C.-T., & Lin, S.-H. (2021). Uremic Toxins and Frailty in Patients with Chronic Kidney Disease: A Molecular Insight. International Journal of Molecular Sciences, 22(12), 6270. https://doi.org/10.3390/ijms22126270