
Cortactin Is Required for Efficient FAK, Src and Abl Tyrosine Kinase Activation and Phosphorylation of Helicobacter pylori CagA


 ,
,  ,
,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
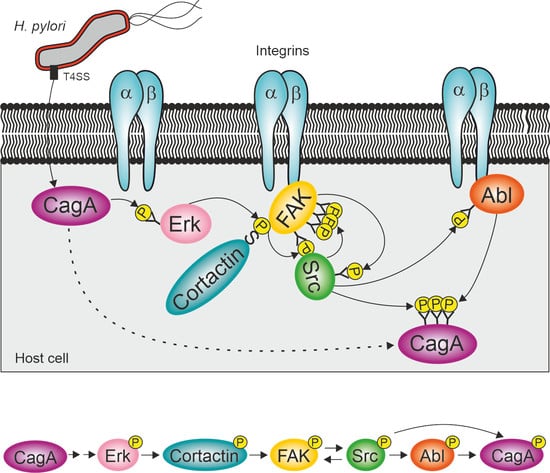
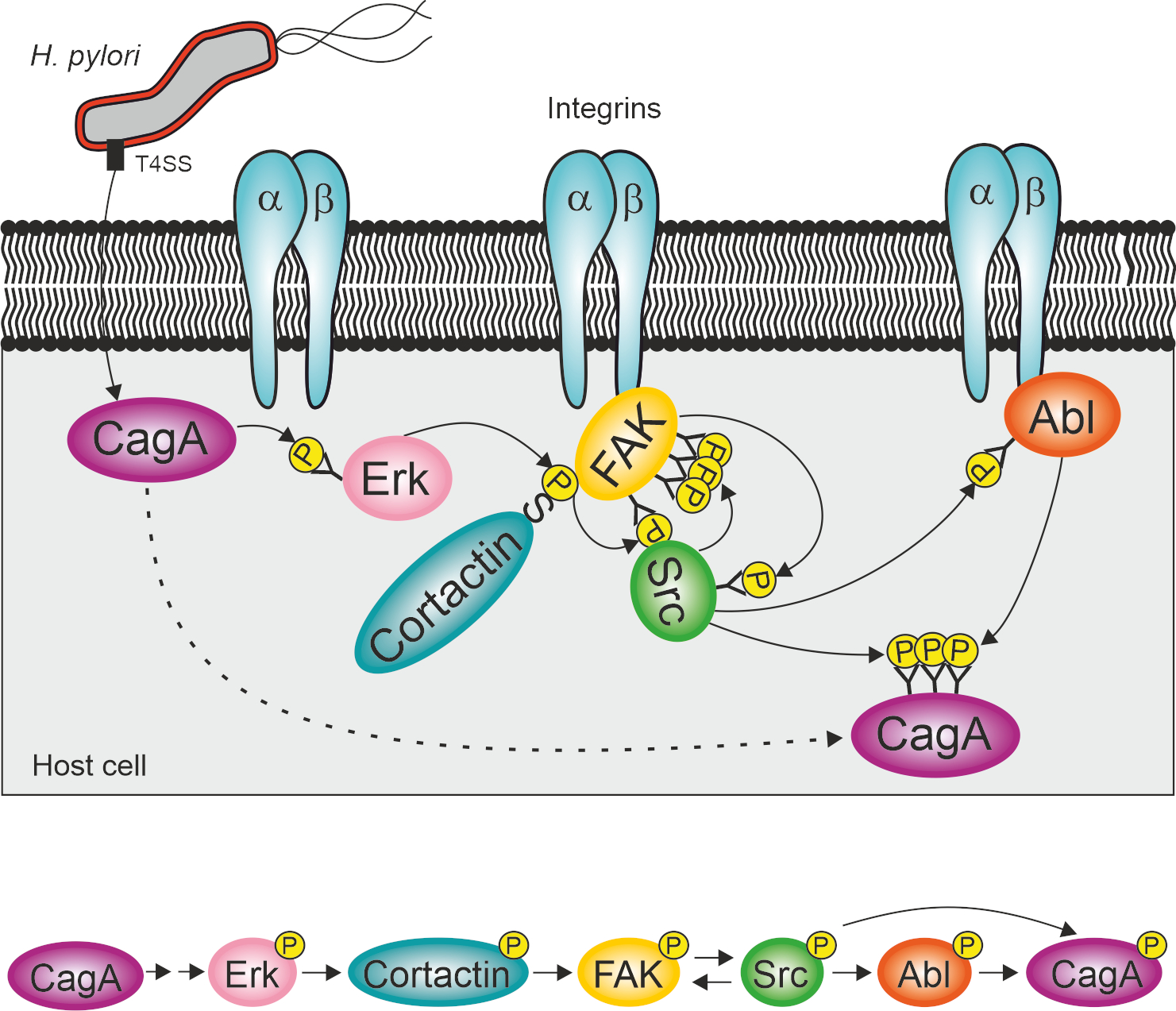
1. Introduction
2. Results
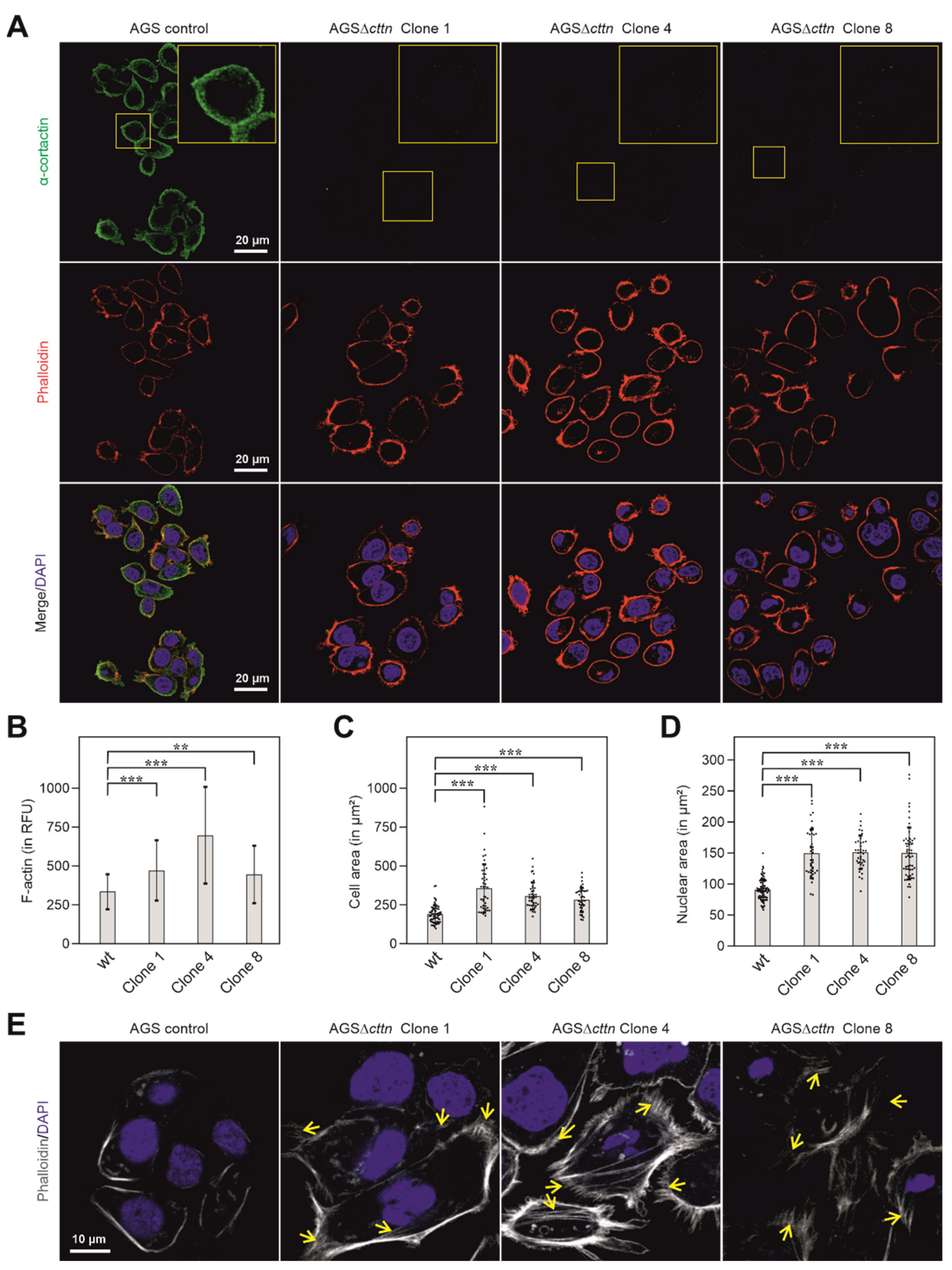
2.1. Cortactin Knockout Does Not Affect Expression of Other Cytoskeletal Proteins, but Increases Actin Stress Fiber Formation and Cell Area
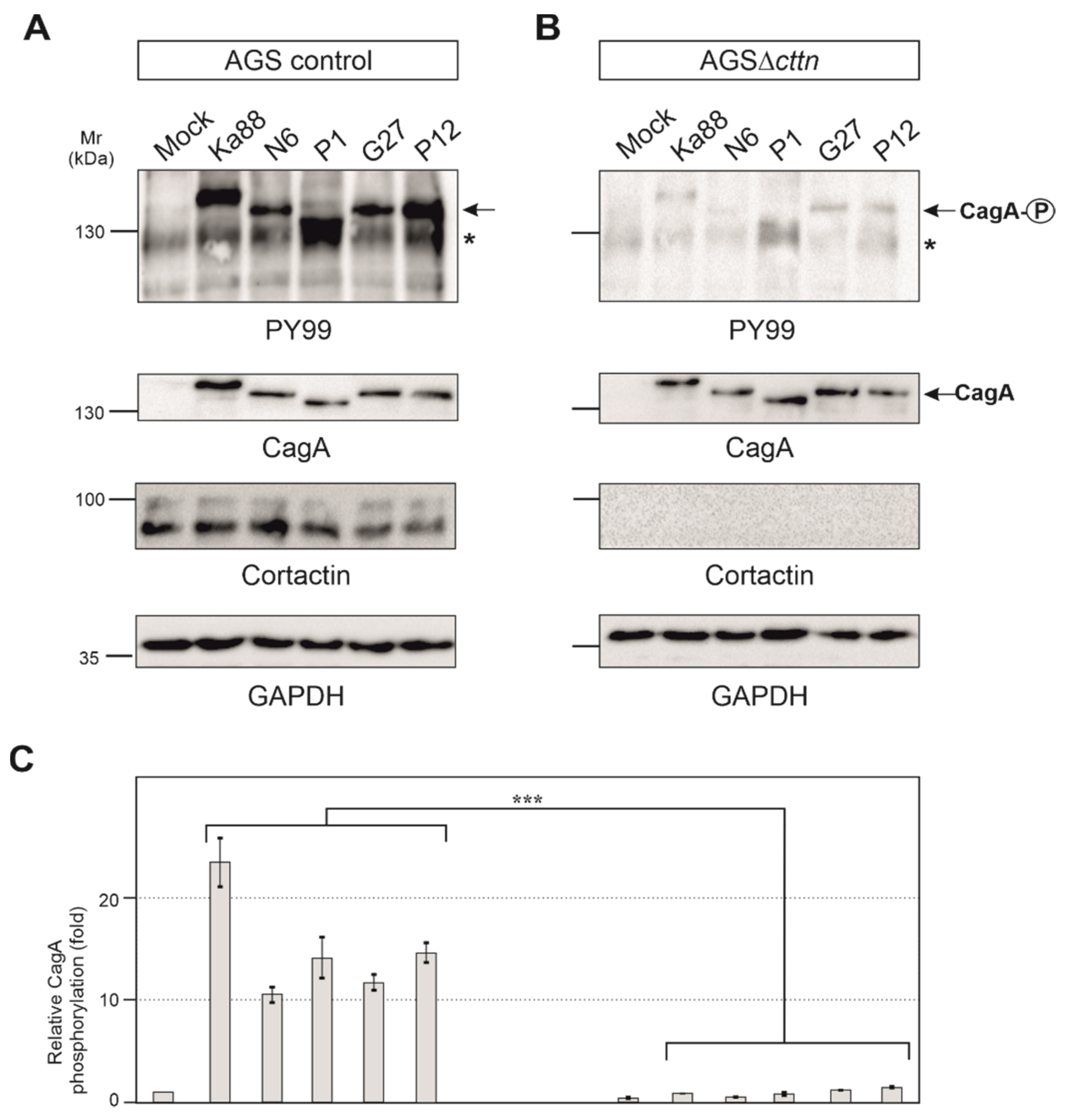
2.2. Efficient Phosphorylation of CagA Is Diminished in AGSΔcttn Cells
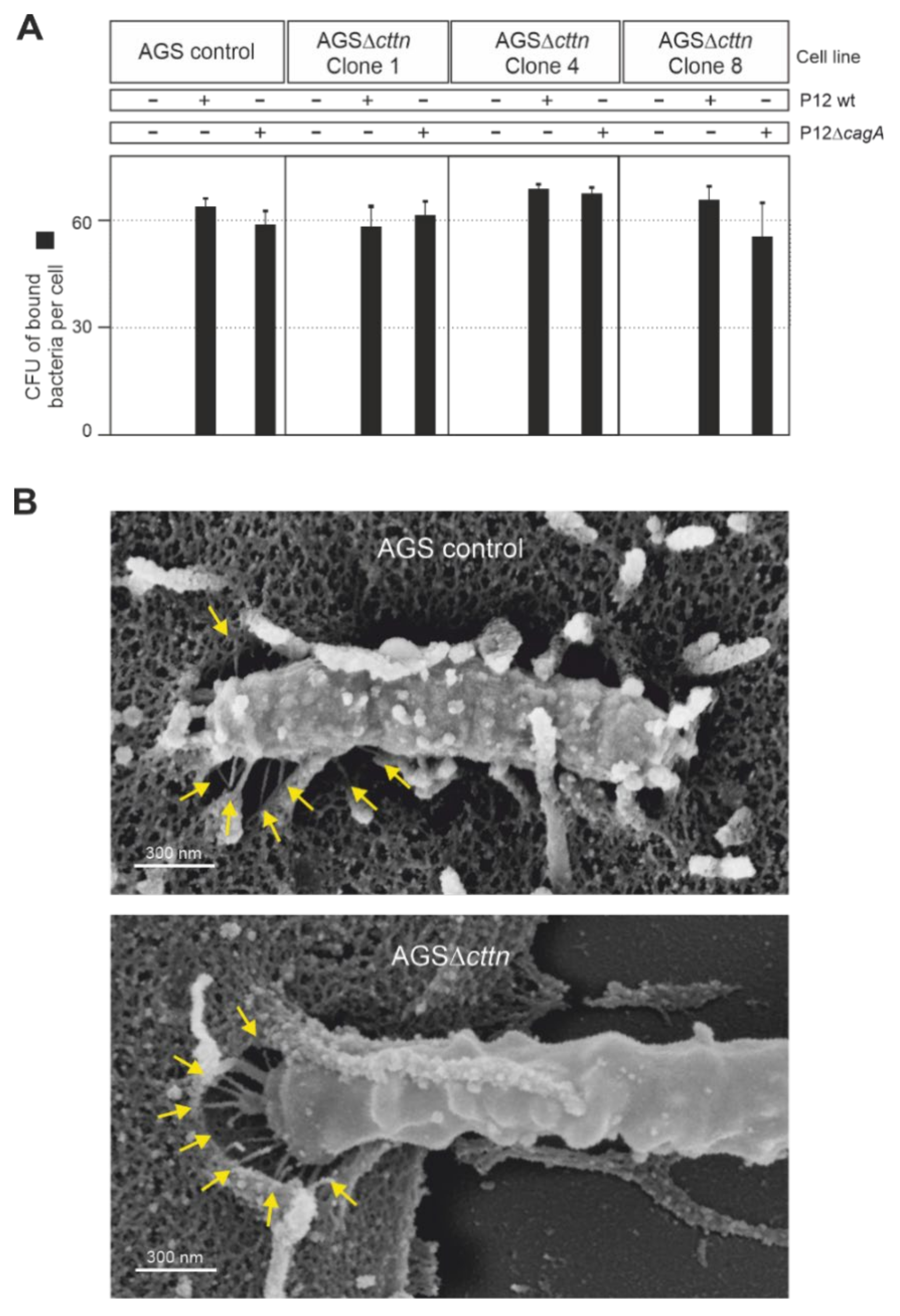
2.3. Bacterial Binding to Cells and T4SS Pilus Formation Are Not Affected by Cortactin Knockout
2.4. Cortactin Is Necessary for Effective Activation of the Kinases FAK, Src and Abl by H. pylori
2.5. Constitutively Active FAK, Src or Abl Rescue CagA Phosphorylation in AGSΔcttn Cells
3. Discussion
4. Materials and Methods
4.1. Cultivation of Eukaryotic Cells
4.2. Generation of Cortactin Knockout Cell Lines
4.3. Cultivation of H. pylori Strains
4.4. Infection of AGS wt and AGSΔcttn Cells with H. pylori
4.5. SDS-PAGE and Immunoblot Analysis
4.6. Immunofluorescence Microscopy
4.7. Electron Microscopy
4.8. Transfection of AGSΔcttn Cells
4.9. Counting of Adherent Bacteria
4.10. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kusters, J.G.; van Vliet, A.H.M.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Salama, N.R.; Hartung, M.L.; Muller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.; Peek, R.M. Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis—A multistep and multifactorial process—1st American cancer society award lecture on cancer epidemiology and prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- Yamaoka, Y.; Graham, D.Y. Helicobacter pylori virulence and cancer pathogenesis. Future Oncol. 2014, 10, 1487–1500. [Google Scholar] [CrossRef]
- McClain, M.S.; Beckett, A.C.; Cover, T.L. Helicobacter pylori vacuolating toxin and gastric cancer. Toxins 2017, 9, 316. [Google Scholar] [CrossRef]
- Hatakeyama, M. Malignant Helicobacter pylori-associated diseases: Gastric cancer and MALT lymphoma. Adv. Exp. Med. Biol. 2019, 1149, 135–149. [Google Scholar] [CrossRef]
- Posselt, G.; Backert, S.; Wessler, S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun. Signal. 2013, 11. [Google Scholar] [CrossRef]
- Schreiber, S.; Konradt, M.; Groll, C.; Scheid, P.; Hanauer, G.; Werling, H.O.; Josenhans, C.; Suerbaum, S. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc. Natl. Acad. Sci. USA 2004, 101, 5024–5029. [Google Scholar] [CrossRef]
- Huang, J.Y.; Sweeney, E.G.; Guillemin, K.; Amieva, M.R. Multiple acid sensors control Helicobacter pylori colonization of the stomach. PLoS Pathog. 2017, 13, e1006118. [Google Scholar] [CrossRef]
- Mobley, H.L.T. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 1996, 10, 57–64. [Google Scholar] [CrossRef]
- Aspholm, M.; Kalia, A.; Ruhl, S.; Schedin, S.; Arnqvist, A.; Linden, S.; Sjostrom, R.; Gerhard, M.; Semino-Mora, C.; Dubois, A.; et al. Helicobacter pylori adhesion to carbohydrates. In Functional Glycomics; Methods in Enzymology; Fukuda, M., Ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2006; Volume 417, pp. 293–339. [Google Scholar]
- Moonens, K.; Hamway, Y.; Neddermann, M.; Reschke, M.; Tegtmeyer, N.; Kruse, T.; Kammerer, R.; Mejías-Luque, R.; Singer, B.B.; Backert, S.; et al. Helicobacter pylori adhesin HopQ disrupts trans dimerization in human CEACAMs. EMBO J. 2018, 37, e98665. [Google Scholar] [CrossRef]
- Pfannkuch, L.; Hurwitz, R.; Traulsen, J.; Sigulla, J.; Poeschke, M.; Matzner, L.; Kosma, P.; Schmid, M.; Meyer, T.F. ADP heptose, a novel pathogen-associated molecular pattern identified in Helicobacter pylori. FASEB J. 2019, 33, 9087–9099. [Google Scholar] [CrossRef]
- Hoy, B.; Lower, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schroder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef]
- Harrer, A.; Boehm, M.; Backert, S.; Tegtmeyer, N. Overexpression of serine protease HtrA enhances disruption of adherens junctions, paracellular transmigration and type IV secretion of CagA by Helicobacter pylori. Gut Pathog. 2017, 9. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Wessler, S.; Necchi, V.; Rohde, M.; Harrer, A.; Rau, T.T.; Asche, C.I.; Boehm, M.; Loessner, H.; Figueiredo, C.; et al. Helicobacter pylori Employs a unique basolateral type IV secretion mechanism for CagA delivery. Cell Host Microbe 2017, 22, 552–560. [Google Scholar] [CrossRef]
- Censini, S.; Lange, C.; Xiang, Z.Y.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef]
- Odenbreit, S.; Puls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000, 287, 1497–1500. [Google Scholar] [CrossRef]
- Backert, S.; Haas, R.; Gerhard, M.; Naumann, M. The Helicobacter pylori type IV secretion system encoded by the cag pathogenicity island: Architecture, function, and signaling. Curr. Top. Microbiol. Immunol 2017, 413, 187–220. [Google Scholar] [CrossRef]
- Tammer, I.; Brandt, S.; Hartig, R.; Konig, W.; Backert, S. Activation of Abl by Helicobacter pylori: A novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 2007, 132, 1309–1319. [Google Scholar] [CrossRef]
- Poppe, M.; Feller, S.M.; Romer, G.; Wessler, S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 2007, 26, 3462–3472. [Google Scholar] [CrossRef]
- Mueller, D.; Tegtmeyer, N.; Brandt, S.; Yamaoka, Y.; De Poire, E.; Sgouras, D.; Wessler, S.; Torres, J.; Smolka, A.; Backert, S. C-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J. Clin. Investig. 2012, 122, 1553–1566. [Google Scholar] [CrossRef]
- Selbach, M.; Paul, F.E.; Brandt, S.; Guye, P.; Daumke, O.; Backert, S.; Dehio, C.; Mann, M. Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe 2009, 5, 397–403. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Neddermann, M.; Asche, C.I.; Backert, S. Subversion of host kinases: A key network in cellular signaling hijacked by Helicobacter pylori CagA. Mol. Microbiol. 2017, 105, 358–372. [Google Scholar] [CrossRef]
- Touati, E. When bacteria become mutagenic and carcinogenic: Lessons from H. pylori. Mutat. Res. 2010, 703, 66–70. [Google Scholar] [CrossRef]
- Knorr, J.; Ricci, V.; Hatakeyama, M.; Backert, S. Classification of Helicobacter pylori virulence factors: Is CagA a toxin or not? Trends Microbiol. 2019, 27, 731–738. [Google Scholar] [CrossRef]
- Sharafutdinov, I.; Backert, S.; Tegtmeyer, N. Cortactin: A major cellular target of the gastric carcinogen Helicobacter pylori. Cancers 2020, 12, 159. [Google Scholar] [CrossRef]
- Wu, H.; Parsons, J.T. Cortactin, an 80/85-kilodalton pp60(src) substrate, is a filamentous actin-binding protein enriched in the cell cortex. J. Cell Biol. 1993, 120, 1417–1426. [Google Scholar] [CrossRef]
- Gallet, C.; Rosa, J.P.; Habib, A.; Lebret, M.; Levy-Toldano, S.; Maclouf, J. Tyrosine phosphorylation of cortactin associated with Syk accompanies thromboxane analogue-induced platelet shape change. J. Biol. Chem. 1999, 274, 23610–23616. [Google Scholar] [CrossRef]
- Uruno, T.; Liu, J.L.; Zhang, P.J.; Fan, Y.X.; Egile, C.; Li, P.; Mueller, S.C.; Zhan, X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 2001, 3, 259–266. [Google Scholar] [CrossRef]
- Weaver, A.M. Cortactin in tumor invasiveness. Cancer Lett. 2008, 265, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell functions of a multifaceted actin-binding protein. Trends Cell Biol. 2018, 28, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Makhov, A.M.; Schafer, D.A.; Bear, J.E. Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell 2008, 134, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Abella, J.V.G.; Galloni, C.; Pernier, J.; Barry, D.J.; Kjaer, S.; Carlier, M.F.; Way, M. Isoform diversity in the Arp2/3 complex determines actin filament dynamics. Nat. Cell Biol. 2016, 18, 76–86. [Google Scholar] [CrossRef]
- Selbach, M.; Backert, S. Cortactin: An Achilles’ heel of the actin cytoskeleton targeted by pathogens. Trends Microbiol. 2005, 13, 181–189. [Google Scholar] [CrossRef]
- Selbach, M.; Moese, S.; Hurwitz, R.; Hauck, C.R.; Meyer, T.F.; Backert, S. The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J. 2003, 22, 515–528. [Google Scholar] [CrossRef]
- Knorr, J.; Backert, S.; Tegtmeyer, N. SHP2-Independent tyrosine dephosphorylation of cortactin and vinculin during infection with Helicobacter pylori. Eur. J. Microbiol. Immunol. 2020, 10, 20–27. [Google Scholar] [CrossRef]
- Campbell, D.H.; Sutherland, R.L.; Daly, R.J. Signaling pathways and structural domains required for phosphorylation of EMS1/cortactin. Cancer Res. 1999, 59, 5376–5385. [Google Scholar]
- Tegtmeyer, N.; Wittelsberger, R.; Hartig, R.; Wessler, S.; Martinez-Quiles, N.; Backert, S. Serine Phosphorylation of cortactin controls focal adhesion kinase activity and cell scattering induced by Helicobacter pylori. Cell Host Microbe 2011, 9, 520–531. [Google Scholar] [CrossRef]
- Backert, S.; Clyne, M.; Tegtmeyer, N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun. Signal. 2011, 9. [Google Scholar] [CrossRef]
- Chang, H.; Chen, D.F.; Ni, B.S.; Zuo, Q.F.; Wang, C.H.; Han, R.; Lan, C.H. Cortactin mediates apoptosis of gastric epithelial cells induced by VacA protein of Helicobacter pylori. Dig. Dis. Sci. 2016, 61, 80–90. [Google Scholar] [CrossRef]
- Sieg, D.J.; Hauck, C.R.; Schlaepfer, D.D. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 1999, 112, 2677–2691. [Google Scholar] [CrossRef]
- Newsome, T.P.; Weisswange, I.; Frischknecht, F.; Way, M. Abl collaborates with Src family kinases to stimulate actin-based motility of vaccinia virus. Cell. Microbiol. 2006, 8, 233–241. [Google Scholar] [CrossRef]
- Tanos, B.; Pendergast, A.M. Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 2006, 281, 32714–32723. [Google Scholar] [CrossRef]
- Schnoor, M.; Lai, F.P.L.; Zarbock, A.; Klaver, R.; Polaschegg, C.; Schulte, D.; Weich, H.A.; Oelkers, J.M.; Rottner, K.; Vestweber, D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability In Vivo. J. Exp. Med. 2011, 208, 1721–1735. [Google Scholar] [CrossRef]
- Tanaka, S.; Masuda, Y.; Harada, A.; Okabe, S. Impaired actin dynamics and suppression of Shank2-mediated spine enlargement in cortactin knockout mice. Microscopy 2020, 69, 44–52. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, H.L.; Blanpied, T.A.; Smith, E.; Zhan, X. Cortactin is implicated in murine zygotic development. Exp. Cell Res. 2010, 316, 848–858. [Google Scholar] [CrossRef]
- Tanaka, S.; Kunii, M.; Harada, A.; Okabe, S. Generation of cortactin floxed mice and cellular analysis of motility in fibroblasts. Genesis 2009, 47, 638–646. [Google Scholar] [CrossRef]
- Lai, F.P.L.; Szczodrak, M.; Oelkers, J.M.; Ladwein, M.; Acconcia, F.; Benesch, S.; Auinger, S.; Faix, J.; Small, J.V.; Polo, S.; et al. Cortactin promotes migration and platelet-derived growth factor-induced actin reorganization by signaling to Rho-GTPases. Mol. Biol. Cell 2009, 20, 3209–3223. [Google Scholar] [CrossRef]
- Kowalski, J.R.; Egile, C.; Gil, S.; Snapper, S.B.; Li, R.; Thomas, S.M. Cortactin regulates cell migration through activation of N-WASP. J. Cell Sci. 2005, 118, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Katsube, T.; Takahisa, M.; Ueda, R.; Hashimoto, N.; Kobayashi, M.; Togashi, S. Cortactin associates with the cell-cell junction protein ZO-1 in both Drosophila and mouse. J. Biol. Chem. 1998, 273, 29672–29677. [Google Scholar] [CrossRef] [PubMed]
- Sauvonnet, N.; Dujeancourt, A.; Dautry-Varsat, A. Cortactin and dynamin are required for the clathrinin-dependent endocytosis of gamma c cytokine receptor. J. Cell Biol. 2005, 168, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Citalan-Madrid, A.F.; Vargas-Robles, H.; Garcia-Ponce, A.; Shibayama, M.; Betanzos, A.; Nava, P.; Salinas-Lara, C.; Rottner, K.; Mennigen, R.; Schnoor, M. Cortactin deficiency causes increased RhoA/ROCK1-dependent actomyosin contractility, intestinal epithelial barrier dysfunction, and disproportionately severe DSS-induced colitis. Mucosal Immunol. 2017, 10, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- MacGrath, S.M.; Koleske, A.J. Cortactin in cell migration and cancer at a glance. J. Cell Sci. 2012, 125, 1621–1626. [Google Scholar] [CrossRef]
- Mezi, S.; Todi, L.; Orsi, E.; Angeloni, A.; Mancini, P. Involvement of the Src-cortactin pathway in migration induced by IGF-1 and EGF in human breast cancer cells. Int. J. Oncol. 2012, 41, 2128–2138. [Google Scholar] [CrossRef]
- Steffen, A.; Ladwein, M.; Dimchev, G.A.; Hein, A.; Schwenkmezger, L.; Arens, S.; Ladwein, K.I.; Margit Holleboom, J.; Schur, F.; Victor Small, J.; et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J. Cell Sci. 2013, 126, 4572–4588. [Google Scholar] [CrossRef]
- Dimchev, V.; Lahmann, I.; Koestler, S.A.; Kage, F.; Dimchev, G.; Steffen, A.; Stradal, T.E.B.; Vauti, F.; Arnold, H.H.; Rottner, K. Induced Arp2/3 complex depletion increases FMNL2/3 formin expression and filopodia formation. Front. Cell Dev. Biol. 2021, 9, 634708. [Google Scholar] [CrossRef]
- Kanellos, G.; Zhou, J.; Patel, H.; Ridgway, R.A.; Huels, D.; Gurniak, C.B.; Sandilands, E.; Carragher, N.O.; Sansom, O.J.; Witke, W.; et al. ADF and Cofilin1 control actin stress fibers, nuclear integrity, and cell survival. Cell Rep. 2015, 13, 1949–1964. [Google Scholar] [CrossRef]
- Costa, P.; Scales, T.M.E.; Ivaska, J.; Parsons, M. Integrin-Specific control of focal adhesion kinase and RhoA regulates membrane protrusion and invasion. PLoS ONE 2013, 8, e74659. [Google Scholar] [CrossRef]
- Orr, A.W.; Pallero, M.A.; Xiong, W.C.; Murphy-Ullrich, J.E. Thrombospondin induces RhoA inactivation through FAK-dependent signaling to stimulate focal adhesion disassembly. J. Biol. Chem. 2004, 279, 48983–48992. [Google Scholar] [CrossRef]
- Nazari, H.; Khaleghian, A.; Takahashi, A.; Harada, N.; Webster, N.J.G.; Nakano, M.; Kishi, K.; Ebina, Y.; Nakaya, Y. Cortactin, an actin binding protein, regulates GLUT4 translocation via actin filament remodeling. Biochemistry 2011, 76, 1262–1269. [Google Scholar] [CrossRef][Green Version]
- Keates, S.; Keates, A.C.; Warny, M.; Peek, R.M.; Murray, P.G.; Kelly, C.P. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag(+) and cag(−) Helicobacter pylori. J. Immunol. 1999, 163, 5552–5559. [Google Scholar]
- Mimuro, H.; Suzuki, T.; Tanaka, J.; Asahi, M.; Haas, R.; Sasakawa, C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol. Cell 2002, 10, 745–755. [Google Scholar] [CrossRef]
- Hirata, Y.; Maeda, S.; Mitsuno, Y.; Tateishi, K.; Yanai, A.; Akanuma, M.; Yoshida, H.; Kawabe, T.; Shiratori, Y.; Omata, M. Helicobacter pylori CagA protein activates serum response element-driven transcription independently of tyrosine phosphorylation. Gastroenterology 2002, 123, 1962–1971. [Google Scholar] [CrossRef]
- Brandt, S.; Kwok, T.; Hartig, R.; Konig, W.; Backert, S. NF-Kappa B activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef]
- Higashi, H.; Nakaya, A.; Tsutsumi, R.; Yokoyama, K.; Fujii, Y.; Ishikawa, S.; Higuchi, M.; Takahashi, A.; Kurashima, Y.; Teishikata, Y.; et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J. Biol. Chem. 2004, 279, 17205–17216. [Google Scholar] [CrossRef]
- Schaller, M.D.; Hildebrand, J.D.; Shannon, J.D.; Fox, J.W.; Vines, R.R.; Parsons, J.T. Autophosphorylation of the focal adhesion kinase, pp125(FAK), directs SH2 dependent binding of pp60(src). Mol. Cell. Biol. 1994, 14, 1680–1688. [Google Scholar] [CrossRef]
- Calalb, M.B.; Polte, T.R.; Hanks, S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity—A role for Src family kinases. Mol. Cell. Biol. 1995, 15, 954–963. [Google Scholar] [CrossRef]
- Wu, L.; Bernard-Trifilo, J.A.; Lim, Y.; Lim, S.T.; Mitra, S.K.; Uryu, S.; Chen, M.; Pallen, C.J.; Cheung, N.K.V.; Mikolon, D.; et al. Distinct FAK-Src activation events promote α5β1 and α4β1 integrin-stimulated neuroblastoma cell motility. Oncogene 2008, 27, 1439–1448. [Google Scholar] [CrossRef]
- Plattner, R.; Kadlec, L.; DeMali, K.A.; Kazlauskas, A.; Pendergast, A.M. C-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999, 13, 2400–2411. [Google Scholar] [CrossRef]
- Selbach, M.; Moese, S.; Hauck, C.R.; Meyer, T.F.; Backert, S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and In Vivo. J. Biol. Chem. 2002, 277, 6775–6778. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The role of Src in solid tumors. Oncologist 2009, 14, 667–678. [Google Scholar] [CrossRef]
- Greuber, E.K.; Smith-Pearson, P.; Wang, J.; Pendergast, A.M. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. [Google Scholar] [CrossRef]
- Parsonnet, J.; Friedman, G.D.; Orentreich, N.; Vogelman, H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997, 40, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Jin, J.S.; Chang, W.K.; Chan, D.C.; Yeh, M.K.; Cherng, S.C.; Lin, L.F.; Sheu, L.F.; Chao, Y.C. Association of cortactin and fascin-1 expression in gastric adenocarcinoma: Correlation with clinicopathological parameters. J. Histochem. Cytochem. 2007, 55, 955–962. [Google Scholar] [CrossRef]
- Wei, J.; Zhao, Z.X.; Li, Y.; Zhou, Z.Q.; You, T.G. Cortactin expression confers a more malignant phenotype to gastric cancer SGC-7901 cells. World J. Gastroenterol. 2014, 20, 3287–3300. [Google Scholar] [CrossRef]
- Costa, L.; Corre, S.; Michel, V.; Le Luel, K.; Fernandes, J.; Ziveri, J.; Jouvion, G.; Danckaert, A.; Mouchet, N.; Da Silva Barreira, D.; et al. USF1 defect drives p53 degradation during Helicobacter pylori infection and accelerates gastric carcinogenesis. Gut 2020, 69, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Blaser, M.J. The role of CagA in the gastric biology of Helicobacter pylori. Cancer Res. 2016, 76, 4028–4031. [Google Scholar] [CrossRef] [PubMed]
- Hartung, M.L.; Gruber, D.C.; Koch, K.N.; Grüter, L.; Rehrauer, H.; Tegtmeyer, N.; Backert, S.; Müller, A.H. H. pylori-induced DNA strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-κB target gene expression. Cell Rep. 2015, 13, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Moese, S.; Selbach, M.; Zimny-Arndt, U.; Jungblut, P.R.; Meyer, T.F.; Backert, S. Identification of a tyrosine-phosphorylated 35 kDa carboxy-terminal fragment (p35CagA) of the Helicobacter pylori CagA protein in phagocytic cells: Processing or breakage? Proteomics 2001, 1, 618–629. [Google Scholar] [CrossRef]
- Backert, S.; Schwarz, T.; Miehlke, S.; Kirsch, C.; Sommer, C.; Kwok, T.; Gerhard, M.; Goebel, U.B.; Lehn, N.; Koenig, W.; et al. Functional analysis of the cag pathogenicity island in Helicobacter pylori isolates from patients with gastritis, peptic ulcer, and gastric cancer. Infect. Immun. 2004, 72, 1043–1056. [Google Scholar] [CrossRef]
- Ferrero, R.L.; Cussac, V.; Courcoux, P.; Labigne, A. Construction of isogenic urease-negative mutants of Helicobacter pylori by allelic exchange. J. Bacteriol. 1992, 174, 4212–4217. [Google Scholar] [CrossRef]
- Baltrus, D.A.; Amieva, M.R.; Covacci, A.; Lowe, T.M.; Merrell, D.S.; Ottemann, K.M.; Stein, M.; Salama, N.R.; Guillemin, K. The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 2009, 191, 447–448. [Google Scholar] [CrossRef]
- Schmitt, W.; Haas, R. Genetic analysis of the Helicobacter pylori vacuolating cytotoxin—Structural similarities with the IGA protease type of exported protein. Mol. Microbiol. 1994, 12, 307–319. [Google Scholar] [CrossRef]
- Haas, R.; Meyer, T.F.; VanPutten, J.P.M. Aflagellated mutants of Helicobacter pylori generated by genetic transformation of naturally competent strains using transposon shuttle mutagenesis. Mol. Microbiol. 1993, 8, 753–760. [Google Scholar] [CrossRef]
- Conradi, J.; Tegtmeyer, N.; Woźna, M.; Wissbrock, M.; Michalek, C.; Gagell, C.; Cover, T.L.; Frank, R.; Sewald, N.; Backert, S. An RGD helper sequence in CagL of Helicobacter pylori assists in interactions with integrins and injection of CagA. Front. Cell Infect. Microbiol. 2012, 2, 70. [Google Scholar] [CrossRef]
- Boehm, M.; Krause-Gruszczynska, M.; Rohde, M.; Tegtmeyer, N.; Takahashi, S.; Oyarzabal, O.A.; Backert, S. Major host factors involved in epithelial cell invasion of Campylobacter jejuni: Role of fibronectin, integrin beta1, FAK, Tiam-1, and DOCK180 in activating Rho GTPase Rac1. Front. Cell. Infect. Microbiol. 2011, 1, 17. [Google Scholar] [CrossRef]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Krause-Gruszczynska, M.; Boehm, M.; Rohde, M.; Tegtmeyer, N.; Takahashi, S.; Buday, L.; Oyarzabal, O.A.; Backert, S. The signaling pathway of Campylobacter jejuni-induced Cdc42 activation: Role of fibronectin, integrin beta1, tyrosine kinases and guanine exchange factor Vav2. Cell Commun. Signal. 2011, 9, 32. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knorr, J.; Sharafutdinov, I.; Fiedler, F.; Soltan Esmaeili, D.; Rohde, M.; Rottner, K.; Backert, S.; Tegtmeyer, N. Cortactin Is Required for Efficient FAK, Src and Abl Tyrosine Kinase Activation and Phosphorylation of Helicobacter pylori CagA. Int. J. Mol. Sci. 2021, 22, 6045. https://doi.org/10.3390/ijms22116045
Knorr J, Sharafutdinov I, Fiedler F, Soltan Esmaeili D, Rohde M, Rottner K, Backert S, Tegtmeyer N. Cortactin Is Required for Efficient FAK, Src and Abl Tyrosine Kinase Activation and Phosphorylation of Helicobacter pylori CagA. International Journal of Molecular Sciences. 2021; 22(11):6045. https://doi.org/10.3390/ijms22116045
Chicago/Turabian StyleKnorr, Jakob, Irshad Sharafutdinov, Florian Fiedler, Delara Soltan Esmaeili, Manfred Rohde, Klemens Rottner, Steffen Backert, and Nicole Tegtmeyer. 2021. "Cortactin Is Required for Efficient FAK, Src and Abl Tyrosine Kinase Activation and Phosphorylation of Helicobacter pylori CagA" International Journal of Molecular Sciences 22, no. 11: 6045. https://doi.org/10.3390/ijms22116045
APA StyleKnorr, J., Sharafutdinov, I., Fiedler, F., Soltan Esmaeili, D., Rohde, M., Rottner, K., Backert, S., & Tegtmeyer, N. (2021). Cortactin Is Required for Efficient FAK, Src and Abl Tyrosine Kinase Activation and Phosphorylation of Helicobacter pylori CagA. International Journal of Molecular Sciences, 22(11), 6045. https://doi.org/10.3390/ijms22116045