
Pathophysiological Roles of Stress-Activated Protein Kinases in Pulmonary Fibrosis

 ,
,  ,
, 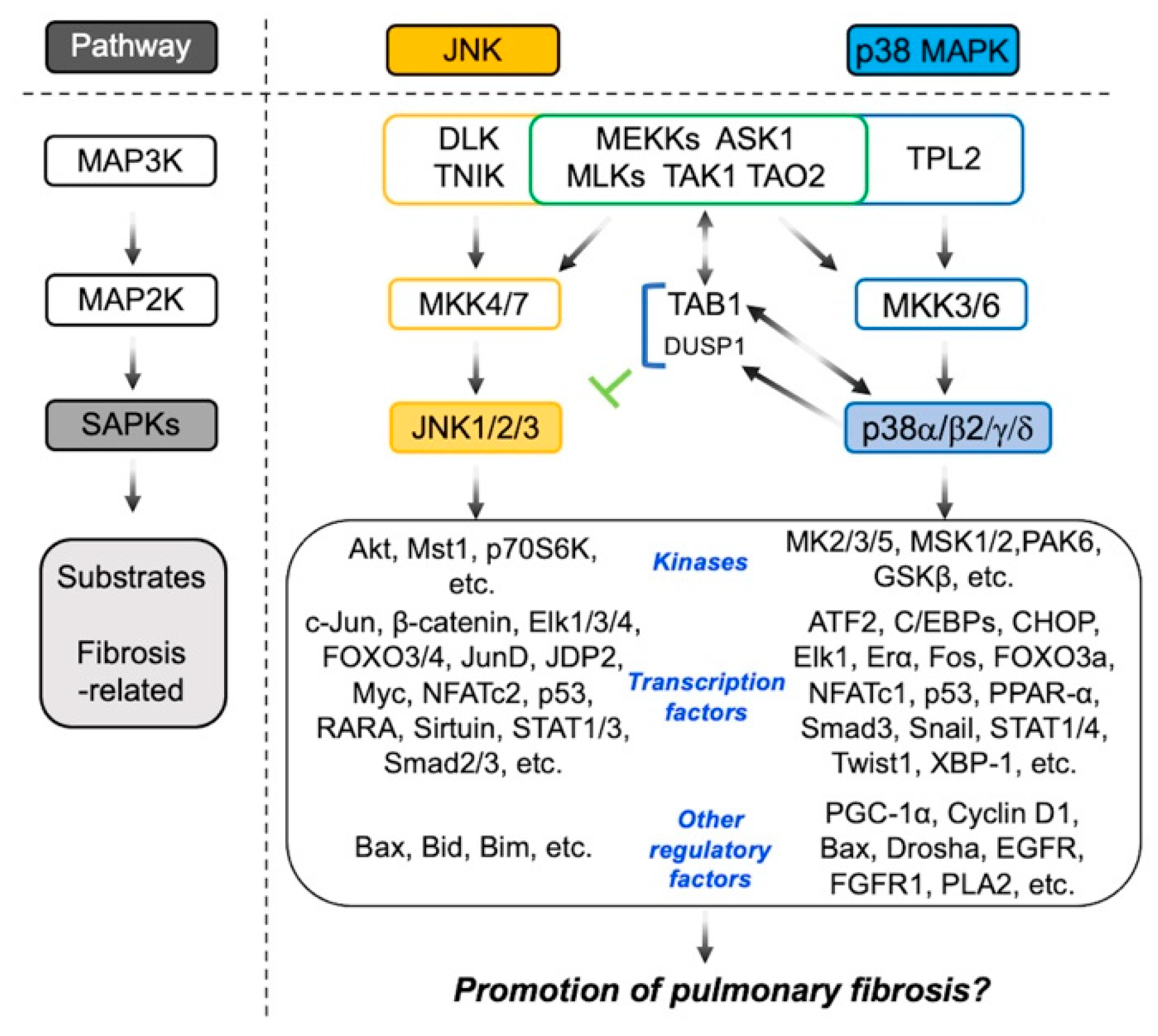{kind=link}
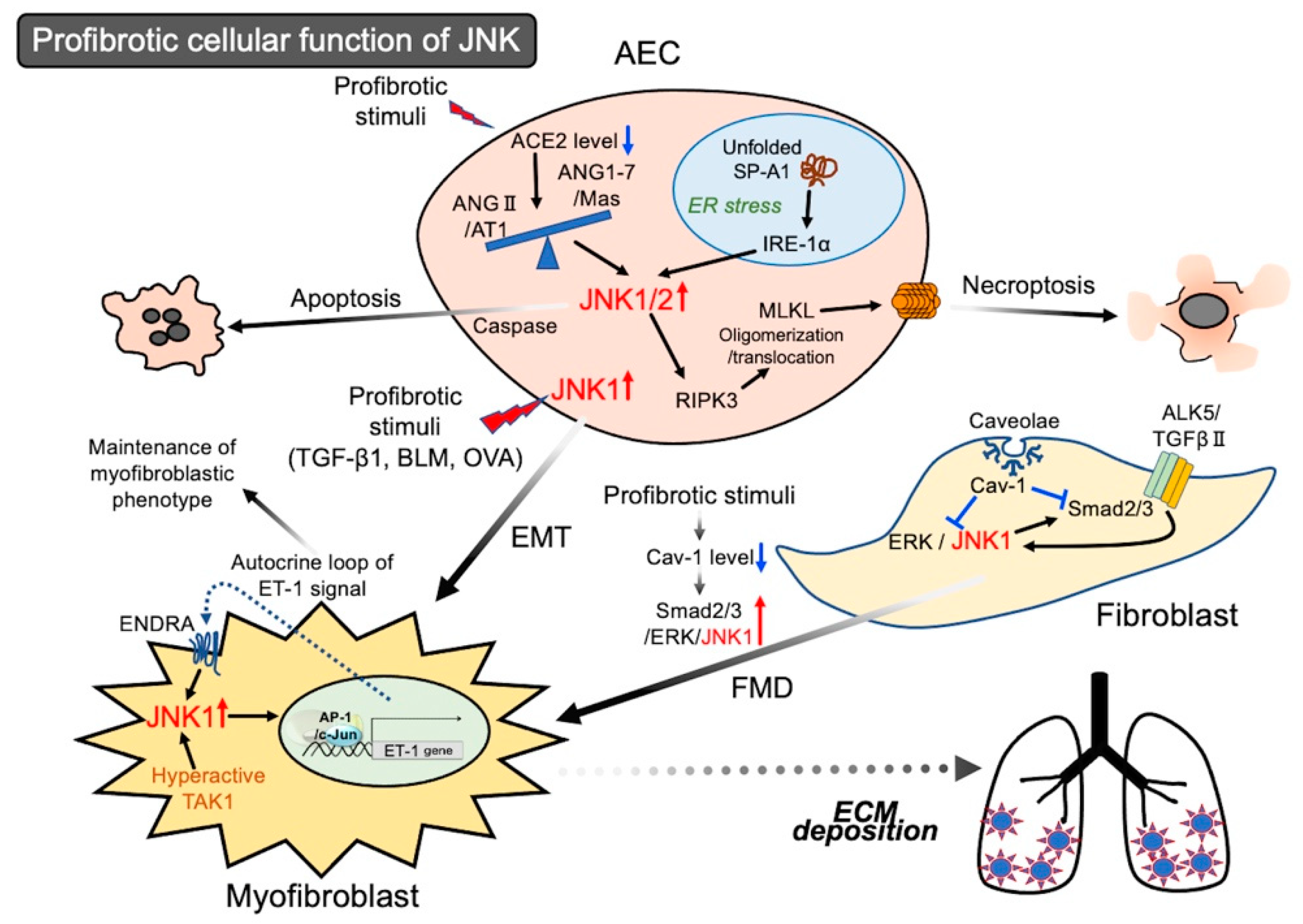{kind=link}
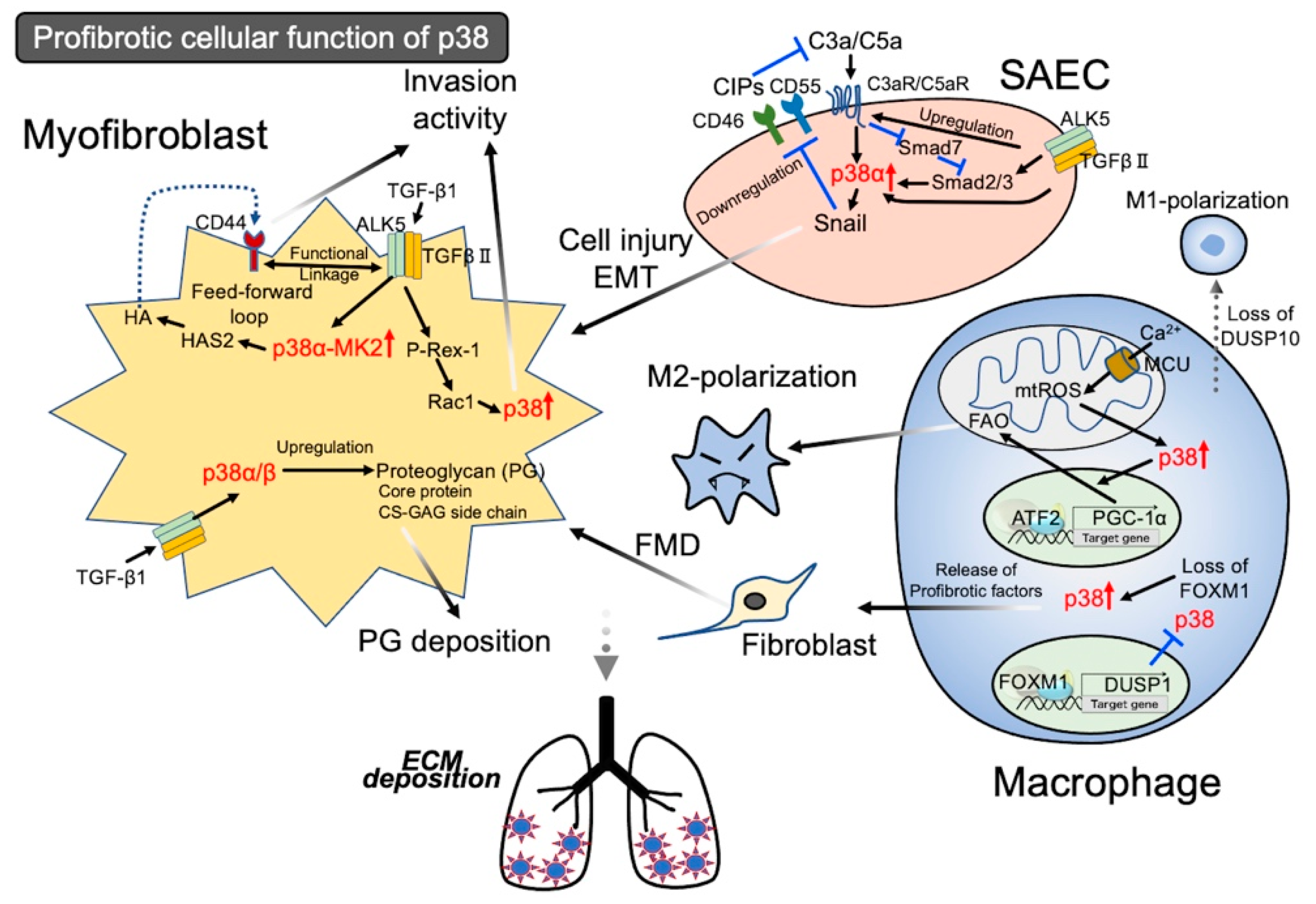{kind=link}
Abstract
1. Introduction
2. Involvement of JNK in the Pathogenesis of Pulmonary Fibrosis
2.1. Activation of JNK in Epithelial Cells Leading to Cell Death
Activation of JNK in Epithelial Cells Leading to EMT
2.2. Activation of JNK in Lung Fibroblasts Leading to Myofibroblastic Phenotypes
3. Involvement of p38 in the Pathogenesis of Pulmonary Fibrosis
3.1. Activation of p38 in Epithelial Cells Leading to Injury
3.1.1. Activation of p38 in Lung Fibroblasts Leads to Profibrotic Characters
3.1.2. Activation of p38 Signal in Lung Fibroblasts Leading to Invasion Activity
3.2. Activation of the p38 Signal in Macrophages Leading to Profibrotic
3.3. Application of Profibrotic Function of p38 Signal to Identify the Therapeutic Target of IPF
4. Is an Anti-SAPK Strategy Effective Against IPF?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef]
- Yue, J.; Lôpez, J.M. Understanding MAPK signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Rehfeldt, S.C.H.; Majolo, F.; Goettert, M.I.; Laufer, S. c-Jun N-terminal kinase inhibitors as potential leads for new therapeutics for Alzheimer’s diseases. Int. J. Mol. Sci. 2020, 21, 9677. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK signaling: Regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, C.; Li, Z.; Guo, W.; Gegner, J.A.; Lin, S.; Han, J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38δ. J. Biol. Chem. 1997, 272, 30122–30128. [Google Scholar] [CrossRef] [PubMed]
- Corre, I.; Paris, F.; Huot, J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget 2017, 8, 55684–55714. [Google Scholar] [CrossRef]
- Cuenda, A.; Sanz-Ezquerro, J.J. p38γ and p38δ: From spectators to key physiological players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Aziz, N.; Cho, J.Y. MKK7, the essential regulator of JNK signaling involved in cancer cell survival: A newly emerging anticancer therapeutic target. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875574. [Google Scholar] [CrossRef]
- Han, J.; Wu, J.; Silke, J. An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F1000Research 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, M.H.; Ying, C.P.; Chen, M.Y. Neferine induces p38 MAPK/JNK1/2 activation to modulate melanoma proliferation, apoptosis, and oxidative stress. Ann. Transl. Med. 2020, 24, 1643. [Google Scholar] [CrossRef]
- Wada, T.; Stepniak, E.; Hui, L.; Leibbrandt, A.; Katada, T.; Nishina, H.; Wagner, E.F.; Penninger, J.M. Antagonistic control of cell fates by JNK and p38-MAPK signaling. Cell Death Diff. 2008, 15, 89–93. [Google Scholar] [CrossRef]
- Ge, B.; Gram, H.; Di Padova, F.; Huang, B.; New, L.; Ulevitch, R.J.; Luo, Y.; Han, J. MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science 2002, 295, 1291–1294. [Google Scholar] [CrossRef]
- Cheung, P.C.; Campbell, D.G.; Nebreda, A.R.; Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J. 2003, 22, 5793–5805. [Google Scholar] [CrossRef] [PubMed]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-translational modifications of the TAK1-TAB complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.R.; Lei, C.Q. TAK1-TABs complex: A central signalosome in inflammatory responses. Front. Immunol. 2020, 11, 608976. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 180033. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, C.; Zhang, Z.K.; Pan, X.; Peng, S.; Lee, W.S.; Lu, A.; Lin, Z.; Zhang, G.; Leung, W.N.; et al. TAK1 inhibition attenuates both inflammation and fibrosis in experimental pneumoconiosis. Cell Discov. 2017, 3, 17023. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Cottin, V. Spectrum of fibrotic lung diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Raghu, G.; Selman, M. Nintedanib and pirfenidone. New antifibrotic treatments indicated for idiopathic pulmonary fibrosis offer hopes and raises questions. Am. J. Respir. Crit. Care Med. 2018, 191, 252–254. [Google Scholar] [CrossRef]
- Carrington, R.; Jordan, S.; Pitchford, S.C.; Page, C.P. Use of animal models in IPF research. Pulm. Pharmacol. Ther. 2018, 51, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L. Mechanisms and consequences of oxidative stress in lung disease: Therapeutic implications for an aging populace. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L642–L653. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Balestro, E.; Calabrese, F.; Turato, G.; Lunardi, F.; Bazzan, E.; Marulli, G.; Biondini, D.; Rossi, E.; Sanduzzi, A.; Rea, F.; et al. Immune inflammation and disease progression in idiopathic pulmonary fibrosis. PLoS ONE 2016, 11, e0154516. [Google Scholar] [CrossRef]
- Yamazaki, R.; Kasuya, Y.; Fujita, T.; Umezawa, H.; Yanagihara, M.; Nakamura, H.; Yoshino, I.; Tatsumi, K.; Murayama, T. Antifibrotic effects of cyclosporine A on TGF-β1–treated lung fibroblasts and lungs from bleomycin-treated mice: Role of hypoxia-inducible factor-1α. FASEB J. 2018, 31, 3359–3371. [Google Scholar] [CrossRef] [PubMed]
- Rubio, K.; Castillo-Negrete, R.; Barreto, G. Non-coding RNAs and nuclear architecture during epithelial-mesenchymal transition in lung cancer and idiopathic pulmonary fibrosis. Cell. Signal. 2020, 70, 109593. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tada, Y.; Gladson, S.; Nishimura, R.; Shimomura, I.; Karasawa, S.; Tatsuki, K.; West, J. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-mesenchymal transition. Respir. Res. 2017, 18, 177. [Google Scholar] [CrossRef] [PubMed]
- Underwood, D.C.; Osborn, R.R.; Bochnowicz, S.; Webb, E.F.; Rieman, D.J.; Lee, J.C.; Romanic, A.M.; Adams, J.L.; Hay, D.W.; Griswold, D.E. SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L895–L902. [Google Scholar] [CrossRef]
- Sisson, T.H.; Mendez, M.; Choi, K.; Subbotina, N.; Courey, A.; Cunningham, A.; Dave, A.; Engelhardt, J.F.; Liu, X.; White, E.S.; et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care. Med. 2010, 181, 254–263. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Diff. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Wu, C.C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef]
- Pihán, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Diff. 2017, 24, 1478–1487. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Gibert, B.; Mehlen, P. Dependence receptors and cancer: Addiction to trophic ligands. Cancer Res. 2016, 75, 5171–5175. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Steighner, R.J.; Povirk, L.F. Bleomycin-induced DNA lesions at mutational hot spots: Implications for the mechanism of double-strand cleavage. Proc. Natl. Acad. Sci. USA 1990, 87, 8350–8354. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Araya, J.; Yoshida, M.; Sakamoto, T.; Koumura, T.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; et al. Involvement of GPx4-regulated lipid peroxidation in idiopathic pulmonary fibrosis pathogenesis. J. Immunol. 2019, 203, 2076–2087. [Google Scholar] [CrossRef]
- Wang, J.; Deng, B.; Liu, Q.; Huang, Y.; Chen, W.; Li, J.; Zhou, Z.; Zhang, L.; Liang, B.; He, J.; et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 2020, 11, 574. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, H.; Soledad-Conrad, V.; Zhuang, J.; Uhal, B.D. Bleomycin induced apoptosis of alveolar epithelial cells requires angiotensin synthesis de novo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L501–L507. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zagariya, A.; Ang, E.; Ibarra-Sunga, O.; Uhal, B.D. Fas-induced apoptosis of alveolar epithelial cells requires angiotensin II generation and receptor interaction. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 277, L1245–L1250. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Alam, G.; Zagariya, A.; Gidea, C.; Pinillos, H.; Lalude, O.; Choudhary, G.; Uhal, B.D. Apoptosis of lung epithelial cells in response to TNF-alpha requires angiotensin II generation de novo. J. Cell Physiol. 2000, 185, 253–259. [Google Scholar] [CrossRef]
- Li, X.; Molina-Molina, M.; Abdul-Hafez, A.; Ramirez, J.; Serrano-Mollar, A.; Xaubet, A.; Uhal, B.D. Extravascular sources of lung angiotensin peptide synthesis in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L887–L895. [Google Scholar] [CrossRef]
- Königshoff, M.; Wilhelm, A.; Jahn, A.; Sedding, D.; Amarie, O.A.; Eul, B.; Seeger, W.; Fink, L.; Günther, A.; Eickelberg, O.; et al. The angiotensin II receptor 2 is expressed and mediates angiotensin II signaling in lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Catarata, M.J.; Ribeiro, R.; Oliveira, M.J.; Cordeiro, C.R.; Medeiros, R. Renin-angiotensin system in lung tumor and microenvironment interactions. Cancers 2020, 12, 1457. [Google Scholar] [CrossRef]
- Bali, A.; Jaggi, A.S. Angiotensin II-triggered kinase signaling cascade in the central nervous system. Rev. Neurosci. 2016, 27, 301–315. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Meng, Y.; Li, T.; Zhou, G.S.; Chen, Y.; Yu, C.H.; Pang, M.X.; Li, W.; Li, Y.; Zhang, W.Y.; Li, X. The angiotensin-converting enzyme 2/angiotensin (1–7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid. Redox Signal. 2015, 2, 241–258. [Google Scholar] [CrossRef]
- Wan, H.; Ma, Q.; Che, J.; Cao, H.; Fei, X.; Qiu, W.; Feng, Y.; Wan, H.; Liu, J.; Zhang, R.; et al. The angiotensin-converting enzyme 2 in tumor growth and tumor-associated angiogenesis in non-small cell lung cancer. Oncol. Rep. 2010, 23, 941–948. [Google Scholar] [CrossRef]
- Li, X.; Molina-Molina, M.; Abdul-Hafez, A.; Xaubet, A.; Uhal, B.D. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L178–L185. [Google Scholar] [CrossRef]
- Uhal, B.D.; Dang, M.; Dang, V.; Llatos, R.; Cano, E.; Abdul-Hafez, A.; Markey, J.; Piasecki, C.C.; Molina-Molina, M. Cell cycle dependence of ACE-2 explains downregulation in idiopathic pulmonary fibrosis. Eur. Respir. J. 2013, 42, 198–210. [Google Scholar] [CrossRef]
- Uhal, B.D.; Li, X.; Xue, A.; Gao, X.; Abdul-Hafez, A. Regulation of alveolar epithelial cell survival by the ACE-2/angiotensin 1–7/Mas axis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L269–L274. [Google Scholar] [CrossRef]
- Takezaki, A.; Tsukumo, S.; Setoguchi, Y.; Ledford, J.G.; Goto, H.; Hosomichi, K.; Uehara, H.; Nishioka, Y.; Yasutomo, K. A homozygous SFTPA1 mutation drives necroptosis of type II alveolar epithelial cells in patients with idiopathic pulmonary fibrosis. J. Exp. Med. 2019, 216, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Phelps, D.S. Surfactant regulation of host defense function in the lung: A question of balance. Pediatr. Pathol. Mol. Med. 2001, 20, 269–292. [Google Scholar] [CrossRef]
- Lin, Z.; Thorenoor, N.; Wu, R.; DiAngelo, S.L.; Ye, M.; Thomas, N.J.; Liao, X.; Lin, T.R.; Warren, S.; Floros, J. Genetic Association of Pulmonary Surfactant Protein Genes, SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD With Cystic Fibrosis. Front. Immunol. 2018, 9, 2256. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Nguyen, V.; Russo, S.J.; Muniswamy, M.; Beers, M.F. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am. J. Respir. Cell Mol. Biol. 2005, 32, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell. Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69084. [Google Scholar] [CrossRef] [PubMed]
- Alcorn, J.F.; Guala, A.S.; van der Velden, J.L.; McElhinney, B.; Irvin, C.G.; Davis, R.J.; Janssen-Heininger, Y.M.W. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-β1. J. Cell Sci. 2008, 121, 1036–1045. [Google Scholar] [CrossRef]
- Alcorn, J.F.; van der Velden, J.L.; Brown, A.L.; McElhinney, B.; Irvin, C.G.; Janssen-Heininger, Y.M.W. c-Jun N-terminal kinase 1 is required for the development of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2009, 40, 422–432. [Google Scholar] [CrossRef]
- Van der Velden, J.L.; Alcorn, J.F.; Chapman, D.G.; ILundblad, L.K.A.; Irvin, C.G.; Davis, R.J.; Butnor, K.; Janssen-Heininger, Y.M.W. Airway epithelial specific deletion of Jun-N-terminal kinase 1 attenuates pulmonary fibrosis in two independent mouse models. PLoS ONE 2020, 15, e0226904. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Schwarz, M.I.; Brown, K.K.; Tooze, J.A.; Colby, T.V.; Waldron, J.A., Jr.; Flint, A.; Thurlbeck, W.; Cherniak, R.M. Idiopathic pulmonary fibrosis: Relationship between histopathologic features and mortality. Am. J. Respir. Crit. Care Med. 2001, 164, 1025–1032. [Google Scholar] [CrossRef]
- Shi-Wen, X.; Chen, Y.; Denton, C.P.; Eastwood, M.; Renzoni, E.A.; Bou-Gharios, G.; Pearson, J.D.; Dashwood, M.; du Bois, R.M.; Black, C.M.; et al. Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol. Biol. Cell 2004, 15, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.M.; Yu, C.C.; Kuo, M.L.; Chen, B.C.; Lin, C.H. Endothelin-1 induces connective tissue growth factor expression in human lung fibroblasts by ETAR-dependent JNK/AP-1 pathway. Biochem. Pharmacol. 2014, 88, 402–411. [Google Scholar] [CrossRef]
- Mayeux, J.D.; Pan, I.Z.; Dechand, J.; Jacobs, J.A.; Jones, T.J.; McKellar, S.H.; Beck, E.; Hatton, N.D.; Ryan, J.J. Management of pulmonary arterial hypertension. Curr. Cardiovasc. Risk Rep. 2021, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Behr, J.; Brown, K.K.; du Bois, R.M.; Lancaster, L.; de Andrade, J.A.; Stähler, G.; Leconte, I.; Roux, S.; Raghu, G. BUILD-1: A randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 75–81. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Brown, K.K.; Raghu, G.; du Bois, R.M.; Lynch, D.A.; Martinez, F.; Valeyre, F.; Leconte, I.; Morganti, A.; Roux, S.; et al. BUILD-3: A randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Lisanti, M.P. The caveolin proteins. Genome Biol. 2004, 5, 214. [Google Scholar] [CrossRef]
- Kruglikov, I.L.; Scherer, P.E. Caveolin-1 as a pathophysiological factor and target in psoriasis. NPJ Aging Mech. Dis. 2019, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef]
- Wang, X.M.; Zhang, Y.; Kim, H.P.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Varsano, S.; Frolkis, I.; Ophir, D. Expression and distribution of cell-membrane complement regulatory glycoproteins along the human respiratory tract. Am. J. Respir. Crit. Care Med. 1995, 152, 1087–1093. [Google Scholar] [CrossRef]
- Gu, H.; Mickler, E.A.; Cummings, O.W.; Sandusky, G.E.; Weber, D.J.; Gracon, A.; Woodruff, T.; Wilkes, D.S.; Vittal, R. Crosstalk between TGF-β1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J. 2014, 28, 4223–4234. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, S.A.; Cavender, D.E.; Beers, S.A.; Lalan, P.; Schafer, P.H.; Malloy, E.A.; Wu, W.; Fahmy, B.; Olini, G.C.; Davis, J.E.; et al. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J. Pharmacol. Exp. Ther. 1999, 291, 680–687. [Google Scholar] [PubMed]
- Ryu, K.J.; Park, S.M.; Park, S.H.; Kim, I.K.; Han, H.; Kim, H.J.; Kim, S.H.; Hong, K.S.; Kim, H.; Kim, M.; et al. p38 stabilizes Snail by suppressing DYRK2-mediated phosphorylation that is required for GSK3β-βTrCP-induced Snail degradation. Cancer Res. 2019, 79, 4135–4148. [Google Scholar] [CrossRef] [PubMed]
- Tsukui, T.; Ueha, S.; Shichino, S.; Inagaki, Y.; Matsushima, K. Intratracheal cell transfer demonstrates the profibrotic potential of resident fibroblasts in pulmonary fibrosis. Am. J. Pathol. 2015, 185, 2939–2948. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, H.; Morgan, L.; Locy, M.L.; Ravi, S.; Deshane, J.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef]
- Ugai, K.; Matsuda, S.; Mikami, H.; Shimada, A.; Misawa, T.; Nakamura, H.; Tatsumi, K.; Hatano, M.; Murayama, T.; Kasuya, Y. Inhibition of the SET8 pathway ameliorates lung fibrosis even through fibroblast dedifferentiation. Front. Mol. Biosci. 2020, 7, 192. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V. Matrix proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef]
- Venkatesan, N.; Ouzzine, M.; Kolb, M.; Netter, P.; Ludwig, M.S. Increased deposition of chondroitin/dermatan sulfate glycosaminoglycan and upregulation of β1,3-glucuronosyltransferase I in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L191–L203. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, N.; Tsuchiya, K.; Kolb, M.; Farkas, L.; Bourhim, M.; Ouzzine, M.; Ludwig, M.S. Glycosyltransferases and glycosaminoglycans in bleomycin and transforming growth factor-β1–induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 50, 583–594. [Google Scholar] [CrossRef]
- Kim, S.I.; Choi, M.E. TGF-β-activated kinase-1: New insights into the mechanism of TGF-β signaling and kidney disease. Kidney Res. Clin. Pract. 2012, 31, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Takekawa, M.; Tatebayashi, K.; Itoh, F.; Adachi, M.; Imai, K.; Saito, H. Smad-dependent GADD45β expression mediates delayed activation of p38 MAP kinase by TGF-β. EMBO J. 2002, 21, 6473–6482. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Liu, N.; Liu, X.; Mena, J.M.; Xie, T.; Geng, Y.; Huan, C.; Zhang, Y.; Taghavifar, F.; Huang, G.; et al. Mitogen-activated protein kinase–activated protein kinase 2 inhibition attenuates fibroblast invasion and severe lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 60, 41–48. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Singleton, P.A.; Zhu, H.; Zhou, B. Hyaluronan promotes signaling interaction between CD44 and the transforming growth factor β receptor I in metastatic breast tumor cells. J. Biol. Chem. 2002, 277, 39703–39712. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qi, L.; Liang, Z.; Song, W.; Liu, Y.; Wang, Y.; Sun, B.; Zhang, B.; Cao, W. Transforming growth factor-β1 induces EMT by the transactivation of epidermal growth factor signaling through HA/CD44 in lung and breast cancer cells. Int. J. Mol. Med. 2015, 36, 113–122. [Google Scholar] [CrossRef]
- Vittal, R.; Fisher, A.; Gu, H.; Mickler, E.A.; Panitch, A.; Lander, C.; Cummings, O.W.; Sandusky, G.E.; Wilkes, D.S. Peptide-mediated inhibition of mitogen-activated protein kinase–activated protein kinase–2 ameliorates bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 47–57. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, S.; Dolado, I.; Beardmore, V.; Shpiro, N.; Marquez, R.; Nebreda, A.R.; Arthur, J.S.C.; Case, L.M.; Tessier-Lavigne, M.; Gaestel, M.; et al. CXCL12 and C5a trigger cell migration via a PAK1/2-p38α MAPK-MAPKAP-K2-HSP27 pathway. Cell. Signal. 2006, 18, 1897–1905. [Google Scholar] [CrossRef]
- Wang, C.; Hockerman, S.; Jacobsen, E.J.; Alippe, Y.; Selness, S.R.; Hope, H.R.; Hirsch, J.L.; Mnich, S.J.; Saabye, M.J.; Hood, W.F.; et al. Selective inhibition of the p38α MAPK–MK2 axis inhibits inflammatory cues including inflammasome priming signals. J. Exp. Med. 2018, 215, 1315–1325. [Google Scholar] [CrossRef]
- Kunschmann, T.; Puder, S.; Fischer, T.; Steffen, A.; Rottner, K.; Mierke, C.T. The small GTPase Rac1 increases cell surface stiffness and enhances 3D migration into extracellular matrices. Sci. Rep. 2019, 9, 7675. [Google Scholar] [CrossRef]
- Liang, Q.; Cheng, N.; Zhang, G.; Liang, Y.; Qian, F.; Wu, D.; Ye, R.D. Identification of P-Rex1 as an anti-inflammatory and anti-fibrogenic target for pulmonary fibrosis. Sci. Rep. 2016, 6, 25785. [Google Scholar] [CrossRef]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.; Dentler, J.; Luttmann, W.; Friedrich, K.; Muller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis—Mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Venosa, A.; Malaviya, R.; Choi, H.; Gow, A.J.; Laskin, J.D.; Laskin, D.L. Characterization of distinct macrophage subpopulations during nitrogen mustard-induced lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 54, 436–446. [Google Scholar] [CrossRef]
- Satoh, T.; Nakagawa, K.; Sugihara, F.; Kuwahara, R.; Ashihara, M.; Yamane, F.; Minowa, Y.; Fukushima, K.; Ebina, I.; Yoshioka, Y.; et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 2017, 541, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Larson-Casey, J.L.; Carter, A.B. Macrophages utilize the mitochondrial calcium uniporter for profibrotic polarization. FASEB J. 2017, 31, 3072–3083. [Google Scholar] [CrossRef]
- Gu, L.; Larson-Casey, J.L.; Andrabib, S.A.; Leeb, J.H.; Meza-Perezc, S.; Randallc, T.D.; Carter, A.B. Mitochondrial calcium uniporter regulates PGC-1α expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol. 2019, 26, 101307. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Goda, C.; Balli, D.; Black, M.; IMilewski, D.; Le, T.; Ustiyan, V.; Ren, X.; Kalinichenko, V.V.; Kalin, T.V. Loss of FOXM1 in macrophages promotes pulmonary fibrosis by activating p38 MAPK signaling pathway. PLoS Genet. 2020, 16, e1008692. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.K.; Abdollah, N.A.; Shafie, N.H.; Yusof, N.M.; Razak, S.R.A. Dual-specificity phosphatase 6 (DUSP6): A review of its molecular characteristics and clinical relevance in cancer. Cancer Biol. Med. 2018, 15, 14–28. [Google Scholar] [CrossRef]
- Xylourgidis, N.; Min, K.; Ahangari, F.; Yu, G.; Herazo-Maya, J.D.; Karampitsakos, T.; Aidinis, V.; Binzenhöfer, L.; Bouros, D.; Bennett, A.M.; et al. Role of dual-specificity protein phosphatase DUSP10/MKP-5 in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L678–L689. [Google Scholar] [CrossRef]
- Matsuda, S.; Kim, J.D.; Sugiyama, F.; Matsuo, Y.; Ishida, J.; Murata, K.; Nakamura, K.; Namiki, K.; Sudo, T.; Kuwaki, T.; et al. Transcriptomic evaluation of pulmonary fibrosis-related genes: Utilization of transgenic mice with modifying p38 signal in the lungs. Int. J. Mol. Sci. 2020, 21, 6746. [Google Scholar] [CrossRef]
- Habiel, D.M.; Espindola, M.S.; Jones, I.C.; Coelho, A.L.; Stripp, B.; Hogaboam, C.M. CCR10+ epithelial cells from idiopathic pulmonary fibrosis lungs drive remodeling. JCI Insight 2018, 3, e122211. [Google Scholar] [CrossRef]
- McDonough, J.E.; Ahangari, F.; Li, Q.; Jain, S.; Verleden, S.E.; Herazo-Maya, J.; Vukmirovic, M.; DeIuliis, G.; Tzouvelekis, A.; Tanabe, N.; et al. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight 2019, 4, e131597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Huang, W.; Ge, X. The role of heat shock proteins in the regulation of fibrotic diseases. Biomed. Pharmacother. 2021, 135, 111067. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.; Shimbori, C.; Yanagihara, T.; Carlson, D.A.; Hughes, P.; Upagupta, C.; Sato, S.; Wheildon, N.; Haystead, T.; Ask, K.; et al. Synergistic role of HSP90α and HSP90β to promote myofibroblast persistence in lung fibrosis. Eur. Respir. J. 2018, 51, 1700386. [Google Scholar] [CrossRef]
- Dong, H.; Luo, L.; Zou, M.; Huang, C.; Wan, X.; Hu, Y.; Le, Y.; Zhao, H.; Li, W.; Zou, F.; et al. Blockade of extracellular heat shock protein 90α by 1G6-D7 attenuates pulmonary fibrosis through inhibiting ERK signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L1006–L1015. [Google Scholar] [CrossRef]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Critical regulation of TGFβ signaling by Hsp90. Proc. Natl. Acad. Sci. USA 2008, 105, 9244–9249. [Google Scholar] [CrossRef] [PubMed]
- Moran, N. p38 kinase inhibitor approved for idiopathic pulmonary fibrosis. Nat. Biotech. 2011, 29, 301. [Google Scholar] [CrossRef] [PubMed]
- Bahudhanapati, H.; Kass, D.J. Unwinding the collagen fibrils: Elucidating the mechanism of pirfenidone and nintedanib in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 57, 10–11. [Google Scholar] [CrossRef]
- Miura, H.; Kondo, Y.; Matsuda, M.; Aoki, K. Cell-to-cell heterogeneity in p38-mediated cross-inhibition of JNK causes stochastic cell death. Cell Rep. 2018, 24, 2658–2668. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Rebecca, F.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasuya, Y.; Kim, J.-D.; Hatano, M.; Tatsumi, K.; Matsuda, S. Pathophysiological Roles of Stress-Activated Protein Kinases in Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 6041. https://doi.org/10.3390/ijms22116041
Kasuya Y, Kim J-D, Hatano M, Tatsumi K, Matsuda S. Pathophysiological Roles of Stress-Activated Protein Kinases in Pulmonary Fibrosis. International Journal of Molecular Sciences. 2021; 22(11):6041. https://doi.org/10.3390/ijms22116041
Chicago/Turabian StyleKasuya, Yoshitoshi, Jun-Dal Kim, Masahiko Hatano, Koichiro Tatsumi, and Shuichi Matsuda. 2021. "Pathophysiological Roles of Stress-Activated Protein Kinases in Pulmonary Fibrosis" International Journal of Molecular Sciences 22, no. 11: 6041. https://doi.org/10.3390/ijms22116041
APA StyleKasuya, Y., Kim, J.-D., Hatano, M., Tatsumi, K., & Matsuda, S. (2021). Pathophysiological Roles of Stress-Activated Protein Kinases in Pulmonary Fibrosis. International Journal of Molecular Sciences, 22(11), 6041. https://doi.org/10.3390/ijms22116041