
Gender-Dependent Phenotype in Polycystic Kidney Disease Is Determined by Differential Intracellular Ca2+ Signals


Abstract
1. Introduction
2. Results
2.1. Male Mice Lacking Expression of Pkd1 Show a Larger Number of Renal Cysts, But Similar Levels of Expression of TMEM16A
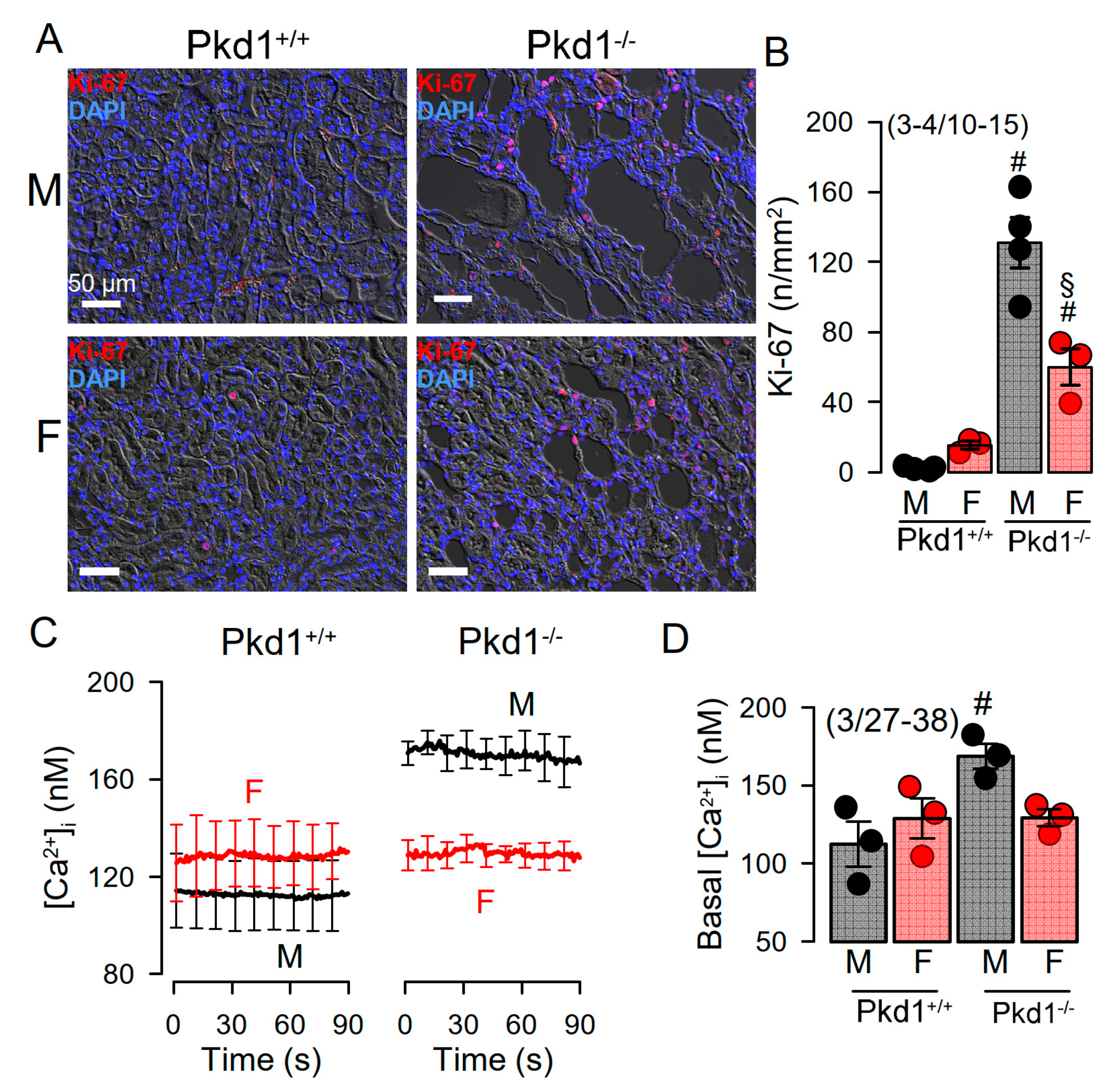
2.2. Cell Proliferation and Basal Ca2+ Levels Are More Enhanced in Renal Epithelial Cells from Pkd1−/− Males Than Pkd1−/− Females
2.3. Testosterone Augments ATP-Induced Whole Cell Currents in Female Pkd1+/+ Cells
2.4. Testosterone Enhances Intracellular Ca2+ Signals
3. Discussion
4. Materials and Methods
4.1. RT-PCR
4.2. Cell Culture
4.3. Renal Medullary Primary Cells Isolation
4.4. Animals and Treatments
4.5. Histologic Analysis, Cystic Index
4.6. Immunohistochemistry
4.7. Western Blotting
4.8. Ki-67 Assay
4.9. Patch Clamp
4.10. Iodide Quenching Experiments
4.11. Measurement of [Ca2+]i
4.12. Materials and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Gabow, P.A.; Johnson, A.M.; Kaehny, W.D.; Kimberling, W.J.; Lezotte, D.C.; Duley, I.T.; Jones, R.H. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int. 1992, 41, 1311–1319. [Google Scholar] [CrossRef]
- Stewart, J.H. End-stage renal failure appears earlier in men than in women with polycystic kidney disease. Am. J. Kidney Dis. 1994, 24, 181–183. [Google Scholar] [CrossRef]
- Johnson, A.M.; Gabow, P.A. Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J. Am. Soc. Nephrol. 1997, 8, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Cowley, B.D., Jr.; Rupp, J.C.; Muessel, M.J.; Gattone, V.H., 2nd. Gender and the effect of gonadal hormones on the progression of inherited polycystic kidney disease in rats. Am. J. Kidney Dis. 1997, 29, 265–272. [Google Scholar] [CrossRef]
- Dubey, R.K.; Jackson, E.K. Estrogen-induced cardiorenal protection: Potential cellular, biochemical, and molecular mechanisms. Am. J. Physiol. Ren. Physiol. 2001, 280, F365–F388. [Google Scholar] [CrossRef] [PubMed]
- Stringer, K.D.; Komers, R.; Osman, S.A.; Oyama, T.T.; Lindsley, J.N.; Anderson, S. Gender hormones and the progression of experimental polycystic kidney disease. Kidney Int. 2005, 68, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Gretz, N.; Ceccherini, I.; Kränzlin, B.; Klöting, I.; Devoto, M.; Rohmeiss, P.; Hocher, B.; Waldherr, R.; Romeo, G. Gender-dependent disease severity in autosomal polycystic kidney disease of rats. Kidney Int. 1995, 48, 496–500. [Google Scholar] [CrossRef][Green Version]
- Grantham, J.J.; Ye, M.; Gattone, V.H., 2nd; Sullivan, L.P. In vitro fluid secretion by epithelium from polycystic kidneys. J Clin. Invest. 1995, 95, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, K.; Devuyst, O.; Schwiebert, E.M.; Wilson, P.D.; Guggino, W.B. A role for CFTR in human autosomal dominant polycystic kidney disease. Am. J. Physiol. 1996, 270, C389–C399. [Google Scholar] [CrossRef]
- Davidow, C.J.; Maser, R.L.; Rome, L.A.; Calvet, J.P.; Grantham, J.J. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 1996, 50, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.; Teschemacher, B.; Schley, G.; Schillers, H.; Eckardt, K.U. Formation of cysts by principal-like MDCK cells depends on the synergy of cAMP- and ATP-mediated fluid secretion. J. Mol. Med. 2011, 89, 251–261. [Google Scholar] [CrossRef]
- Yang, B.; Sonawane, N.D.; Zhao, D.; Somlo, S.; Verkman, A.S. Small-Molecule CFTR Inhibitors Slow Cyst Growth in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2008, 19, 1300–1310. [Google Scholar] [CrossRef]
- Magenheimer, B.S.; St John, P.L.; Isom, K.S.; Abrahamson, D.R.; De Lisle, R.C.; Wallace, D.P.; Maser, R.L.; Grantham, J.J.; Calvet, J.P. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(-) Co-transporter-dependent cystic dilation. J. Am. Soc. Nephrol. 2006, 17, 3424–3437. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Glockner, J.F.; Rossetti, S.; Babovich-Vuksanovic, D.; Harris, P.C.; Torres, V.E. Autosomal dominant polycystic kidney disease coexisting with cystic fibrosis. J. Nephrol. 2006, 19, 529–534. [Google Scholar] [PubMed]
- O’Sullivan, D.A.; Torres, V.E.; Gabow, P.A.; Thibodeau, S.N.; King, B.F.; Bergstralh, E.J. Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 1998, 32, 976–983. [Google Scholar] [CrossRef]
- Persu, A.; Devuyst, O.; Lannoy, N.; Materne, R.; Brosnahan, G.; Gabow, P.A.; Pirson, Y.; Verellen-Dumoulin, C. CF gene and cystic fibrosis transmembrane conductance regulator expression in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2000, 11, 2285–2296. [Google Scholar] [CrossRef]
- Wilson, P.D.; Hovater, J.S.; Casey, C.C.; Fortenberry, J.A.; Schwiebert, E.M. ATP release mechanisms in primary cultures of epithelia derived from the cysts of polycystic kidneys. J. Am. Soc. Nephrol. 1999, 10, 218–229. [Google Scholar] [CrossRef]
- Schwiebert, E.M.; Wallace, D.P.; Braunstein, G.M.; King, S.R.; Peti-Peterdi, J.; Hanaoka, K.; Guggino, W.B.; Guay-Woodford, L.M.; Bell, P.D.; Sullivan, L.P.; et al. Autocrine extracellular purinergic signaling in epithelial cells derived from polycystic kidneys. Am. J. Physiol. Ren. Physiol. 2002, 282, F763–F775. [Google Scholar] [CrossRef]
- Kraus, A.; Grampp, S.; Goppelt-Struebe, M.; Schreiber, R.; Kunzelmann, K.; Peters, D.J.; Leipziger, J.; Schley, G.; Schodel, J.; Eckardt, K.U.; et al. P2Y2R is a direct target of HIF-1alpha and mediates secretion-dependent cyst growth of renal cyst-forming epithelial cells. Purinergic Signal. 2016, 12, 687–695. [Google Scholar] [CrossRef]
- Buchholz, B.; Faria, D.; Schley, G.; Schreiber, R.; Eckardt, K.U.; Kunzelmann, K. Anoctamin 1 induces calcium-activated chloride secretion and tissue proliferation in polycystic kidney disease. Kidney Int. 2014, 85, 1058–1067. [Google Scholar] [CrossRef]
- Buchholz, B.; Schley, G.; Faria, D.; Kroening, S.; Willam, C.; Schreiber, R.; Klanke, B.; Burzlaff, N.; Kunzelmann, K.; Eckardt, K.U. Hypoxia-Inducible Factor-1a Causes Renal Cyst Expansion through Calcium-Activated Chloride Secretion. J. Am. Soc. Nephrol. 2014, 25, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Buchholz, B.; Schreiber, R.; Kunzelmann, K. TMEM16A drives renal cyst growth by augmenting Ca(2+) signaling in M1 cells. J. Mol. Med. 2020, 98, 659–671. [Google Scholar] [CrossRef]
- Cabrita, I.; Kraus, A.; Scholz, J.K.; Skoczynski, K.; Schreiber, R.; Kunzelmann, K.; Buchholz, B. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat. Commun. 2020, 11, 4320. [Google Scholar] [CrossRef]
- Cabrita, I.; Talbi, K.; Kunzelmann, K.; Schreiber, R. Loss of PKD1 and PKD2 share common effects on intracellular Ca2+ signaling. Cell Calcium 2021, 97, 102413. [Google Scholar] [CrossRef]
- Cha, J.Y.; Wee, J.; Jung, J.; Jang, Y.; Lee, B.; Hong, G.S.; Chang, B.C.; Choi, Y.L.; Shin, Y.K.; Min, H.Y.; et al. Anoctamin 1 (TMEM16A) is essential for testosterone-induced prostate hyperplasia. Proc. Natl. Acad. Sci. USA 2015, 112, 9722–9727. [Google Scholar] [CrossRef]
- Martinez-Pinna, J.; Soriano, S.; Tudurí, E.; Nadal, A.; de Castro, F. A Calcium-Dependent Chloride Current Increases Repetitive Firing in Mouse Sympathetic Neurons. Front. Physiol. 2018, 9, 508. [Google Scholar] [CrossRef]
- Chen, D.; Meng, W.; Shu, L.; Liu, S.; Gu, Y.; Wang, X.; Feng, M. ANO1 in urethral SMCs contributes to sex differences in urethral spontaneous tone. Am. J. Physiol Ren. Physiol 2020. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.B. The gender gap in cystic fibrosis survival. J. Gend. Specif. Med. JGSM Off. J. Partnersh. Women’s Health Columbia 1999, 2, 47–51. [Google Scholar]
- Sweezey, N.; Tchepichev, S.; Gagnon, S.; Fertuck, K.; O’Brodovich, H. Female gender hormones regulate mRNA levels and function of the rat lung epithelial Na channel. Am. J. Physiol 1998, 274, C379–C386. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Harvey, B.J. Estrogen and the cystic fibrosis gender gap. Steroids 2014, 81, 4–8. [Google Scholar] [CrossRef]
- Kraus, A.; Peters, D.J.M.; Klanke, B.; Weidemann, A.; Willam, C.; Schley, G.; Kunzelmann, K.; Eckardt, K.U.; Buchholz, B. HIF-1alpha promotes cyst progression in a mouse model of autosomal dominant polycystic kidney disease. Kidney Int. 2018, 94, 887–899. [Google Scholar] [CrossRef]
- Lantinga-van Leeuwen, I.S.; Leonhard, W.N.; van de Wal, A.; Breuning, M.H.; Verbeek, S.; de Heer, E.; Peters, D.J. Transgenic mice expressing tamoxifen-inducible Cre for somatic gene modification in renal epithelial cells. Genesis 2006, 44, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Nagao, S.; Kusaka, M.; Nishii, K.; Marunouchi, T.; Kurahashi, H.; Takahashi, H.; Grantham, J. Androgen receptor pathway in rats with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2052–2062. [Google Scholar] [CrossRef]
- Gaeggeler, H.P.; Gonzalez-Rodriguez, E.; Jaeger, N.F.; Loffing-Cueni, D.; Norregaard, R.; Loffing, J.; Horisberger, J.D.; Rossier, B.C. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J. Am. Soc. Nephrol. 2005, 16, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Blok, L.J.; Perry, J.E.; Lindzey, J.K.; Tindall, D.J. Calcium regulation of androgen receptor expression in the human prostate cancer cell line LNCaP. Endocrinology 1995, 136, 2172–2178. [Google Scholar] [CrossRef]
- Estrada, M.; Liberona, J.L.; Miranda, M.; Jaimovich, E. Aldosterone- and testosterone-mediated intracellular calcium response in skeletal muscle cell cultures. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E132–E139. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Honisch, S.; Liu, G.; Schmidt, S.; Alkahtani, S.; AlKahtane, A.A.; Stournaras, C.; Lang, F. Up-regulation of Orai1 expression and store operated Ca(2+) entry following activation of membrane androgen receptors in MCF-7 breast tumor cells. BMC Cancer 2015, 15, 995. [Google Scholar] [CrossRef]
- Steinsapir, J.; Socci, R.; Reinach, P. Effects of androgen on intracellular calcium of LNCaP cells. Biochem. Biophys. Res. Commun. 1991, 179, 90–96. [Google Scholar] [CrossRef]
- Sun, Y.H.; Gao, X.; Tang, Y.J.; Xu, C.L.; Wang, L.H. Androgens induce increases in intracellular calcium via a G protein-coupled receptor in LNCaP prostate cancer cells. J. Androl. 2006, 27, 671–678. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Ibarra, C.; Estrada, M.; Chiong, M.; Soto, D.; Parra, V.; Diaz-Araya, G.; Jaimovich, E.; Lavandero, S. Testosterone induces an intracellular calcium increase by a nongenomic mechanism in cultured rat cardiac myocytes. Endocrinology 2006, 147, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Berry, P.A.; Birnie, R.; Droop, A.P.; Maitland, N.J.; Collins, A.T. The calcium sensor STIM1 is regulated by androgens in prostate stromal cells. Prostate 2011, 71, 1646–1655. [Google Scholar] [CrossRef]
- Sheridan, J.T.; Gilmore, R.C.; Watson, M.J.; Archer, C.B.; Tarran, R. 17β-Estradiol inhibits phosphorylation of stromal interaction molecule 1 (STIM1) protein: Implication for store-operated calcium entry and chronic lung diseases. J. Biol. Chem. 2013, 288, 33509–33518. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamanaka, K.; Miyamoto, Y.; Waki, H.; Gouraud, S. Trpv4 involvement in the sex differences in blood pressure regulation in spontaneously hypertensive rats. Physiol. Genom. 2018, 50, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yu, K.; Perez-Cornejo, P.; Cui, Y.; Arreola, J.; Hartzell, H.C. Voltage- and calcium-dependent gating of TMEM16A/Ano1 chloride channels are physically coupled by the first intracellular loop. Proc. Natl. Acad. Sci. USA 2011, 108, 8891–8896. [Google Scholar] [CrossRef]
- Jin, X.; Shah, S.; Liu, Y.; Zhang, H.; Lees, M.; Fu, Z.; Lippiat, J.D.; Beech, D.J.; Sivaprasadarao, A.; Baldwin, S.A.; et al. Activation of the Cl- Channel ANO1 by Localized Calcium Signals in Nociceptive Sensory Neurons Requires Coupling with the IP3 Receptor. Sci. Signal. 2013, 6, ra73. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Benedetto, R.; Fonseca, A.; Wanitchakool, P.; Sirianant, L.; Skryabin, B.V.; Schenk, L.K.; Pavenstadt, H.; Schreiber, R.; Kunzelmann, K. Differential effects of anoctamins on intracellular calcium signals. FASEB J. 2017, 31, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, L.; Caputo, A.; Ubby, I.; Bussani, E.; Zegarra-Moran, O.; Ravazzolo, R.; Pagani, F.; Galietta, L.J. Regulation of TMEM16A chloride channel properties by alternative splicing. J. Biol. Chem. 2009, 284, 33360–33368. [Google Scholar] [CrossRef] [PubMed]
- Lerias, J.; Pinto, M.; Benedetto, R.; Schreiber, R.; Amaral, M.; Aureli, M.; Kunzelmann, K. Compartmentalized crosstalk of CFTR and TMEM16A (ANO1) through EPAC1 and ADCY1. Cell Signal. 2018, 44, 10–19. [Google Scholar] [CrossRef] [PubMed]
- AlDehni, F.; Spitzner, M.; Martins, J.R.; Barro Soria, R.; Schreiber, R.; Kunzelmann, K. Role of bestrophin for proliferation and in epithelial to mesenchymal transition. J. Am. Soc. Nephrol. 2009, 20, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Lantinga-van Leeuwen, I.S.; Leonhard, W.N.; van der Wal, A.; Breuning, M.H.; de Heer, E.; Peters, D.J. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum. Mol. Genet. 2007, 16, 3188–3196. [Google Scholar] [CrossRef] [PubMed]
- Greger, R.; Kunzelmann, K. Simultaneous recording of the cell membrane potential and properties of the cell attached membrane of HT29 colon carcinoma and CF-PAC cells. Pflügers Arch. 1991, 419, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Uliyakina, I.; Kongsuphol, P.; Warth, R.; Mirza, M.; Martins, J.R.; Kunzelmann, K. Expression and Function of Epithelial Anoctamins. J. Biol. Chem. 2010, 285, 7838–7845. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Accession Number | Primer | Size (bp) |
|---|---|---|
| Tmem16a NM_001242349.2 | s: 5′-GTGACAAGACCTGCAGCTAC as: 5′-GCTGCAGCTGTGGAGATTC | 406 |
| Cftr NM_021050.2 | s: 5′-GAATCCCCAGCTTATCCACG as: 5′-CTTCACCATCATCTTCCCTAG | 544 |
| Pkd1 NM_013630.2 | s: 5′-CTTCTACTTTGCCCATGAGG as: 5′-CTTCTACTTGCACCTCTGTC | 473 |
| Esr1 NM_007956.5 | s: 5′-CTCAAGATGCCCATGGAGAG as: 5′-GTTTCCTTTCTCGTTACTGCTG | 441 |
| Esr2 NM_207707.1 | s: 5′-GACCTACGCAAGACATGGAG as: 5′-CTTGGACTAGTAACAGGGCTG | 436 |
| Esrra NM_007953.2 | s: 5′-CAGGGCAGTGGGAAGCTAG as: 5′-GCTACTGCCAGAGGTCCAG | 362 |
| Esrrb NM_011934.4 | s: 5′-GTGGTATCATGGAGGACTCC as: 5′-GTCAATGGCTTTTTAGCAGGTG | 388 |
| Esrrg NM_011935.3 | s: 5′-CAGCACCATCGTAGAGGATC as: 5′-CATGGCATAGATCTTCTCTGG | 442 |
| Gapdh NM_001289726 | s: 5′-GTATTGGGCGCCTGGTCAC as: 5′-CTCCTGGAAGATGGTGATGG | 200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talbi, K.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. Gender-Dependent Phenotype in Polycystic Kidney Disease Is Determined by Differential Intracellular Ca2+ Signals. Int. J. Mol. Sci. 2021, 22, 6019. https://doi.org/10.3390/ijms22116019
Talbi K, Cabrita I, Schreiber R, Kunzelmann K. Gender-Dependent Phenotype in Polycystic Kidney Disease Is Determined by Differential Intracellular Ca2+ Signals. International Journal of Molecular Sciences. 2021; 22(11):6019. https://doi.org/10.3390/ijms22116019
Chicago/Turabian StyleTalbi, Khaoula, Inês Cabrita, Rainer Schreiber, and Karl Kunzelmann. 2021. "Gender-Dependent Phenotype in Polycystic Kidney Disease Is Determined by Differential Intracellular Ca2+ Signals" International Journal of Molecular Sciences 22, no. 11: 6019. https://doi.org/10.3390/ijms22116019
APA StyleTalbi, K., Cabrita, I., Schreiber, R., & Kunzelmann, K. (2021). Gender-Dependent Phenotype in Polycystic Kidney Disease Is Determined by Differential Intracellular Ca2+ Signals. International Journal of Molecular Sciences, 22(11), 6019. https://doi.org/10.3390/ijms22116019