
Comparative Transcriptome-Based Mining of Senescence-Related MADS, NAC, and WRKY Transcription Factors in the Rapid-Senescence Line DLS-91 of Brassica rapa

 , , ,
, , , 
Abstract
1. Introduction
2. Results
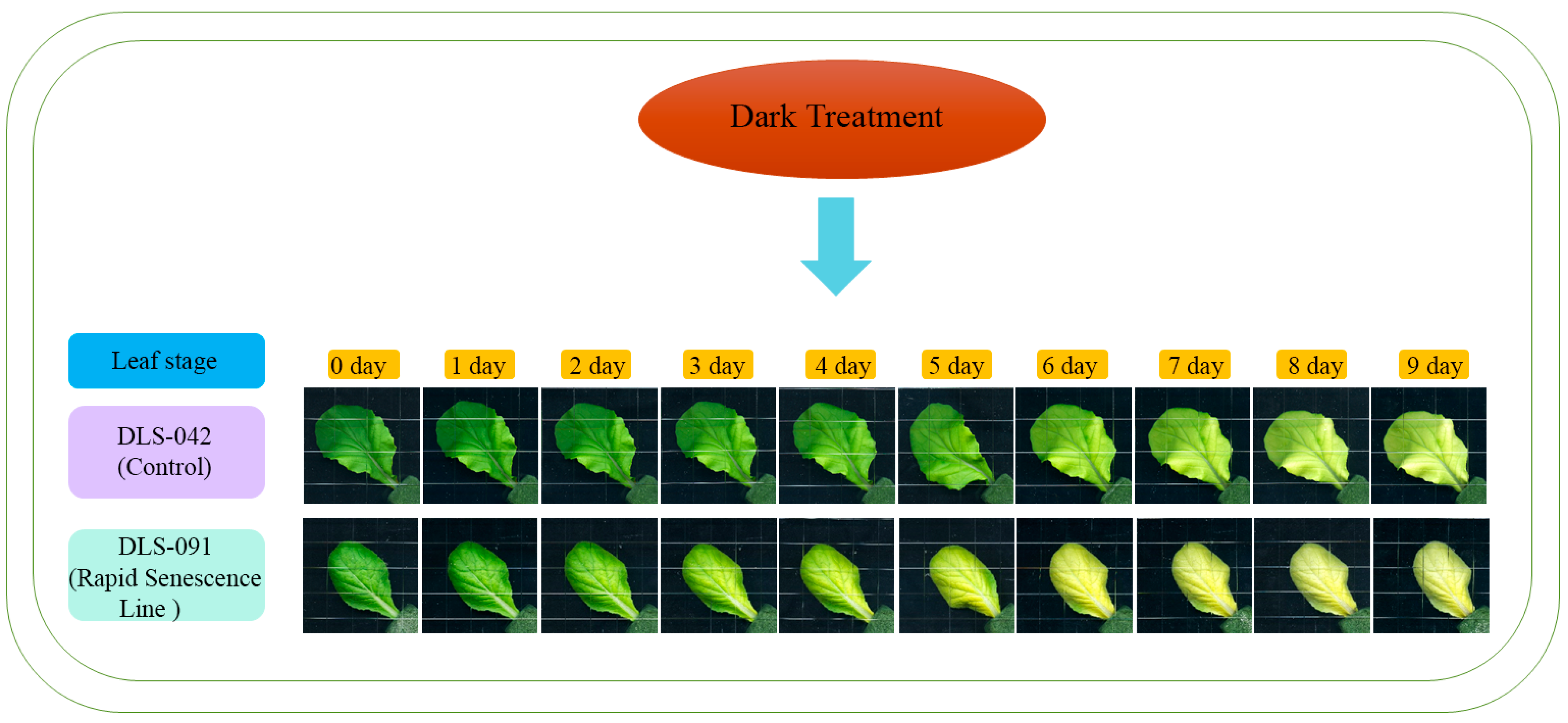
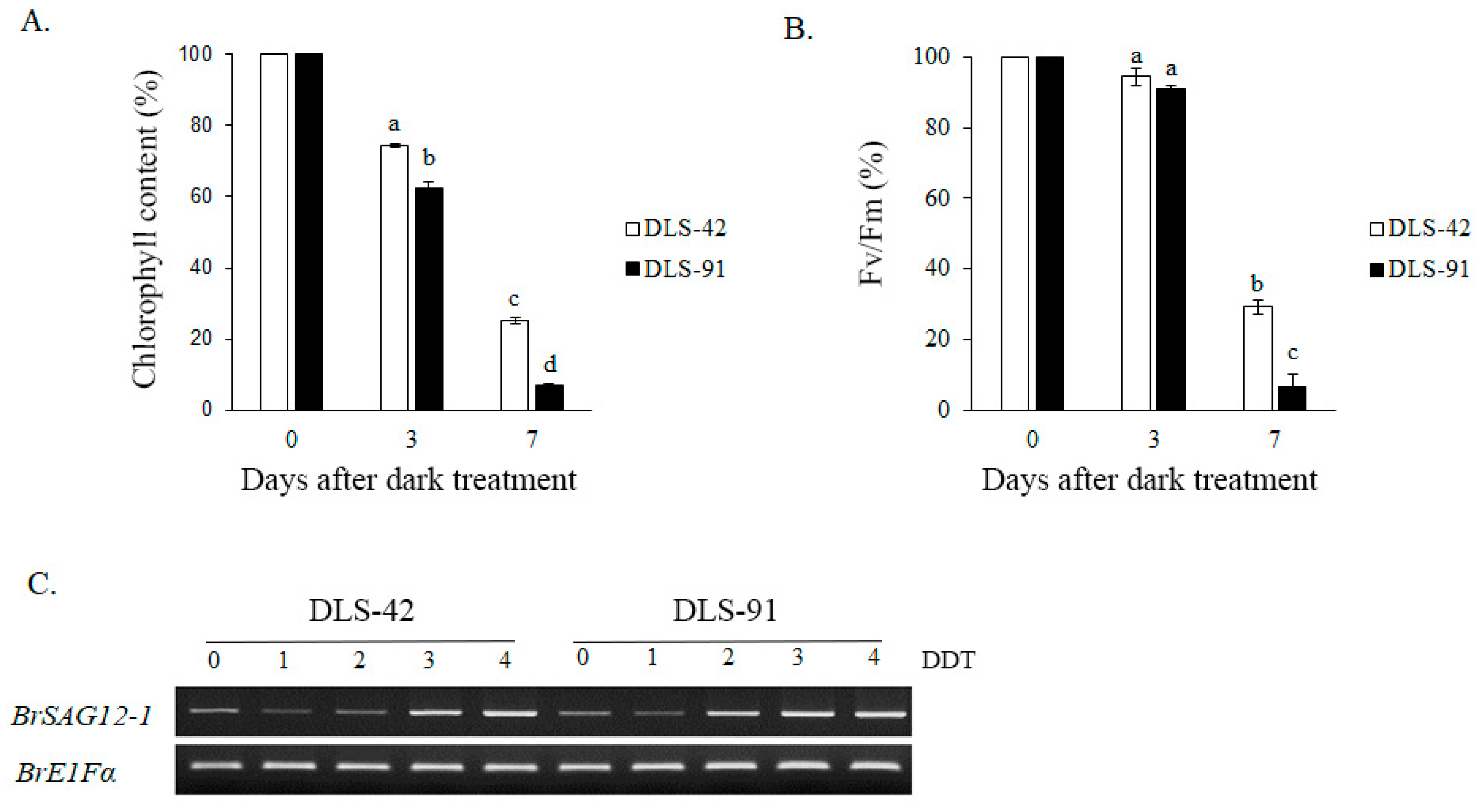
2.1. DLS-91 Exhibited More Rapid Leaf Senescence than the Control Line during Dark Treatment
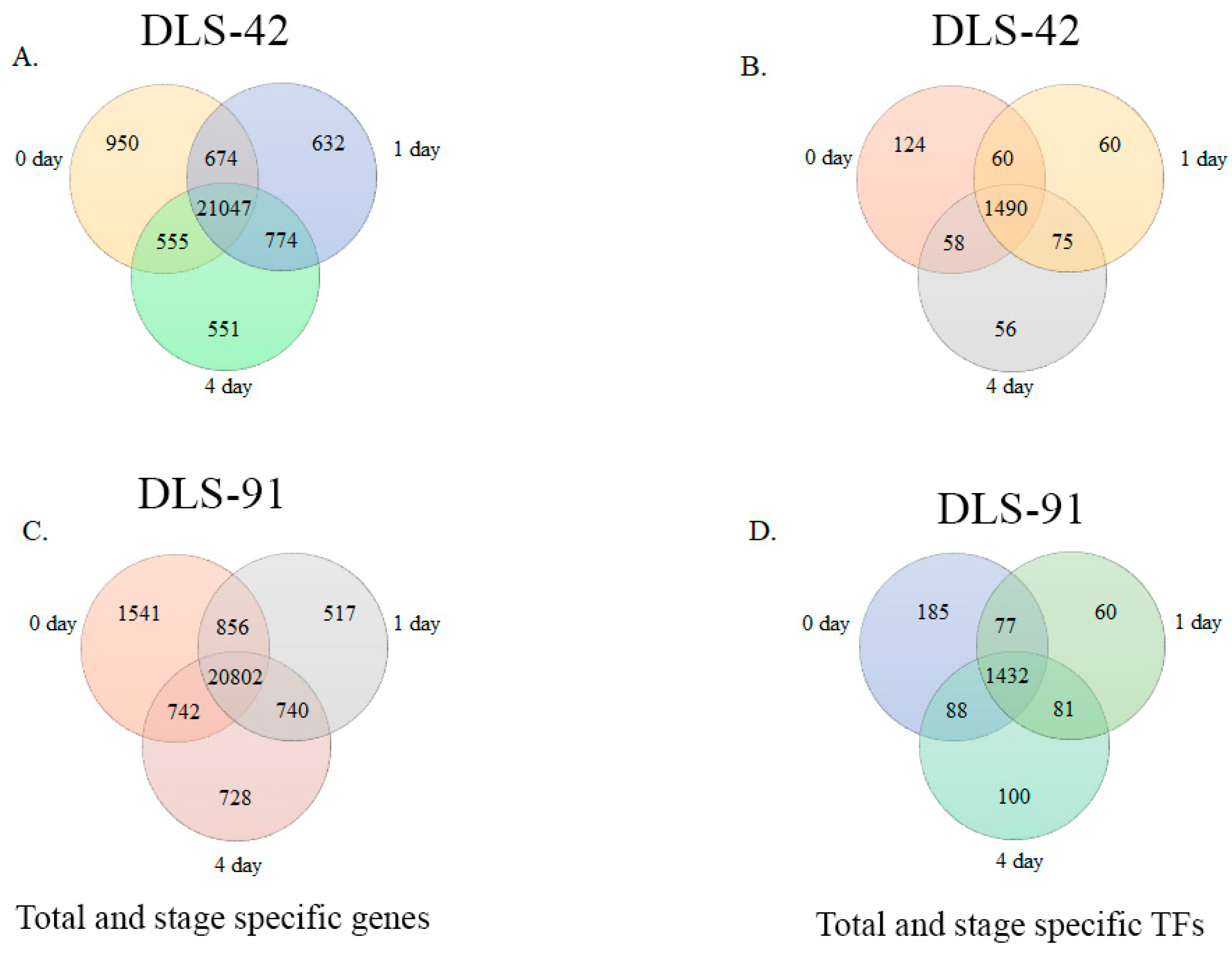
2.2. Assessment of DLS-42 and DLS-91 RNA Sequence Data and Identification of Stage- and Genotype-Specific Genes
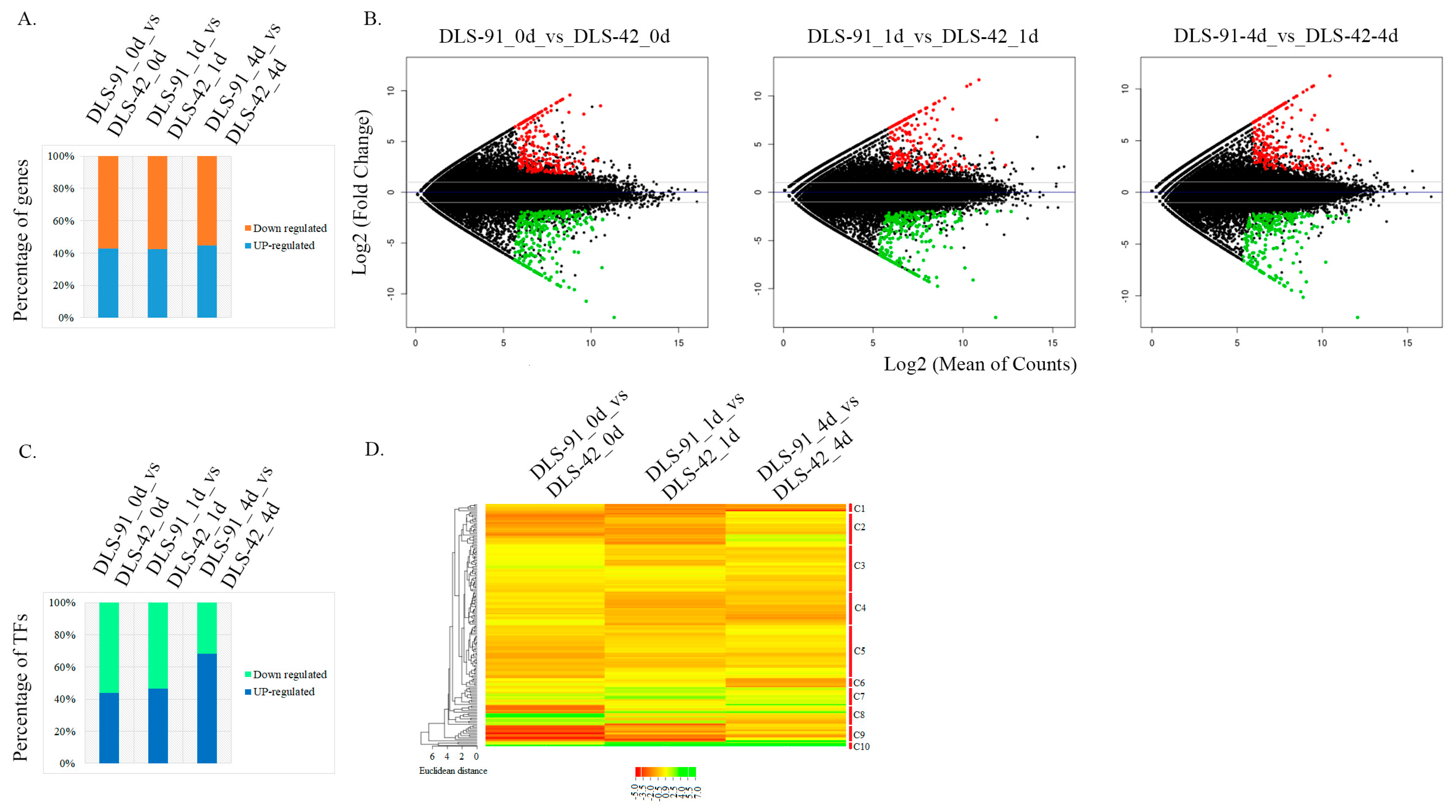
2.3. Identification of DEGs and TF Genes in 0-, 1-, and 4-Day Dark-Treated Samples
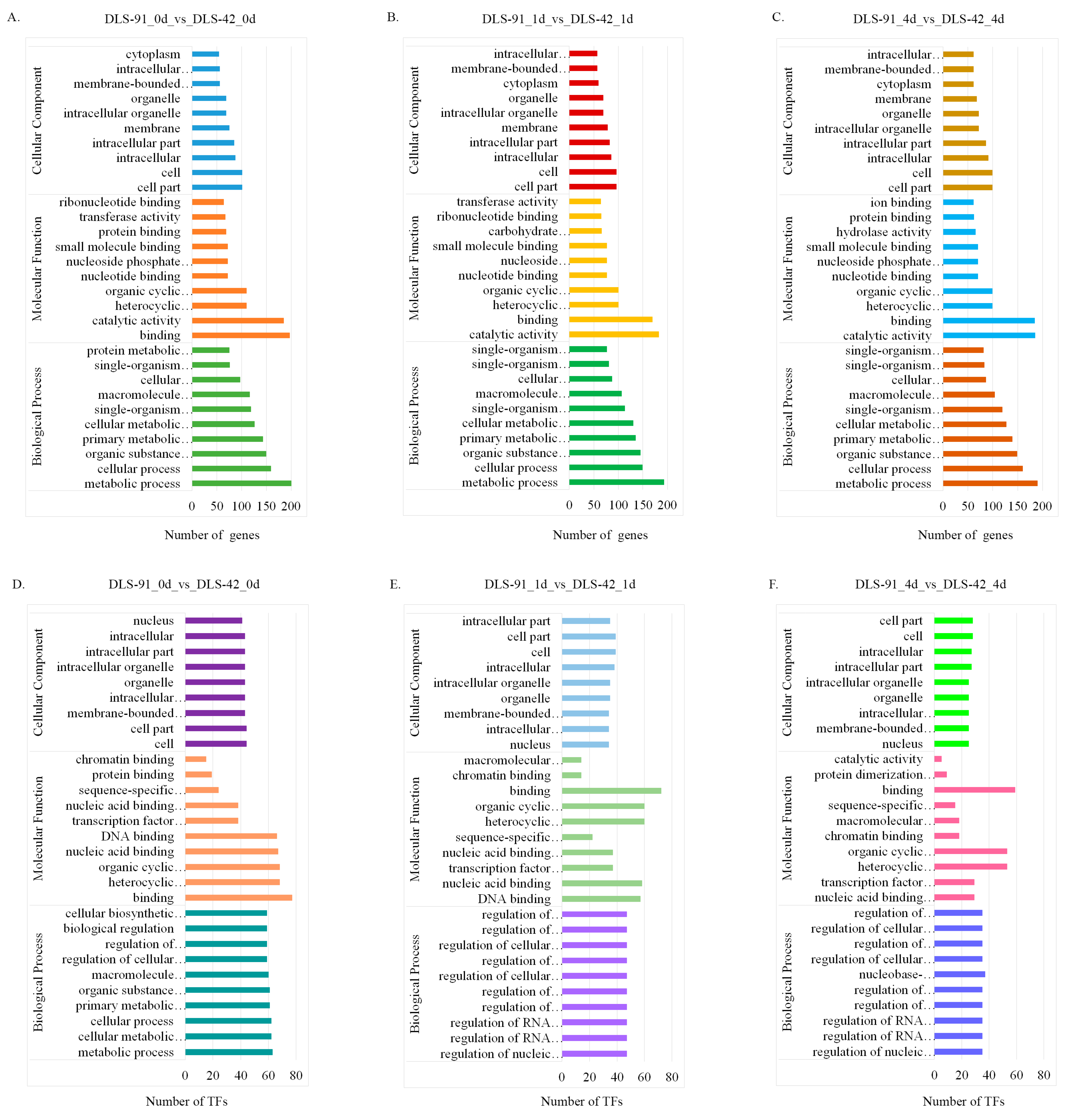
2.4. Functional Enrichment Analysis of DEGs
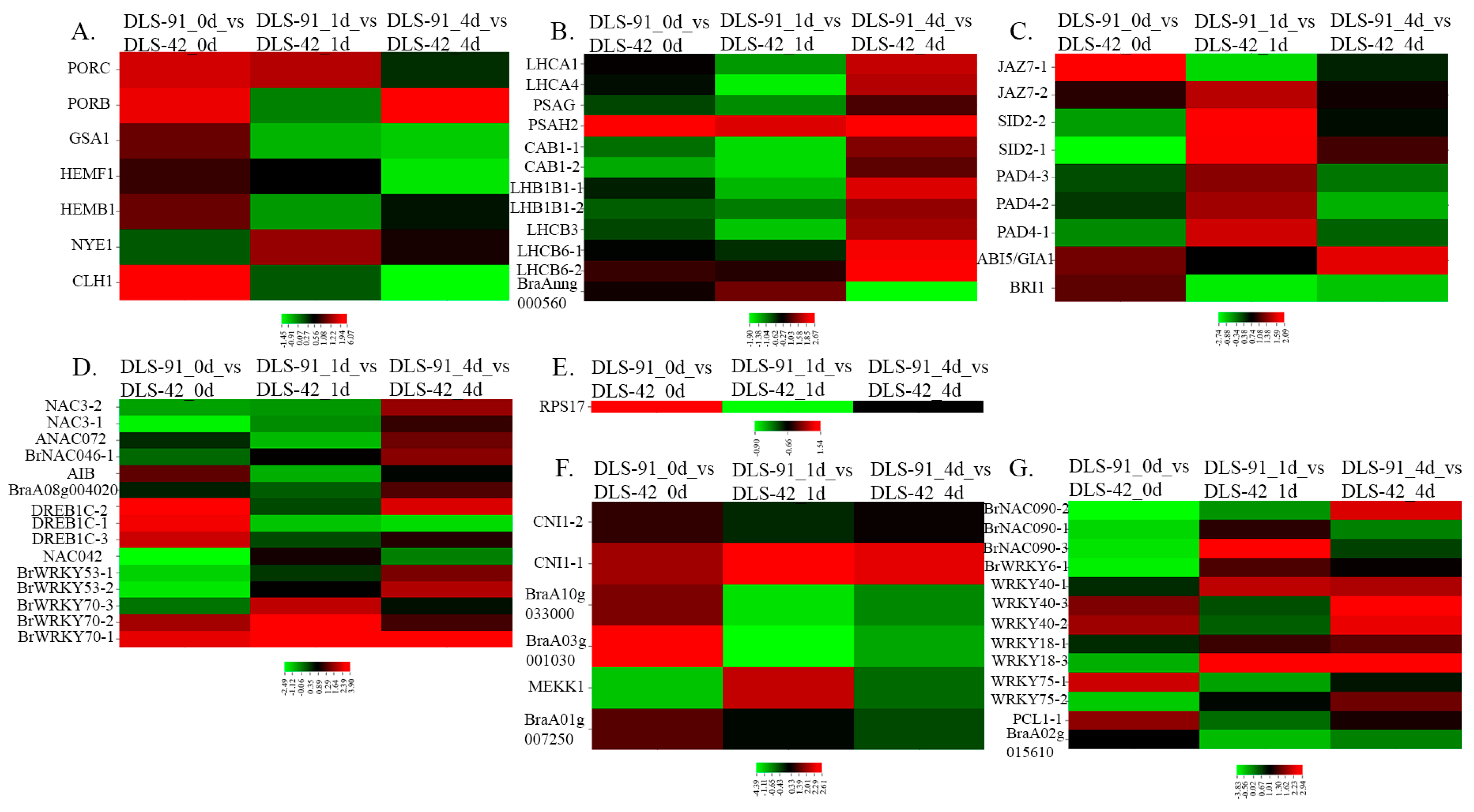
2.5. Transcript Abundance of Genes with Various Functions during Senescence
2.6. Identification of TFs Involved in Senescence
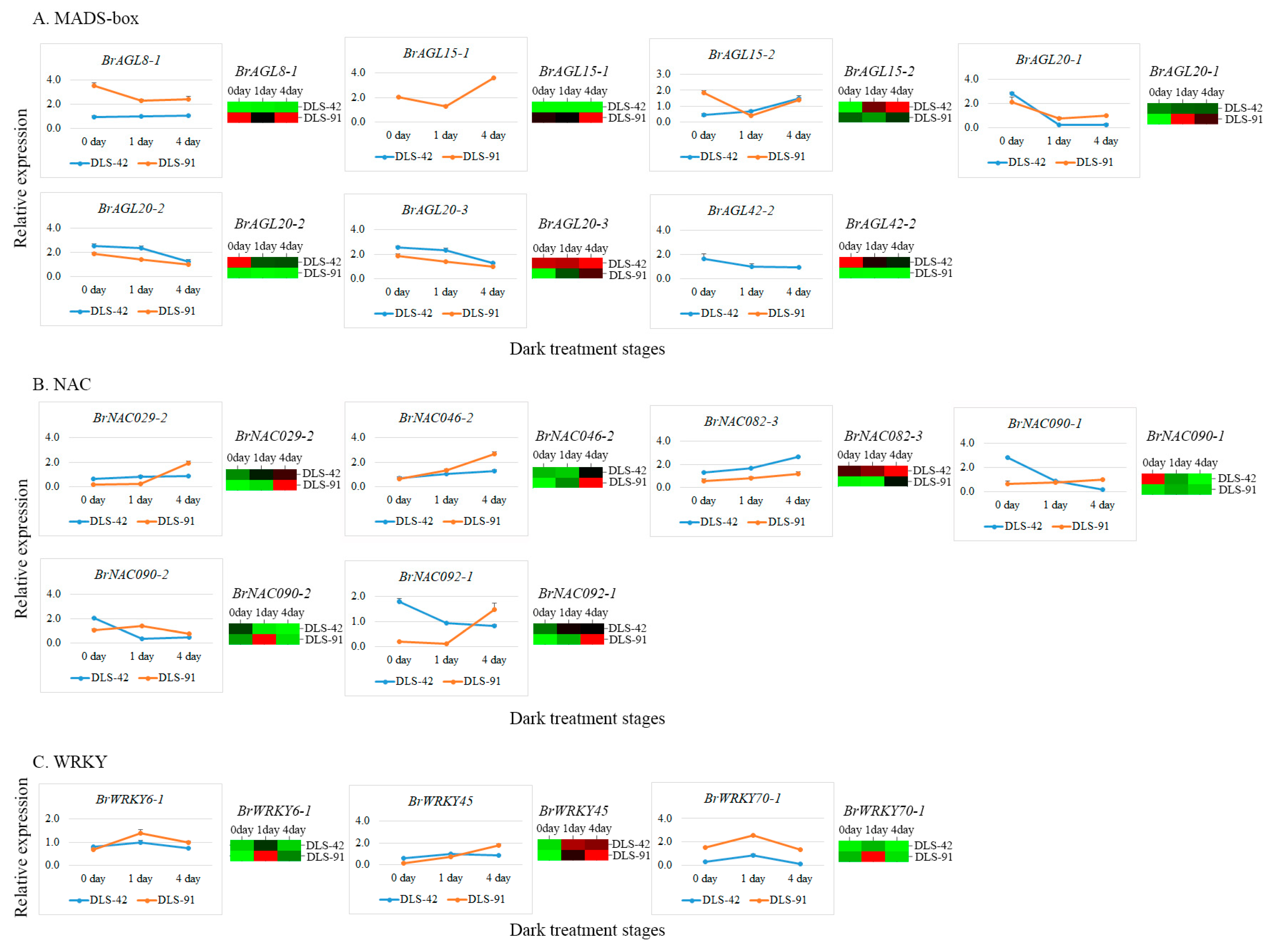
2.7. qRT-PCR Validation of MADS, NAC, and WRKY TFs
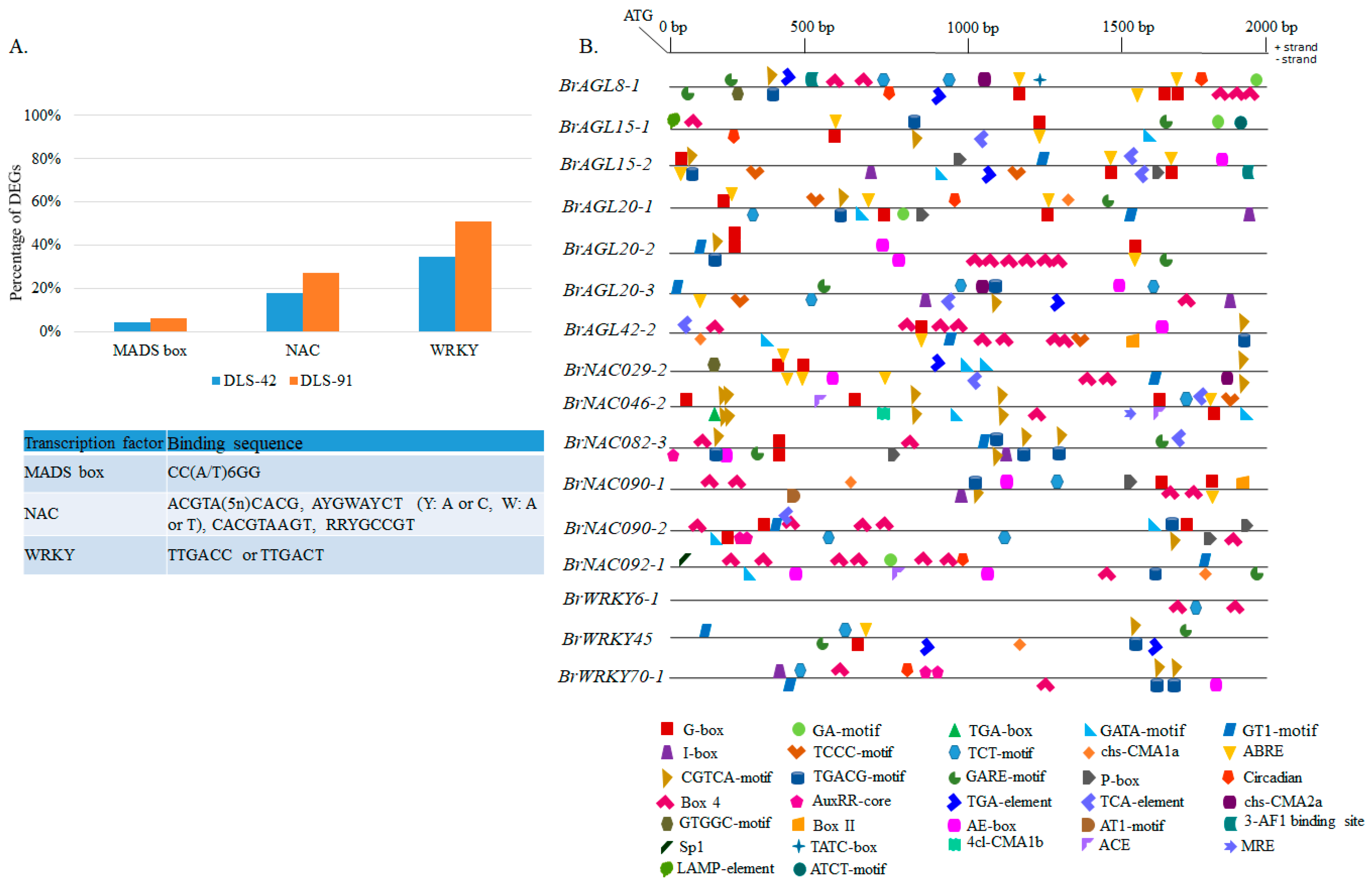
2.8. Genomic Analysis of MADS, NAC, and WRKY TFs
2.9. Prediction of MADS, NAC, and WRKY Binding Sites in DEGs and Identification of Other Cis-Elements in Promoter Regions of Selected TFs
3. Discussion
3.1. Physiological and Molecular Responses of DLS-91 during DLS
3.2. Roles of MADS, NAC, and WRKY TFs in Various Processes Related to Senescence
4. Materials and Methods
4.1. Estimation of Chlorophyll Content and Selection of Plant Materials
4.2. Photochemical Efficiency
4.3. RNA Sequencing and Transcript Annotation
4.4. Identification of DEGs and TFs and GO Annotation
4.5. Validation of Transcript Abundance by qRT-PCR
4.6. Promoter Analysis
4.7. Genomic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DDT | Days after dark treatment |
| DEGs | Differentially expressed genes |
| TFs | transcription factors |
| SAGs | Senescence associated genes |
| FPKM | Fragments per kilo base of transcript per million fragments mapped |
| FC | Fold change |
| MA plot | Bland–Altman plot |
| GO | Gene ontology |
References
- Song, Y.; Yang, C.; Gao, S.; Zhang, W.; Li, L.; Kuai, B. Age-triggered and dark-induced leaf senescence require the bHLH transcription factors PIF3, 4, and 5. Mol. Plant 2014, 7, 1776–1787. [Google Scholar] [CrossRef]
- Sarwat, M.; Naqvi, A.R.; Ahmad, P.; Ashraf, M.; Akram, N.A. Phytohormones and microRNAs as sensors and regulators of leaf senescence: Assigning macro roles to small molecules. Biotechnol. Adv. 2013, 31, 1153–1171. [Google Scholar] [CrossRef]
- Maillard, A.; Diquélou, S.; Billard, V.; Laîné, P.; Garnica, M.; Prudent, M.; Garcia-Mina, J.M.; Yvin, J.C.; Ourry, A. Leaf mineral nutrient remobilization during leaf senescence and modulation by nutrient deficiency. Front. Plant Sci. 2015, 6, 317. [Google Scholar] [CrossRef]
- Bengoa Luoni, S.; Astigueta, F.H.; Nicosia, S.; Moschen, S.; Fernandez, P.; Heinz, R. Transcription Factors Associated with Leaf Senescence in Crops. Plants 2019, 8, 411. [Google Scholar] [CrossRef] [PubMed]
- Buchanan-Wollaston, V.; Earl, S.; Harrison, E.; Mathas, E.; Navabpour, S.; Page, T.; Pink, D. The molecular analysis of leaf senescence—A genomics approach. Plant Biotechnol. J. 2003, 1, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Sharma, V.; Kishore, K. Leaf senescence: An overview. Indian J. Plant Physiol. 2016, 21, 225–238. [Google Scholar] [CrossRef]
- Gan, S. Leaf Senescence as an Important Target for Improving Crop Production. Adv. Crop Sci. Technol. 2014, 2, 116–118. [Google Scholar] [CrossRef]
- Lim, K.T.; Kim, J.; Chung, J.H. Development of Long-Term Storage Technology for Chinese Cabbage—Physiological Characteristics of Postharvest Freshness in a Cooler with a Monitoring and Control Interface. J. Biosyst. Eng. 2014, 39, 194–204. [Google Scholar] [CrossRef]
- Tan, X.-L.; Zhao, Y.-T.; Shan, W.; Kuang, J.-F.; Lu, W.-J.; Su, X.-G.; Tao, N.-G.; Lakshmanan, P.; Chen, J.-Y. Melatonin delays leaf senescence of postharvest Chinese flowering cabbage through ROS homeostasis. Food Res. Int. 2020, 138, 109790. [Google Scholar] [CrossRef]
- Buchanan-Wollaston, V.; Page, T.; Harrison, E.; Breeze, E.; Lim, P.O.; Nam, H.G.; Lin, J.-F.; Wu, S.-H.; Swidzinski, J.; Ishizaki, K.; et al. Comparative transcriptome analysis reveals significant differences in gene expression and signalling pathways between developmental and dark/starvation-induced senescence in Arabidopsis. Plant J. 2005, 42, 567–585. [Google Scholar] [CrossRef]
- Sobieszczuk-Nowicka, E.; Wrzesinski, T.; Bagniewska-Zadworna, A.; Kubala, S.; Rucinska-Sobkowiak, R.; Polcyn, W.; Misztal, L.; Mattoo, A.K. Physio-Genetic Dissection of Dark-Induced Leaf Senescence and Timing Its Reversal in Barley. Plant Physiol. 2018, 178, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.M.; Amasino, R.M. Senescence is induced in individually darkened Arabidopsis leaves, but inhibited in whole darkened plants. Plant Physiol. 2001, 127, 876–886. [Google Scholar] [CrossRef]
- Park, S.-Y.; Yu, J.-W.; Park, J.-S.; Li, J.; Yoo, S.-C.; Lee, N.-Y.; Lee, S.-K.; Jeong, S.-W.; Seo, H.S.; Koh, H.-J.; et al. The Senescence-Induced Staygreen Protein Regulates Chlorophyll Degradation. Plant Cell 2007, 19, 1649–1664. [Google Scholar] [CrossRef] [PubMed]
- Noodén, L.D.; Guiamét, J.J.; John, I. Senescence mechanisms. Physiol. Plantarum 1997, 101, 746–753. [Google Scholar] [CrossRef]
- Ougham, H.; Hörtensteiner, S.; Armstead, I.; Donnison, I.; King, I.; Thomas, H.; Mur, L. The control of chlorophyll catabolism and the status of yellowing as a biomarker of leaf senescence. Plant Biol. 2008, 10, 4–14. [Google Scholar] [CrossRef]
- Pruzinská, A.; Tanner, G.; Aubry, S.; Anders, I.; Moser, S.; Müller, T.; Ongania, K.-H.; Kräutler, B.; Youn, J.-Y.; Liljegren, S.J.; et al. Chlorophyll breakdown in senescent Arabidopsis leaves. Characterization of chlorophyll catabolites and of chlorophyll catabolic enzymes involved in the degreening reaction. Plant Physiol. 2005, 139, 52–63. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Park, S.Y.; Paek, N.C. The Divergent Roles of STAYGREEN (SGR) Homologs in Chlorophyll Degradation. Mol. Cells 2015, 38, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Hörtensteiner, S. Chlorophyll degradation during senescence. Annu. Rev. Plant Biol. 2006, 57, 55–77. [Google Scholar] [CrossRef]
- Kusaba, M.; Ito, H.; Morita, R.; Iida, S.; Sato, Y.; Fujimoto, M.; Kawasaki, S.; Tanaka, R.; Hirochika, H.; Nishimura, M.; et al. Rice NON-YELLOW COLORING1 Is Involved in Light-Harvesting Complex II and Grana Degradation during Leaf Senescence. Plant Cell 2007, 19, 1362–1375. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Ohta, H.; Okawa, K.; Iwamatsu, A.; Shimada, H.; Masuda, T.; Takamiya, K. Cloning of chlorophyllase, the key enzyme in chlorophyll degradation: Finding of a lipase motif and the induction by methyl jasmonate. Proc. Natl. Acad. Sci. USA 1999, 96, 15362–15367. [Google Scholar] [CrossRef]
- Pružinská, A.; Tanner, G.; Anders, I.; Roca, M.; Hörtensteiner, S. Chlorophyll breakdown: Pheophorbide a oxygenase is a Rieske-type iron–sulfur protein, encoded by the accelerated cell death 1 gene. Proc. Natl. Acad. Sci. USA 2003, 100, 15259–15264. [Google Scholar] [CrossRef] [PubMed]
- Pružinská, A.; Anders, I.; Aubry, S.; Schenk, N.; Tapernoux-Lüthi, E.; Müller, T.; Kräutler, B.; Hörtensteiner, S. In Vivo Participation of Red Chlorophyll Catabolite Reductase in Chlorophyll Breakdown. Plant Cell 2007, 19, 369–387. [Google Scholar] [CrossRef]
- Suzuki, Y.; Amano, T.; Shioi, Y. Characterization and Cloning of the Chlorophyll-Degrading Enzyme Pheophorbidase from Cotyledons of Radish. Plant Physiol. 2006, 140, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Armstead, I.; Donnison, I.; Aubry, S.; Harper, J.; Hörtensteiner, S.; James, C.; Mani, J.; Moffet, M.; Ougham, H.; Roberts, L.; et al. Cross-species identification of Mendel’s I locus. Science 2007, 315, 73. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Gordon, M.I.; Amarasinghe, V.; Strauss, S.H. Extensive transcriptome changes during seasonal leaf senescence in field-grown black cottonwood (Populus trichocarpa Nisqually-1). Sci. Rep. 2020, 10, 6581. [Google Scholar] [CrossRef]
- Moschen, S.; Marino, J.; Nicosia, S.; Higgins, J.; Alseekh, S.; Astigueta, F.; Bengoa Luoni, S.; Rivarola, M.; Fernie, A.R.; Blanchet, N.; et al. Exploring gene networks in two sunflower lines with contrasting leaf senescence phenotype using a system biology approach. BMC Plant Biol. 2019, 19, 446. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, M.; Tian, L.; Wang, S.; Wu, L.; Ku, L.; Zhang, J.; Song, X.; Liu, H.; Chen, Y. Global transcriptome analysis of the maize (Zea mays L.) inbred line 08LF during leaf senescence initiated by pollination-prevention. PLoS ONE 2017, 12, e0185838. [Google Scholar] [CrossRef]
- Kim, J.H.; Woo, H.R.; Kim, J.; Lim, P.O.; Lee, I.C.; Choi, S.H.; Hwang, D.; Nam, H.G. Trifurcate Feed-Forward Regulation of Age-Dependent Cell Death Involving miR164 in Arabidopsis. Science 2009, 323, 1053–1057. [Google Scholar] [CrossRef]
- Balazadeh, S.; Siddiqui, H.; Allu, A.D.; Matallana-Ramirez, L.P.; Caldana, C.; Mehrnia, M.; Zanor, M.I.; Köhler, B.; Mueller-Roeber, B. A gene regulatory network controlled by the NAC transcription factor ANAC092/AtNAC2/ORE1 during salt-promoted senescence. Plant J. 2010, 62, 250–264. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Y.; Turečková, V.; Xue, G.-P.; Fernie, A.R.; Mueller-Roeber, B.; Balazadeh, S. The NAC Transcription Factor SlNAP2 Regulates Leaf Senescence and Fruit Yield in Tomato. Plant Physiol. 2018, 177, 1286–1302. [Google Scholar] [CrossRef]
- Oda-Yamamizo, C.; Mitsuda, N.; Sakamoto, S.; Ogawa, D.; Ohme-Takagi, M.; Ohmiya, A. The NAC transcription factor ANAC046 is a positive regulator of chlorophyll degradation and senescence in Arabidopsis leaves. Sci. Rep. 2016, 6, 23609. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Nam, H.G.; Lim, P.O. Regulatory network of NAC transcription factors in leaf senescence. Curr. Opin. Plant Biol. 2016, 33, 48–56. [Google Scholar] [CrossRef]
- Huang, R.; Liu, D.; Huang, M.; Ma, J.; Li, Z.; Li, M.; Sui, S. CpWRKY71, a WRKY Transcription Factor Gene of Wintersweet (Chimonanthus praecox), Promotes Flowering and Leaf Senescence in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 5325. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Cui, X.; Zhao, P.; Sun, M.; Yang, B.; Deyholos, M.K.; Li, Y.; Zhao, X.; Jiang, Y.Q. WRKY42 transcription factor positively regulates leaf senescence through modulating SA and ROS synthesis in Arabidopsis thaliana. Plant J. 2020, 104, 171–184. [Google Scholar] [CrossRef]
- Chen, L.; Xiang, S.; Chen, Y.; Li, D.; Yu, D. Arabidopsis WRKY45 Interacts with the DELLA Protein RGL1 to Positively Regulate Age-Triggered Leaf Senescence. Mol. Plant 2017, 10, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Li, L.; Wei, H.; Wang, H.; Su, J.; Guo, Y.; Yu, S. Identification of the group IIa WRKY subfamily and the functional analysis of GhWRKY17 in upland cotton (Gossypium hirsutum L.). PLoS ONE 2018, 13, e0191681. [Google Scholar] [CrossRef]
- Gu, L.; Dou, L.; Guo, Y.; Wang, H.; Li, L.; Wang, C.; Ma, L.; Wei, H.; Yu, S. The WRKY transcription factor GhWRKY27 coordinates the senescence regulatory pathway in upland cotton (Gossypium hirsutum L.). BMC Plant Biol. 2019, 19, 116. [Google Scholar] [CrossRef]
- Gu, L.; Wei, H.; Wang, H.; Su, J.; Yu, S. Characterization and functional analysis of GhWRKY42, a group IId WRKY gene, in upland cotton (Gossypium hirsutum L.). BMC Genet. 2018, 19, 48. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Ma, Q.; Zhang, C.; Wang, C.; Wei, H.; Wang, H.; Yu, S. The Cotton GhWRKY91 Transcription Factor Mediates Leaf Senescence and Responses to Drought Stress in Transgenic Arabidopsis thaliana. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef]
- Zhao, M.-M.; Zhang, X.-W.; Liu, Y.-W.; Li, K.; Tan, Q.; Zhou, S.; Wang, G.; Zhou, C.-J. A WRKY transcription factor, TaWRKY42-B, facilitates initiation of leaf senescence by promoting jasmonic acid biosynthesis. BMC Plant Biol. 2020, 20, 444. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, X.; Yang, B.; Xu, S.; Wei, X.; Zhao, P.; Niu, F.; Sun, M.; Wang, C.; Cheng, H. WRKY55 transcription factor positively regulates leaf senescence and the defense response by modulating the transcription of genes implicated in the biosynthesis of reactive oxygen species and salicylic acid in Arabidopsis. Development 2020, 147. [Google Scholar] [CrossRef]
- Guo, P.; Li, Z.; Huang, P.; Li, B.; Fang, S.; Chu, J.; Guo, H. A Tripartite Amplification Loop Involving the Transcription Factor WRKY75, Salicylic Acid, and Reactive Oxygen Species Accelerates Leaf Senescence. Plant Cell 2017, 29, 2854–2870. [Google Scholar] [CrossRef]
- Fan, Z.-Q.; Tan, X.-L.; Shan, W.; Kuang, J.-F.; Lu, W.-J.; Chen, J.-Y. BrWRKY65, a WRKY Transcription Factor, Is Involved in Regulating Three Leaf Senescence-Associated Genes in Chinese Flowering Cabbage. Int. J. Mol. Sci. 2017, 18, 1228. [Google Scholar]
- Rameneni, J.J.; Dhandapani, V.; Paul, P.; Im, S.; Oh, M.-H.; Choi, S.R.; Lim, Y.P. Genome-wide identification, characterization, and comparative phylogeny analysis of MADS-box transcription factors in Brassica rapa. Genes Genom. 2014, 36, 509–525. [Google Scholar] [CrossRef]
- Xie, Q.; Hu, Z.; Zhu, Z.; Dong, T.; Zhao, Z.; Cui, B.; Chen, G. Overexpression of a novel MADS-box gene SlFYFL delays senescence, fruit ripening and abscission in tomato. Sci. Rep. 2014, 4, 4367. [Google Scholar] [CrossRef] [PubMed]
- Breeze, E.; Harrison, E.; McHattie, S.; Hughes, L.; Hickman, R.; Hill, C.; Kiddle, S.; Kim, Y.-S.; Penfold, C.A.; Jenkins, D.; et al. High-Resolution Temporal Profiling of Transcripts during Arabidopsis Leaf Senescence Reveals a Distinct Chronology of Processes and Regulation. Plant Cell 2011, 23, 873–894. [Google Scholar] [CrossRef]
- Fang, S.-C.; Fernandez, D.E. Effect of Regulated Overexpression of the MADS Domain Factor AGL15 on Flower Senescence and Fruit Maturation. Plant Physiol. 2002, 130, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Wang, Y.; Huang, S.; Dong, S.; Liu, Z.; Feng, H. Investigation of genes associated with petal variations between diploid and autotetraploid in Chinese cabbage (Brassica rapa L. ssp. pekinensis) by RNA-seq and sRNA-seq. Mol. Genet. Genom. 2020, 295, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, S.J.; Lee, I.H.; Chu, H.; Penfold, C.A.; Kim, J.H.; Buchanan-Wollaston, V.; Nam, H.G.; Woo, H.R.; Lim, P.O. Comparative transcriptome analysis in Arabidopsis ein2/ore3 and ahk3/ore12 mutants during dark-induced leaf senescence. J. Exp. Bot. 2018, 69, 3023–3036. [Google Scholar] [CrossRef]
- James, M.; Poret, M.; Masclaux-Daubresse, C.; Marmagne, A.; Coquet, L.; Jouenne, T.; Chan, P.; Trouverie, J.; Etienne, P. SAG12, a Major Cysteine Protease Involved in Nitrogen Allocation during Senescence for Seed Production in Arabidopsis thaliana. Plant Cell Physiol. 2018, 59, 2052–2063. [Google Scholar] [CrossRef]
- Aubry, S.; Fankhauser, N.; Ovinnikov, S.; Pruzinska, A.; Stirnemann, M.; Zienkiewicz, K.; Herrfurth, C.; Feussner, I.; Hortensteiner, S. Pheophorbide a May Regulate Jasmonate Signaling during Dark-Induced Senescence. Plant Physiol. 2020, 182, 776–791. [Google Scholar] [CrossRef]
- Hinckley, W.E.; Brusslan, J.A. Gene Expression Changes Occurring at Bolting Time are Associated with Leaf Senescence in Arabidopsis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Eckhardt, U.; Grimm, B.; Hortensteiner, S. Recent advances in chlorophyll biosynthesis and breakdown in higher plants. Plant Mol. Biol. 2004, 56, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.L.; Li, Z.; Guo, H. Genetic Network between Leaf Senescence and Plant Immunity: Crucial Regulatory Nodes and New Insights. Plants 2020, 9, 495. [Google Scholar] [CrossRef]
- Wang, H.-L.; Guo, H.; Li, Z. Gene network analysis of senescence-associated genes in annual plants and comparative assessment of aging in perennials and animals. Transl. Med. Aging 2019, 3, 6–13. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.H.; Lyu, J.I.; Woo, H.R.; Lim, P.O. New insights into the regulation of leaf senescence in Arabidopsis. J. Exp. Bot. 2018, 69, 787–799. [Google Scholar] [CrossRef]
- Li, Z.; Peng, J.; Wen, X.; Guo, H. Gene network analysis and functional studies of senescence-associated genes reveal novel regulators of Arabidopsis leaf senescence. J. Integr. Plant Biol. 2012, 54, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.R.; Kim, H.J.; Nam, H.G.; Lim, P.O. Plant leaf senescence and death-regulation by multiple layers of control and implications for aging in general. J. Cell Sci. 2013, 126, 4823–4833. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Zou, D.; Zhao, Y.; Wang, H.-L.; Zhang, Y.; Xia, X.; Luo, J.; Guo, H.; Zhang, Z. LSD 3.0: A comprehensive resource for the leaf senescence research community. Nucleic Acids Res. 2019, 48, D1069–D1075. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Liu, X.; Yu, J.; Ji, Y.; Liu, J.; Cheng, L.; Zhou, J. Up-regulation of autophagy by low concentration of salicylic acid delays methyl jasmonate-induced leaf senescence. Sci. Rep. 2020, 10, 11472. [Google Scholar] [CrossRef]
- Su, M.; Huang, G.; Zhang, Q.; Wang, X.; Li, C.; Tao, Y.; Zhang, S.; Lai, J.; Yang, C.; Wang, Y. The LEA protein, ABR, is regulated by ABI5 and involved in dark-induced leaf senescence in Arabidopsis thaliana. Plant Sci. 2016, 247, 93–103. [Google Scholar] [CrossRef]
- Qi, T.; Wang, J.; Huang, H.; Liu, B.; Gao, H.; Liu, Y.; Song, S.; Xie, D. Regulation of Jasmonate-Induced Leaf Senescence by Antagonism between bHLH Subgroup IIIe and IIId Factors in Arabidopsis. Plant Cell 2015, 27, 1634–1649. [Google Scholar] [CrossRef] [PubMed]
- Pourtau, N.; Marès, M.; Purdy, S.; Quentin, N.; Ruël, A.; Wingler, A. Interactions of abscisic acid and sugar signalling in the regulation of leaf senescence. Planta 2004, 219, 765–772. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, X.; Ren, J.; Qiu, K.; Li, Z.; Xie, Z.; Gao, J.; Zhou, X.; Kuai, B. Suppressor of Overexpression of CO 1 Negatively Regulates Dark-Induced Leaf Degreening and Senescence by Directly Repressing Pheophytinase and Other Senescence-Associated Genes in Arabidopsis. Plant Physiol. 2017, 173, 1881–1891. [Google Scholar] [CrossRef]
- Chen, M.K.; Hsu, W.H.; Lee, P.F.; Thiruvengadam, M.; Chen, H.I.; Yang, C.H. The MADS box gene, FOREVER YOUNG FLOWER, acts as a repressor controlling floral organ senescence and abscission in Arabidopsis. Plant J. 2011, 68, 168–185. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.H.; Kim, J.; Kim, J.J.; Hong, S.; Kim, J.; Kim, J.H.; Woo, H.R.; Hyeon, C.; Lim, P.O.; et al. Time-evolving genetic networks reveal a NAC troika that negatively regulates leaf senescence in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, e4930–e4939. [Google Scholar] [CrossRef]
- Lee, H.N.; Lee, K.H.; Kim, C.S. Abscisic acid receptor PYRABACTIN RESISTANCE-LIKE 8, PYL8, is involved in glucose response and dark-induced leaf senescence in Arabidopsis. Biochem. Biophys. Res. Commun. 2015, 463, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gan, S. AtNAP, a NAC family transcription factor, has an important role in leaf senescence. Plant J. 2006, 46, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, K.; El-Kereamy, A.; Kim, S.H.; Nambara, E.; Rothstein, S.J. ANAC032 Positively Regulates Age-Dependent and Stress-Induced Senescence in Arabidopsis thaliana. Plant Cell Physiol. 2016, 57, 2029–2046. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.D.; Seo, P.J.; Yoon, H.K.; Park, C.M. The Arabidopsis NAC transcription factor VNI2 integrates abscisic acid signals into leaf senescence via the COR/RD genes. Plant Cell 2011, 23, 2155–2168. [Google Scholar] [CrossRef]
- Tsutsui, T.; Kato, W.; Asada, Y.; Sako, K.; Sato, T.; Sonoda, Y.; Kidokoro, S.; Yamaguchi-Shinozaki, K.; Tamaoki, M.; Arakawa, K.; et al. DEAR1, a transcriptional repressor of DREB protein that mediates plant defense and freezing stress responses in Arabidopsis. J. Plant Res. 2009, 122, 633–643. [Google Scholar] [CrossRef]
- Vainonen, J.P.; Jaspers, P.; Wrzaczek, M.; Lamminmäki, A.; Reddy, R.A.; Vaahtera, L.; Brosché, M.; Kangasjärvi, J. RCD1-DREB2A interaction in leaf senescence and stress responses in Arabidopsis thaliana. Biochem. J. 2012, 442, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Chao, Q.; Rothenberg, M.; Solano, R.; Roman, G.; Terzaghi, W.; Ecker, J.R. Activation of the ethylene gas response pathway in Arabidopsis by the nuclear protein ETHYLENE-INSENSITIVE3 and related proteins. Cell 1997, 89, 1133–1144. [Google Scholar] [CrossRef]
- Chang, C.; Kwok, S.F.; Bleecker, A.B.; Meyerowitz, E.M. Arabidopsis ethylene-response gene ETR1: Similarity of product to two-component regulators. Science 1993, 262, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Hall, B.P.; Gao, Z.; Schaller, G.E. A strong constitutive ethylene-response phenotype conferred on Arabidopsis plants containing null mutations in the ethylene receptors ETR1 and ERS1. BMC Plant Biol. 2007, 7, 3. [Google Scholar] [CrossRef]
- Robatzek, S.; Somssich, I.E. Targets of AtWRKY6 regulation during plant senescence and pathogen defense. Genes Dev. 2002, 16, 1139–1149. [Google Scholar] [CrossRef]
- Miao, Y.; Zentgraf, U. The antagonist function of Arabidopsis WRKY53 and ESR/ESP in leaf senescence is modulated by the jasmonic and salicylic acid equilibrium. Plant Cell 2007, 19, 819–830. [Google Scholar] [CrossRef]
- Besseau, S.; Li, J.; Palva, E.T. WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J. Exp. Bot. 2012, 63, 2667–2679. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, S.; Zhou, B.; Luo, X.; Zhou, X.F.; Cai, B.; Jin, Y.H.; Niu, D.; Lin, J.; Cao, X. The Histone H3K4 Demethylase JMJ16 Represses Leaf Senescence in Arabidopsis. Plant Cell 2019, 31, 430–443. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.J.; Vu, Q.T.; Jung, S.; McClung, C.R.; Hong, S.; Nam, H.G. Circadian control of ORE1 by PRR9 positively regulates leaf senescence in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, 8448–8453. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Arteca, R.N.; Pell, E.J. Senescence-associated gene expression during ozone-induced leaf senescence in Arabidopsis. Plant Physiol. 1999, 120, 1015–1024. [Google Scholar] [CrossRef]
- Quirino, B.F.; Reiter, W.-D.; Amasino, R.D. One of two tandem Arabidopsis genes homologous to monosaccharide transporters is senescence-associated. Plant Mol. Biol. 2001, 46, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, A.; Grzyb, J.; Hermanowicz, P.; Zgłobicki, P.; Łabuz, J.; Strzałka, W.; Dziga, D.; Banaś, A.K. Arabidopsis Phototropins Participate in the Regulation of Dark-Induced Leaf Senescence. Int. J. Mol. Sci. 2021, 22, 1836. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Jeong, J.; Kang, M.-Y.; Kim, J.; Paek, N.-C.; Choi, G. Phytochrome-interacting transcription factors PIF4 and PIF5 induce leaf senescence in Arabidopsis. Nat. Commun. 2014, 5, 4636. [Google Scholar] [CrossRef]
- Oh, S.A.; Park, J.H.; Lee, G.I.; Paek, K.H.; Park, S.K.; Nam, H.G. Identification of three genetic loci controlling leaf senescence in Arabidopsis thaliana. Plant J. 1997, 12, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Ito, T.; Inoue, R.; Masuda, Y.; Nagashima, Y.; Kozuka, T.; Kusaba, M. Genetic Interaction Among Phytochrome, Ethylene and Abscisic Acid Signaling During Dark-Induced Senescence in Arabidopsis thaliana. Front. Plant Sci. 2020, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.M.; Gan, S.S.; Quirino, B.; Amasino, R.M. A comparison of the expression patterns of several senescence-associated genes in response to stress and hormone treatment. Plant Mol. Biol. 1998, 37, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.-S.; Amasino, R.M. Identification of a promoter region responsible for the senescence-specific expression of SAG12. Plant Mol. Biol. 1999, 41, 181–194. [Google Scholar] [CrossRef]
- Balazadeh, S.; Riano-Pachon, D.M.; Mueller-Roeber, B. Transcription factors regulating leaf senescence in Arabidopsis thaliana. Plant Biol. I 2008, 10, 63–75. [Google Scholar] [CrossRef]
- Koyama, T. The roles of ethylene and transcription factors in the regulation of onset of leaf senescence. Front. Plant Sci. 2014, 5, 650. [Google Scholar] [CrossRef]
- Jan, S.; Abbas, N.; Ashraf, M.; Ahmad, P. Roles of potential plant hormones and transcription factors in controlling leaf senescence and drought tolerance. Protoplasma 2019, 256, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Balanzà, V.; Martínez-Fernández, I.; Ferrándiz, C. Sequential action of FRUITFULL as a modulator of the activity of the floral regulators SVP and SOC1. J. Exp. Bot. 2014, 65, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Mandel, M.A.; Yanofsky, M.F. The Arabidopsis AGL8 MADS box gene is expressed in inflorescence meristems and is negatively regulated by APETALA1. Plant Cell 1995, 7, 1763–1771. [Google Scholar]
- Kaufmann, K.; Muiño, J.M.; Jauregui, R.; Airoldi, C.A.; Smaczniak, C.; Krajewski, P.; Angenent, G.C. Target Genes of the MADS Transcription Factor SEPALLATA3: Integration of Developmental and Hormonal Pathways in the Arabidopsis Flower. PLoS Biol. 2009, 7, e1000090. [Google Scholar] [CrossRef]
- Fernandez, D.E.; Heck, G.R.; Perry, S.E.; Patterson, S.E.; Bleecker, A.B.; Fang, S.C. The embryo MADS domain factor AGL15 acts postembryonically. Inhibition of perianth senescence and abscission via constitutive expression. Plant Cell 2000, 12, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Podzimska-Sroka, D.; O’Shea, C.; Gregersen, P.L.; Skriver, K. NAC Transcription Factors in Senescence: From Molecular Structure to Function in Crops. Plants 2015, 4, 412–448. [Google Scholar] [CrossRef] [PubMed]
- Rameneni, J.J.; Choi, S.R.; Chhapekar, S.S.; Kim, M.S.; Singh, S.; Yi, S.Y.; Oh, S.H.; Kim, H.; Lee, C.Y.; Oh, M.-H.; et al. Red Chinese Cabbage Transcriptome Analysis Reveals Structural Genes and Multiple Transcription Factors Regulating Reddish Purple Color. Int. J. Mol. Sci. 2020, 21, 2901. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 1–6. [Google Scholar] [CrossRef]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. Brassica rapa Genome Sequencing Project C. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Simon Anders, W.H. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2014, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Category | Gene Count | Expression Value (Log2 Fold Change Range) | Reference | ||
|---|---|---|---|---|---|
| DLS-91_0d_vs_ DLS-42_0d | DLS-91_1d_vs_ DLS-42_1d | DLS-91_4d_vs_ DLS-42_4d | |||
| Chlorophyll biosynthesis genes | 44 | 1.94~−1.28 | 1.30~−3.79 | 2~−2.96 | [5] |
| Chlorophyll degradation genes | 11 | 6.07~−1.88 | 1.22~−1.35 | 1.02~−1.45 | [5] |
| Subunits of photosystems I genes | 13 | 2.67~−0.81 | 1.85~−1.76 | 2.16~−1.71 | [5] |
| Subunits of photosystems II genes | 42 | 0.73~−5.40 | 1.35~−2.50 | 2.25~−2.85 | [5] |
| Regulators that act through changes in chromatin | 11 | 0.58~−0.42 | 0.63~−0.71 | 0.76~−0.52 | [57,59] |
| Regulators that act on the transcriptional level | 45 | 2.78~−5.12 | 2.85~−2.23 | 3.90~−3.92 | [57,59] |
| Regulators that act at the translational level | 1 | 1.54 | −0.90 | −0.65 | [59] |
| Regulators that act at the post-translational level | 25 | 2.61~−5.35 | 2.60~−4.74 | 2.29~−6.03 | [57,59] |
| Phytohormone signal components in leaf senescence and plant immunity | 29 | 1.66~−5.72 | 2.09~−5.53 | 1.59~−5.95 | [55] |
| List of the genes that are involved in leaf senescence and plant immunity. | 33 | 2.18~−3.83 | 2.94~−3.31 | 2.51~−2.93 | [58] |
| TFs Associated with Leaf Senescence in Crops | 27 | 0.5~−2.58 | 1.33~−2.67 | 2.03~−1.15 | [3] |
| List of genes that delay leaf senescence | 119 | 4.59~−1.72 | 4.64~−3.78 | 5.63~−5.44 | [3,58] |
| List of gene involved in leaf color change | 74 | 4.47~−6.84 | 4.55~−5.99 | 3.48~−6.22 | [58] |
| Previously reported senescence up regulated genes during dark-induced and age-triggered senescence | 1218 | 7.16~−7.42 | 7.90~−8.03 | 8.27~−6.83 | [5] |
| Previously reported senescence down regulated genes during dark-induced and age-triggered senescence | 1310 | 8.09~−6.31 | 7.52~−5.75 | 8.18~−5.87 | [5] |
| Given Name | Gene Id V 3.0 | DLS-42_0d | DLS-42_1d | DLS-42_4d | DLS-91_0d | DLS-91_1d | DLS-91_4d | Chromosome | Gene Start | Gene End | Effect on Senescence | Source | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BrAGL8-1 | BraA02g042750 | 1.63 | 2.06 | 2.46 | 26.29 | 14.48 | 27.51 | A02 | 25192486 | 25195876 | Promote | Mutant | [46] |
| BrAGL15-1 | BraA09g025840 | 0.00 | 0.00 | 0.00 | 9.40 | 8.36 | 16.52 | A09 | 17735929 | 17736685 | Delay | Transgene | [47] |
| BrAGL15-2 | BraA10g025090 | 0.16 | 0.98 | 1.33 | 0.51 | 0.39 | 0.61 | A10 | 12699925 | 12701540 | Delay | Transgene | [47] |
| BrAGL20-1 | BraA03g023790 | 0.98 | 1.19 | 1.23 | 0.00 | 3.97 | 2.48 | A03 | 10918286 | 10920672 | Promote/Delay | Mutant | [65] |
| BrAGL20-2 | BraA04g031640 | 85.00 | 29.39 | 30.50 | 3.71 | 1.66 | 2.40 | A04 | 18723546 | 18725960 | Promote/Delay | Mutant | [65] |
| BrAGL20-3 | BraA05g005370 | 14.22 | 13.90 | 16.29 | 3.07 | 7.51 | 11.53 | A05 | 2530305 | 2532747 | Promote/Delay | Mutant | [65] |
| BrAGL42-2 | BraA09g007170 | 32.44 | 18.26 | 13.67 | 0.17 | 0.12 | 0.04 | A09 | 3574619 | 3579987 | Delay | Transgene | [47] |
| BrNAC029-2 | BraA07g034350 | 95.51 | 170.36 | 235.98 | 10.82 | 34.48 | 377.56 | A07 | 21638992 | 21639971 | Promote | Mutant/Transgene | [10,69] |
| BrNAC046-2 | BraA05g042320 | 1.66 | 1.41 | 4.08 | 0.68 | 2.27 | 7.89 | A05 | 24529701 | 24531081 | Promote | Mutant/Transgene | [33] |
| BrNAC082-3 | BraA10g031140 | 56.95 | 60.77 | 71.15 | 32.97 | 32.16 | 49.86 | A10 | 14081664 | 14083371 | Delay | Mutant/Transgene | [10,71] |
| BrNAC090-1 | BraA03g010320 | 12.98 | 2.45 | 0.09 | 0.77 | 2.05 | 1.09 | A03 | 4319782 | 4321055 | Delay | Mutant/Transgene | [67] |
| BrNAC090-2 | BraA10g018990 | 1.05 | 0.19 | 0.09 | 0.57 | 2.68 | 0.24 | A10 | 9862356 | 9863361 | Delay | Mutant/Transgene | [67] |
| BrNAC092-1 | BraA04g012020 | 16.54 | 33.84 | 31.40 | 0.35 | 9.03 | 61.35 | A04 | 7538248 | 7539477 | Promote | Mutant | [30,31] |
| BrWRKY6-1 | BraA03g063940 | 0.16 | 0.65 | 0.16 | 0.02 | 1.64 | 0.41 | Scaffold000096 | 241928 | 244473 | Promote | Mutant | [77] |
| BrWRKY45 | BraAnng005520 | 9.18 | 76.23 | 70.30 | 3.02 | 56.72 | 95.61 | Scaffold000217 | 68236 | 68758 | Promote | Mutant | [78] |
| BrWRKY70-1 | BraA04g004020 | 8.24 | 48.80 | 1.22 | 47.34 | 343.66 | 20.64 | A04 | 2296461 | 2297781 | Delay | Mutant | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, S.Y.; Rameneni, J.J.; Lee, M.; Song, S.G.; Choi, Y.; Lu, L.; Lee, H.; Lim, Y.P. Comparative Transcriptome-Based Mining of Senescence-Related MADS, NAC, and WRKY Transcription Factors in the Rapid-Senescence Line DLS-91 of Brassica rapa. Int. J. Mol. Sci. 2021, 22, 6017. https://doi.org/10.3390/ijms22116017
Yi SY, Rameneni JJ, Lee M, Song SG, Choi Y, Lu L, Lee H, Lim YP. Comparative Transcriptome-Based Mining of Senescence-Related MADS, NAC, and WRKY Transcription Factors in the Rapid-Senescence Line DLS-91 of Brassica rapa. International Journal of Molecular Sciences. 2021; 22(11):6017. https://doi.org/10.3390/ijms22116017
Chicago/Turabian StyleYi, So Young, Jana Jeevan Rameneni, Myungjin Lee, Seul Gi Song, Yuri Choi, Lu Lu, Hyeokgeun Lee, and Yong Pyo Lim. 2021. "Comparative Transcriptome-Based Mining of Senescence-Related MADS, NAC, and WRKY Transcription Factors in the Rapid-Senescence Line DLS-91 of Brassica rapa" International Journal of Molecular Sciences 22, no. 11: 6017. https://doi.org/10.3390/ijms22116017
APA StyleYi, S. Y., Rameneni, J. J., Lee, M., Song, S. G., Choi, Y., Lu, L., Lee, H., & Lim, Y. P. (2021). Comparative Transcriptome-Based Mining of Senescence-Related MADS, NAC, and WRKY Transcription Factors in the Rapid-Senescence Line DLS-91 of Brassica rapa. International Journal of Molecular Sciences, 22(11), 6017. https://doi.org/10.3390/ijms22116017