
Molecular Details of the Frataxin–Scaffold Interaction during Mitochondrial Fe–S Cluster Assembly


 ,
,
Abstract
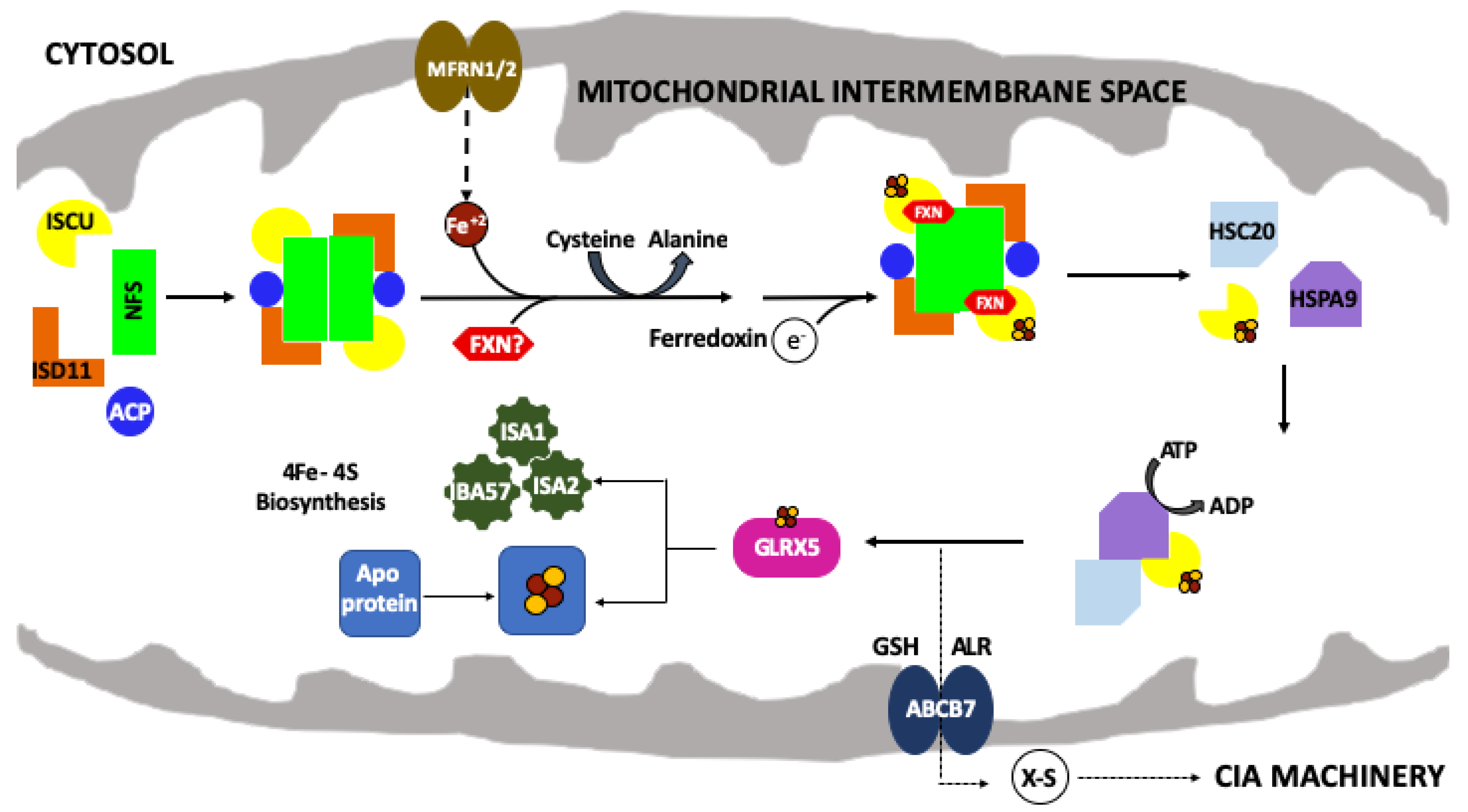
1. Introduction
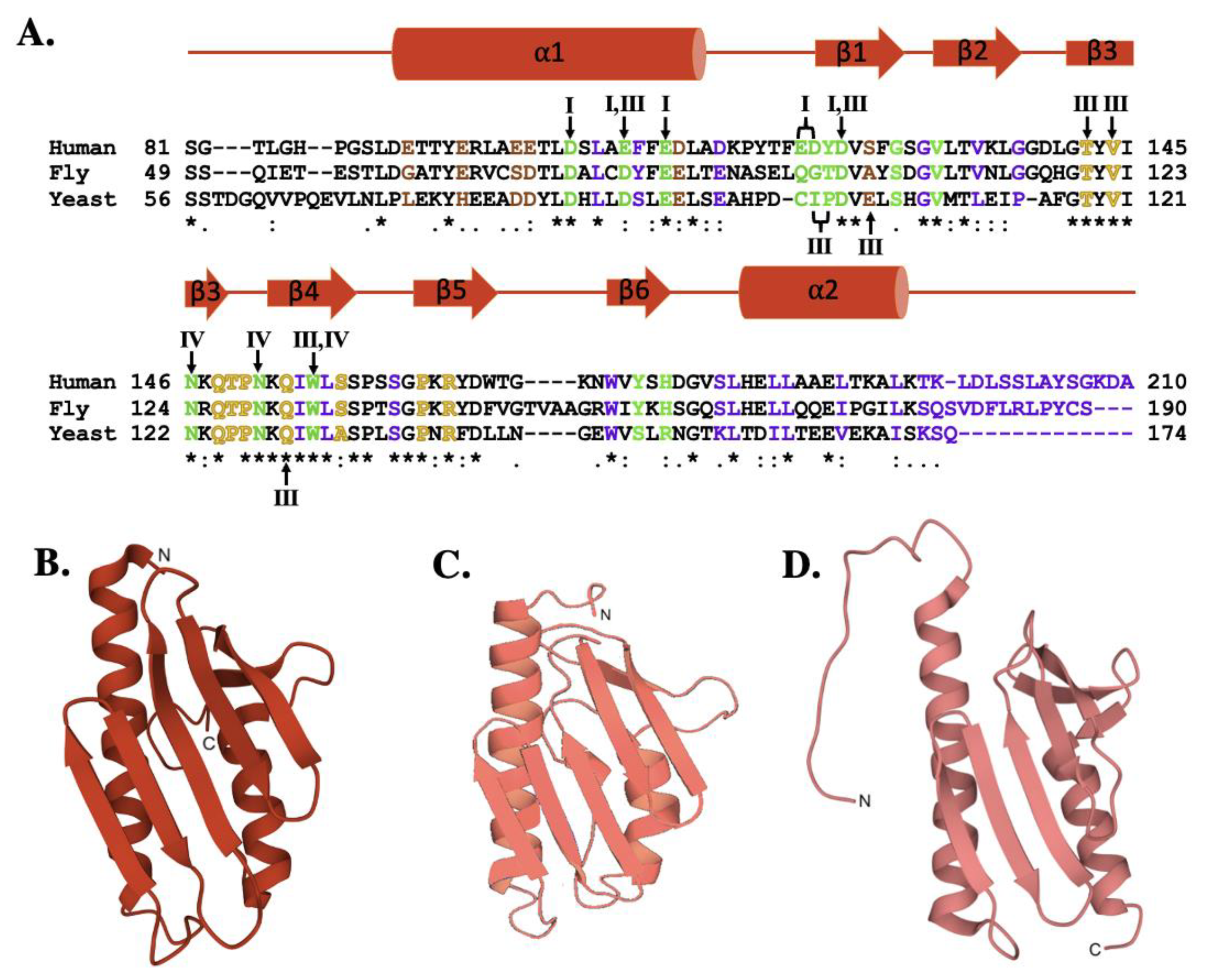
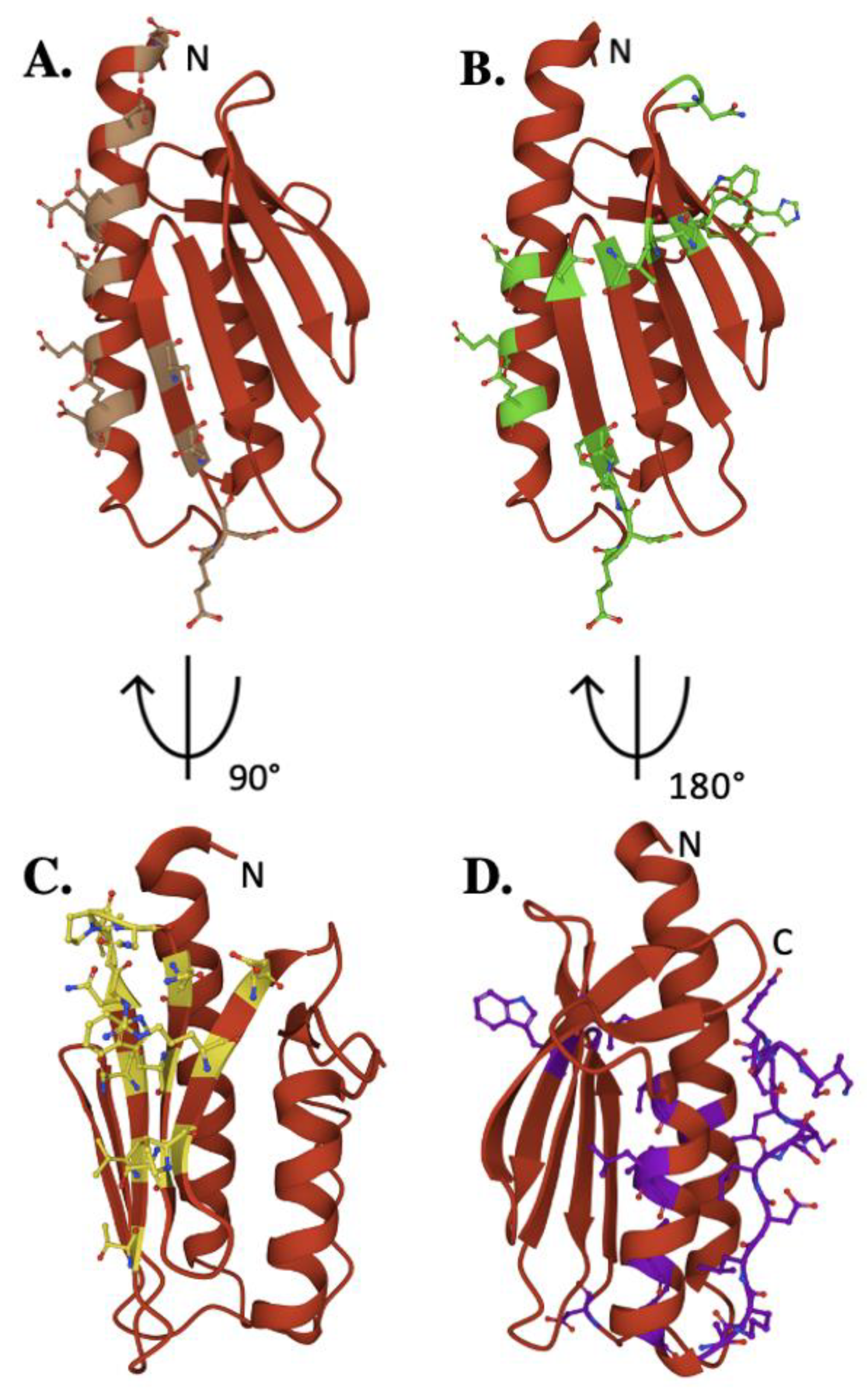
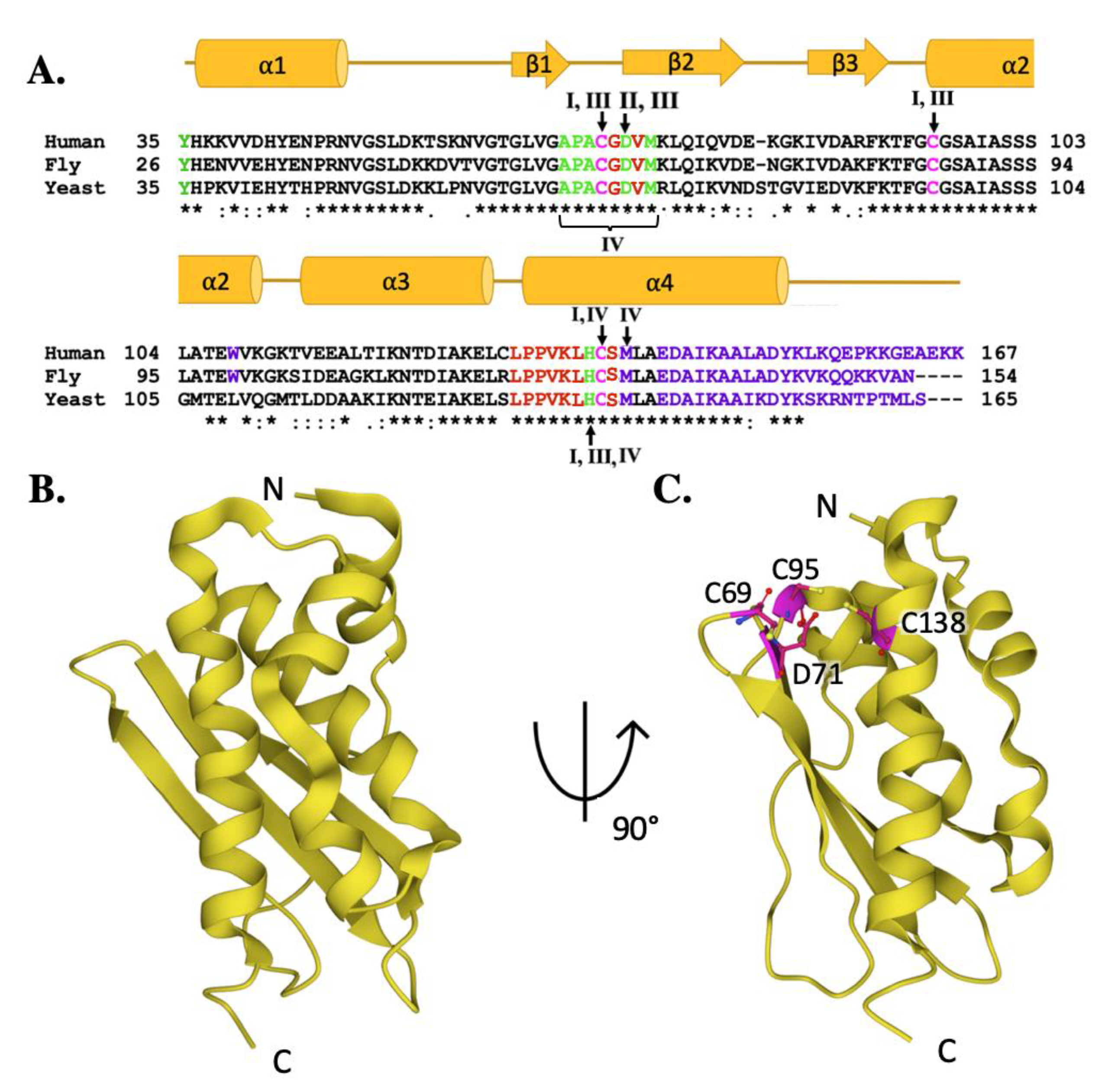
2. The Frataxin Protein
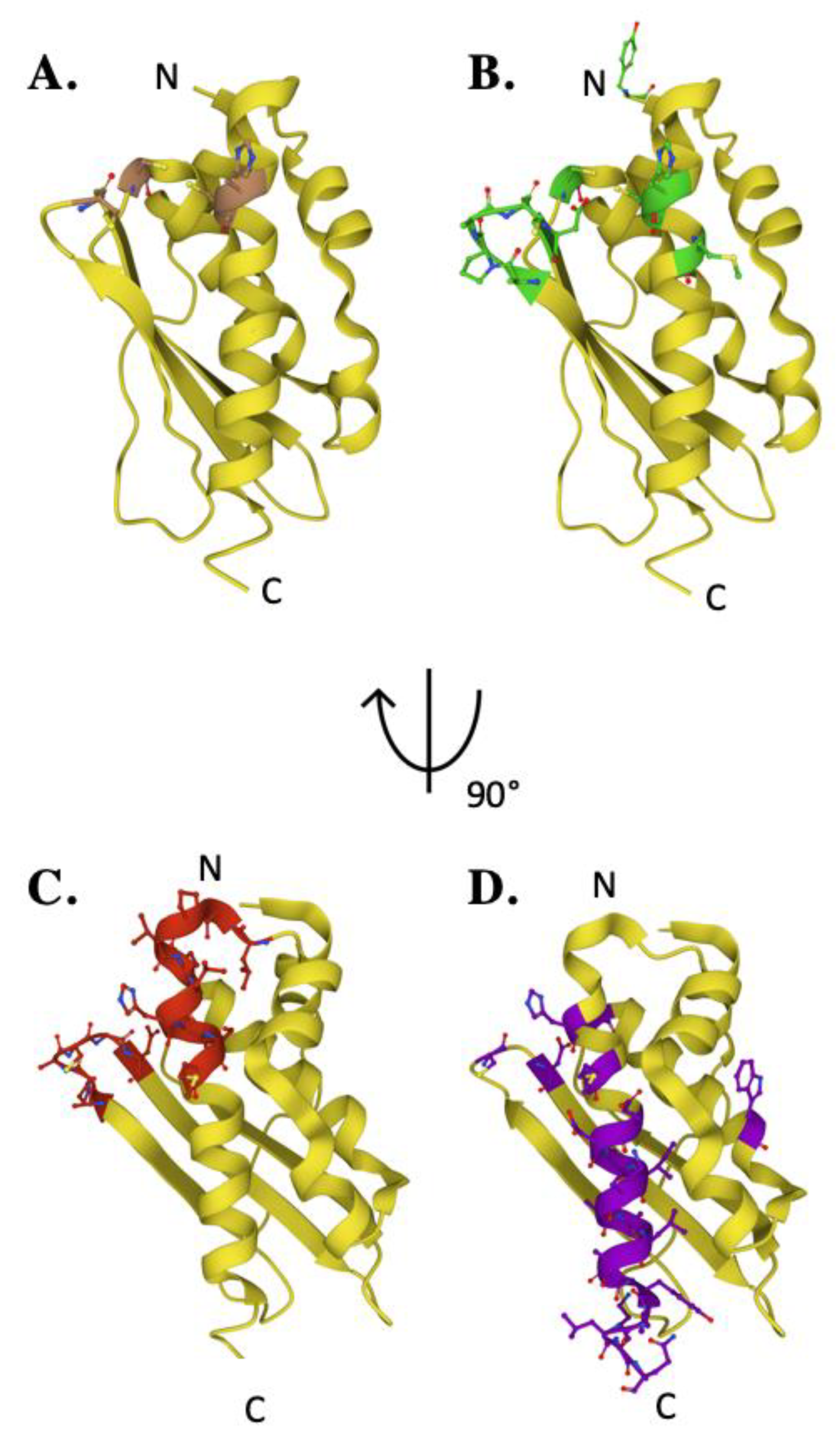
3. The Scaffold Protein
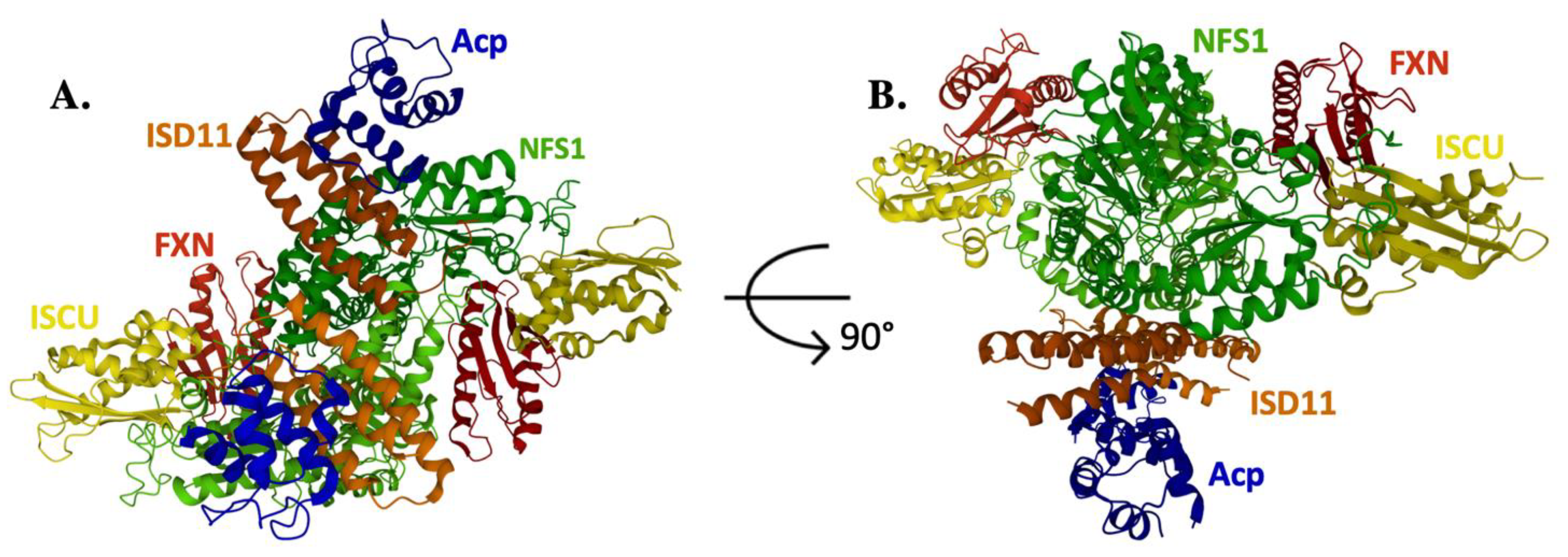
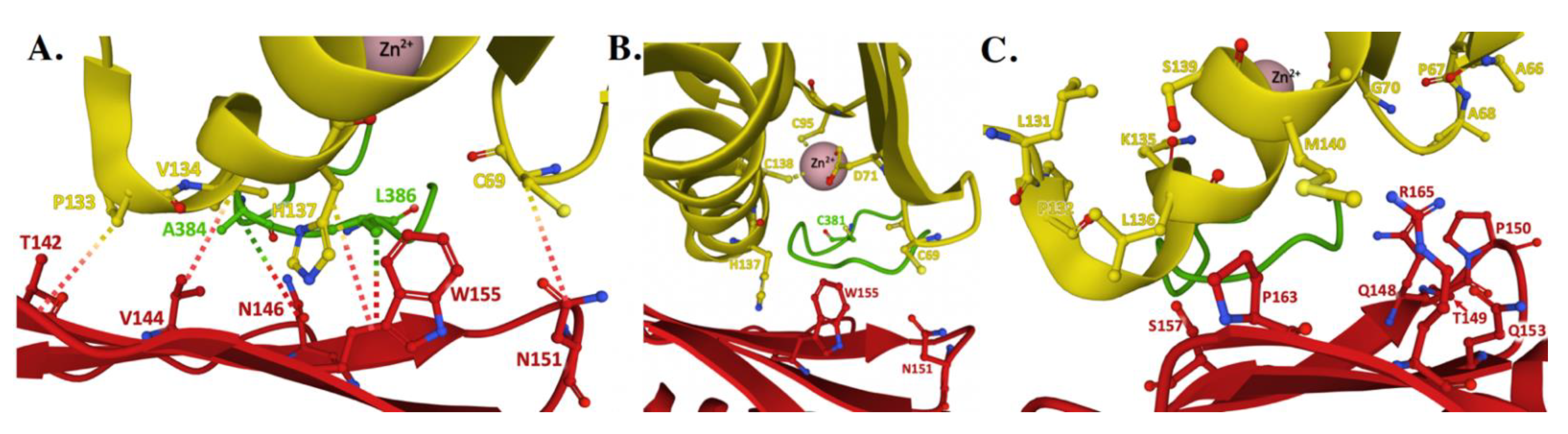
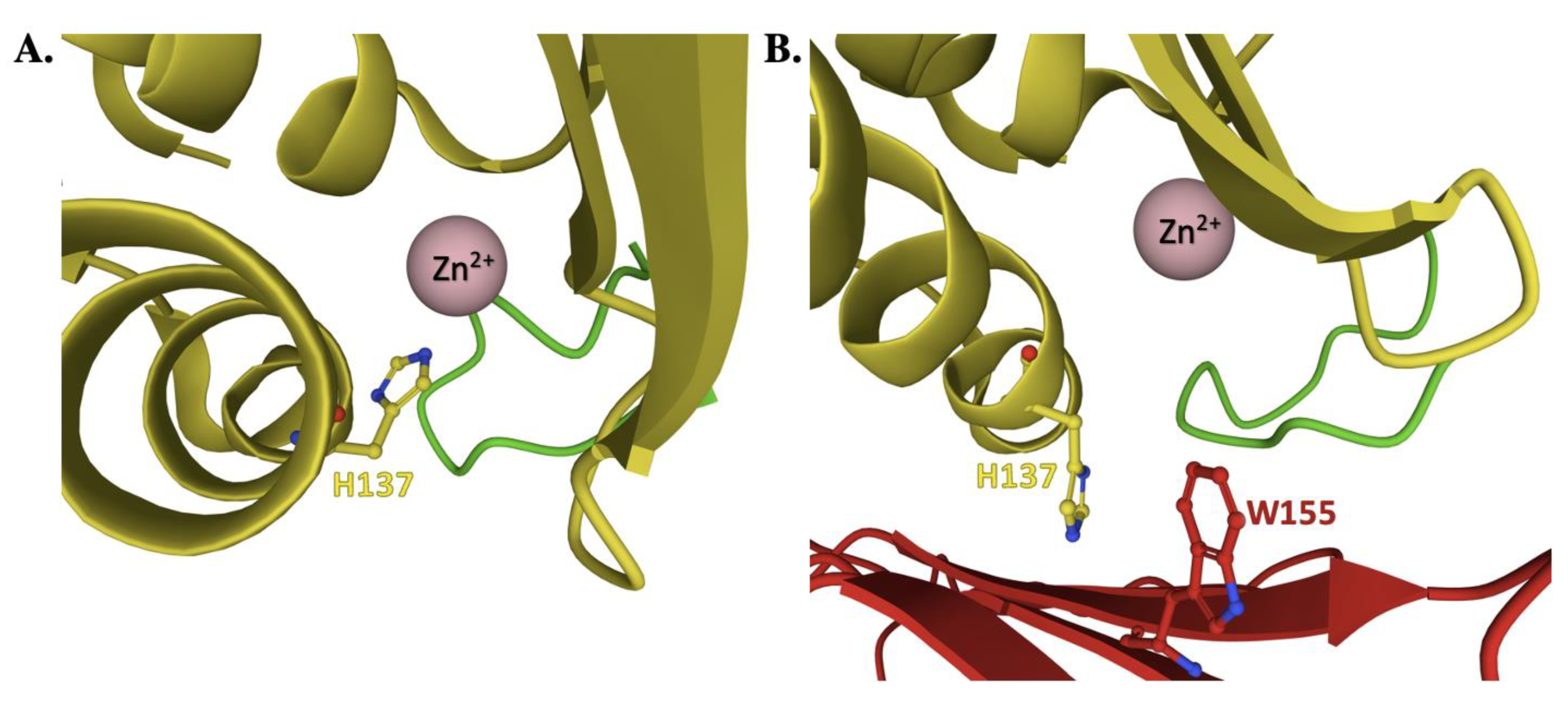
4. Frataxin/Scaffold Interactions
5. Discussion and Health Relevance
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braymer, J.J.; Freibert, S.A.; Rakwalska-Bange, M.; Lill, R. Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118863. [Google Scholar] [CrossRef]
- Srour, B.; Gervason, S.; Monfort, B.; D’Autreaux, B. Mechanism of Iron-Slufur Cluster Assembly, In the Intimacy of Iron and SUlfur Encounter. Inorganics 2020, 8, 55. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Mettert, E.L.; Kiley, P.J. How Is Fe-S Cluster Formation Regulated? Annu. Rev. Microbiol. 2015, 69, 505–526. [Google Scholar] [CrossRef]
- Beinert, H.; Holm, R.H.; Munck, E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R. Inorganic Biochemistry of Iron Metabolism: From Molecular Mechanisms to Clinical Consequences, 2nd ed.; John Whiler & Sons, Ltd.: West Sussex, UK, 2001. [Google Scholar]
- Fassbinder, J.W.; Stanjek, H.; Vali, H. Occurrence of magnetic bacteria in soil. Nature 1990, 343, 161–163. [Google Scholar] [CrossRef]
- Baussier, C.; Fakroun, S.; Aubert, C.; Dubrac, S.; Mandin, P.; Py, B.; Barras, F. Making iron-sulfur cluster: Structure, regulation and evolution of the bacterial ISC system. Adv. Microb. Physiol. 2020, 76, 1–39. [Google Scholar] [CrossRef]
- Garcia, P.S.; Gribaldo, S.; Py, B.; Barras, F. The SUF system: An ABC ATPase-dependent protein complex with a role in Fe-S cluster biogenesis. Res. Microbiol. 2019, 170, 426–434. [Google Scholar] [CrossRef]
- Outten, F.W. Recent advances in the Suf Fe-S cluster biogenesis pathway: Beyond the Proteobacteria. Biochim. Biophys. Acta 2015, 1853, 1464–1469. [Google Scholar] [CrossRef]
- Beilschmidt, L.K.; Puccio, H.M. Mammalian Fe-S cluster biogenesis and its implication in disease. Biochimie 2014, 100, 48–60. [Google Scholar] [CrossRef]
- Schilke, B.; Voisine, C.; Beinert, H.; Craig, E. Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomydes cervisiae. Proc. Natl. Acad. Sci. USA 1999, 96, 10206–10211. [Google Scholar] [CrossRef] [PubMed]
- Ciofi-Baffoni, S.; Nasta, V.; Banci, L. Protein networks in the maturation of human iron-sulfur proteins. Metallomics 2018, 10, 49–72. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Jain, A.; Rouault, T.A. Mammalian iron-sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr. Opin. Chem. Biol. 2020, 55, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Van Vranken, J.G.; Jeong, M.Y.; Wei, P.; Chen, Y.C.; Gygi, S.P.; Winge, D.R.; Rutter, J. The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Boniecki, M.T.; Freibert, S.A.; Muhlenhoff, U.; Lill, R.; Cygler, M. Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. Interactions of iron-bound frataxin with ISCU and ferredoxin on the cysteine desulfurase complex leading to Fe-S cluster assembly. J. Inorg. Biochem. 2018, 183, 107–116. [Google Scholar] [CrossRef]
- Cory, S.A.; Van Vranken, J.G.; Brignole, E.J.; Patra, S.; Winge, D.R.; Drennan, C.L.; Rutter, J.; Barondeau, D.P. Structure of human Fe-S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP-ISD11 interactions. Proc. Natl. Acad. Sci. USA 2017, 114, E5325–E5334. [Google Scholar] [CrossRef]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef]
- Shi, R.; Proteau, A.; Villarroya, M.; Moukadiri, I.; Zhang, L.; Trempe, J.F.; Matte, A.; Armengod, M.E.; Cygler, M. Structural basis for Fe-S cluster assembly and tRNA thiolation mediated by IscS protein-protein interactions. PLoS Biol. 2010, 8, 1–18. [Google Scholar] [CrossRef]
- Herrera, M.G.; Noguera, M.E.; Sewell, K.E.; Agudelo Suarez, W.A.; Capece, L.; Klinke, S.; Santos, J. Structure of the Human ACP-ISD11 Heterodimer. Biochemistry 2019, 58, 4596–4609. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Adrover, M.; Puglisi, R.; Yan, R.; Temussi, P.A.; Pastore, A. The role of zinc in the stability of the marginally stable IscU scaffold protein. Protein Sci. Publ. Protein Soc. 2014, 23, 1208–1219. [Google Scholar] [CrossRef]
- Liu, J.; Oganesyan, N.; Shin, D.H.; Jancarik, J.; Yokota, H.; Kim, R.; Kim, S.H. Structural characterization of an iron-sulfur cluster assembly protein IscU in a zinc-bound form. Proteins 2005, 59, 875–881. [Google Scholar] [CrossRef]
- Ramelot, T.A.; Cort, J.R.; Goldsmith-Fischman, S.; Kornhaber, G.J.; Xiao, R.; Shastry, R.; Acton, T.B.; Honig, B.; Montelione, G.T.; Kennedy, M.A. Solution NMR structure of the iron-sulfur cluster assembly protein U (IscU) with zinc bound at the active site. J. Mol. Biol. 2004, 344, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Tonelli, M.; Kim, T.; Markley, J.L. Three-dimensional structure and determinants of stability of the iron-sulfur cluster scaffold protein IscU from Escherichia coli. Biochemistry 2012, 51, 5557–5563. [Google Scholar] [CrossRef] [PubMed]
- Ramazzotti, A.; Vanmansart, V.; Foury, F. Mitochondrial functional interactions between frataxin and Isu1p, the iron-sulfur cluster scaffold protein, in Saccharomyces cerevisiae. FEBS Lett. 2004, 557, 215–220. [Google Scholar] [CrossRef]
- Wang, T.; Craig, E.A. Binding of yeast frataxin to the scaffold for Fe-S cluster biogenesis, Isu. J. Biol. Chem. 2008, 283, 12674–12679. [Google Scholar] [CrossRef]
- Cook, J.D.; Kondapalli, K.C.; Rawat, S.; Childs, W.C.; Murugesan, Y.; Dancis, A.; Stemmler, T.L. Molecular details of the yeast frataxin-Isu1 interaction during mitochondrial Fe-S cluster assembly. Biochemistry 2010, 49, 8756–8765. [Google Scholar] [CrossRef]
- Rodrigues, A.V.; Kandegedara, A.; Rotondo, J.A.; Dancis, A.; Stemmler, T.L. Iron loading site on the Fe-S cluster assembly scaffold protein is distinct from the active site. Biometals 2015, 28, 567–576. [Google Scholar] [CrossRef]
- Dzul, S.P.; Rocha, A.G.; Rawat, S.; Kandegedara, A.; Kusowski, A.; Pain, J.; Murari, A.; Pain, D.; Dancis, A.; Stemmler, T.L. In vitro characterization of a novel Isu homologue from Drosophila melanogaster for de novo FeS-cluster formation. Metallomics 2017, 9, 48–60. [Google Scholar] [CrossRef]
- Lewis, B.E.; Mason, Z.; Rodrigues, A.V.; Nuth, M.; Dizin, E.; Cowan, J.A.; Stemmler, T.L. Unique roles of iron and zinc binding to the yeast Fe-S cluster scaffold assembly protein “Isu1”. Metallomics 2019, 11, 1820–1835. [Google Scholar] [CrossRef]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Muller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Muhlenhoff, U.; Richhardt, N.; Ristow, M.; Kispal, G.; Lill, R. The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol. Genet. 2002, 11, 2025–2036. [Google Scholar] [CrossRef]
- Rotig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Chen, O.S.; Hemenway, S.; Kaplan, J. Inhibition of Fe-S cluster biosynthesis decreases mitochondrial iron export: Evidence that Yfh1p affects Fe-S cluster synthesis. Proc. Natl. Acad. Sci. USA 2002, 99, 12321–12326. [Google Scholar] [CrossRef] [PubMed]
- Duby, G.; Foury, F.; Ramazzotti, A.; Herrmann, J.; Lutz, T. A non-essential function for yeast frataxin in iron-sulfur cluster assembly. Hum. Mol. Genet. 2002, 11, 2635–2643. [Google Scholar] [CrossRef]
- Cavadini, P.; O’Neill, H.A.; Benada, O.; Isaya, G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002, 11, 217–227. [Google Scholar] [CrossRef]
- Aloria, K.; Schilke, B.; Andrew, A.; Craig, E.A. Iron-induced oligomerization of yeast frataxin homologue Yfh1 is dispensable in vivo. EMBO Rep. 2004, 5, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.D.; Bencze, K.Z.; Jankovic, A.D.; Crater, A.K.; Busch, C.N.; Bradley, P.B.; Stemmler, A.J.; Spaller, M.R.; Stemmler, T.L. Monomeric Yeast Frataxin Is an Iron-Binding Protein. Biochemistry 2006, 45, 7767–7777. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, K.C.; Kok, N.M.; Dancis, A.; Stemmler, T.L. Drosophila Frataxin: An Iron Chaperone during Cellular Fe−S Cluster Bioassembly. Biochemistry 2008, 47, 6917–6927. [Google Scholar] [CrossRef]
- Subramanian, P.; Rodrigues, A.V.; Ghimire-Rijal, S.; Stemmler, T.L. Iron chaperones for mitochondrial Fe-S cluster biosynthesis and ferritin iron storage. Curr. Opin. Chem. Biol. 2011, 15, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Gordon, D.M.; Pain, J.; Stemmler, T.L.; Dancis, A.; Pain, D. Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe-S cluster scaffold protein with frataxin-bypassing ability acts similarly. J. Biol. Chem. 2013, 288, 36773–36786. [Google Scholar] [CrossRef]
- Patra, S.; Barondeau, D.P. Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc. Natl. Acad. Sci. USA 2019, 116, 19421–19430. [Google Scholar] [CrossRef]
- Yoon, H.; Golla, R.; Lesuisse, E.; Pain, J.; Donald, J.E.; Lyver, E.R.; Pain, D.; Dancis, A. Mutation in the Fe-S scaffold protein Isu bypasses frataxin deletion. Biochem. J. 2012, 441, 473–480. [Google Scholar] [CrossRef]
- Yoon, H.; Knight, S.A.B.; Pandey, A.; Pain, J.; Zhang, Y.; Pain, D.; Dancis, A. Frataxin-bypassing Isu1: Characterization of the bypass activity in cells and mitochondria. Biochem. J. 2015, 459, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Adinolfi, S.; Pastore, A.; Laue, T.M.; Dennis Chasteen, N. Iron binding and oxidation kinetics in frataxin CyaY of Escherichia coli. J. Mol. Biol. 2004, 341, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 1–6. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- Bencze, K.Z.; Kondapalli, K.C.; Cook, J.D.; McMahon, S.; Millan-Pacheco, C.; Pastor, N.; Stemmler, T.L. The structure and function of frataxin. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 269–291. [Google Scholar] [CrossRef]
- Huang, J.; Dizin, E.; Cowan, J.A. Mapping iron binding sites on human frataxin: Implications for cluster assembly on the ISU Fe-S cluster scaffold protein. J. Biol. Inorg. Chem. 2008, 13, 825–836. [Google Scholar] [CrossRef]
- Bellanda, M.; Maso, L.; Doni, D.; Bortolus, M.; De Rosa, E.; Lunardi, F.; Alfonsi, A.; Noguera, M.E.; Herrera, M.G.; Santos, J.; et al. Exploring iron-binding to human frataxin and to selected Friedreich ataxia mutants by means of NMR and EPR spectroscopies. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 140254. [Google Scholar] [CrossRef] [PubMed]
- Doni, D.; Passerini, L.; Audran, G.; Marque, S.R.A.; Schulz, M.; Santos, J.; Costantini, P.; Bortolus, M.; Carbonera, D. Effects of Fe(2+)/Fe(3+) Binding to Human Frataxin and Its D122Y Variant, as Revealed by Site-Directed Spin Labeling (SDSL) EPR Complemented by Fluorescence and Circular Dichroism Spectroscopies. Int. J. Mol. Sci. 2020, 21, 9619. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Pastore, A.; Trincal, M. Acidic residues of yeast frataxin have an essential role in Fe-S cluster assembly. EMBO Rep. 2007, 8, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Nair, M.; Politou, A.; Bayer, E.; Martin, S.; Temussi, P.; Pastore, A. The Factors Governing the Thermal Stability of Frataxin Orthologues: How To Increase a Protein’s Stability. Biochemistry 2004, 43, 6511–6518. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Cowan, J.A. Structural, Mechanistic and Coordination Chemistry of Relevance to the Biosynthesis of Iron-Sulfur and Related Iron Cofactors. Coord. Chem. Rev. 2011, 255, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.R.; Pastore, C.; Adinolfi, S.; Pastore, A.; Gomes, C.M. Dynamics, stability and iron-binding activity of frataxin clinical mutants. FEBS J. 2008, 275, 3680–3690. [Google Scholar] [CrossRef]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhoffer-Donze, M.; Puccio, H. Mammalian Frataxin: An Essential Function for Cellular Viability through an Interaction with a Preformed ISCU/NFS1/ISD11 Iron-Sulfur Assembly Complex. PLoS ONE 2011, 6, e16199. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, S.; De Smet, S.; Foury, F. Frataxin interacts with Isu1 through a conserved tryptophan in its beta-sheet. Hum. Mol. Genet. 2010, 19, 276–286. [Google Scholar] [CrossRef][Green Version]
- Tsai, C.L.; Bridwell-Rabb, J.; Barondeau, D.P. Friedreich’s Ataxia Variants I154F and W155R diminish Frataxin-Based Activation of the Iron-Sulfur Cluster Assembly Complex. Biochemistry 2011, 50, 6478–6487. [Google Scholar] [CrossRef]
- Clark, E.; Butler, J.S.; Isaacs, C.J.; Napierala, M.; Lynch, D.R. Selected missense mutations impair frataxin processing in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2017, 4, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.R.; Wang, T.; Craig, E.A.; Gomes, C.M. Iron-binding activity in yeast frataxin entails a trade off with stability in the alpha1/beta1 acidic ridge region. Biochem. J. 2010, 426, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, M.; Pasqup, A.; Novak, L.; Toto, A.; Gianni, S.; Mantuano, E.; Veneziano, L.; Minicozzi, V.; Pastore, A.; Puglisi, R.; et al. Characterization of human frataxin missense variants in cancer tissues. Hum. Mutat. 2019, 40, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Faggianelli, N.; Puglisi, R.; Veneziano, L.; Romano, S.; Frontali, M.; Vannocci, T.; Fortuni, S.; Testi, R.; Pastore, A. Analyzing the Effects of a G137V Mutation in the FXN Gene. Front. Mol. Neurosci. 2015, 8. [Google Scholar] [CrossRef]
- Shan, Y.; Napoli, E.; Cortopassi, G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum. Mol. Genet. 2007, 16, 929–941. [Google Scholar] [CrossRef]
- Roman, E.A.; Faraj, S.E.; Gallo, M.; Salvay, A.G.; Ferreiro, D.U.; Santos, J. Protein stability and dynamics modulation: The case of human frataxin. PLoS ONE 2012, 7, e45743. [Google Scholar] [CrossRef]
- Faraj, S.E.; Roman, E.A.; Aran, M.; Gallo, M.; Santos, J. The alteration of the C-terminal region of human frataxin distorts is structural dynamics and function. FEBS J. 2014, 281, 3397–3419. [Google Scholar] [CrossRef]
- Adinolfi, S.; Iannuzzi, C.; Prischi, F.; Pastore, C.; Iametti, S.; Martin, S.R.; Bonomi, F.; Pastore, A. Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009, 16, 390–396. [Google Scholar] [CrossRef]
- Mochel, F.; Knight, M.A.; Tong, W.H.; Hernandez, D.; Ayyad, K.; Taivassalo, T.; Andersen, P.M.; Singleton, A.; Rouault, T.A.; Fischbeck, K.H.; et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 2008, 82, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Watson, H.M.; Gentry, L.E.; Asuru, A.P.; Wang, Y.; Marcus, S.; Busenlehner, L.S. Heterotrifunctional Chemical Cross-Linking Mass Spectrometry Confirms Physical Interaction between Human Frataxin and ISU. Biochemistry 2012, 51, 6889–6891. [Google Scholar] [CrossRef]
- Nuth, M.; Yoon, T.; Cowan, J.A. Iron-sulfur cluster biosynthesis: Characterization of iron nucleation sites for assembly of the [2Fe-2S]2+ cluster core in IscU proteins. J. Am. Chem. Soc. 2002, 124, 8774–8775. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Fuzery, A.K.; Tonelli, M.; Ta, D.T.; Westler, W.M.; Vickery, L.E.; Markley, J.L. Structure and dynamics of the iron-sulfur cluster assembly scaffold protein IscU and its interaction with the cochaperone HscB. Biochemistry 2009, 48, 6062–6071. [Google Scholar] [CrossRef]
- Tanaka, N.; Yuda, E.; Fujishiro, T.; Hirabayashi, K.; Wada, K.; Takahashi, Y. Identification of IscU residues critical for de novo iron-sulfur cluster assembly. Mol. Microbiol. 2019, 112, 1769–1783. [Google Scholar] [CrossRef]
- Majewska, J.; Ciesielski, S.J.; Schilke, B.; Kominek, J.; Blenska, A.; Delewski, W.; Song, J.-Y.; Marszalek, J.; Craig, E.A.; Dutkiewicz, R. Binding of the Chaperone Jac1 Protein and Cysteine Desulfurase Nfs1 to the Iron-Sulfur Scaffold Isu Protein is Mutually Exclusive. J. Biol. Chem. 2013, 288, 29134–29142. [Google Scholar] [CrossRef]
- Cai, K.; Frederick, R.O.; Kim, J.H.; Reinen, N.M.; Tonelli, M.; Markley, J.L. Human Mitochondrial Chaperone (mtHSP70) and Cysteine Desulfurase (NFS1) Bind Preferentially to the Disordered Conformation, Whereas Co-chaperone (HSC20) Binds to the Structured Conformation of the Iron-Sulfur Cluster Scaffold Protein (ISCU). J. Biol. Chem. 2013, 288, 28755–28770. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.V. Characterization of Initial Iron Binding Location and the Structure/Iron Binding Site on S. cereviseiae and D. melanogaster Frataxin. Ph.D. Thesis, Wayne State University, Detroit, MI, USA, 2014; pp. 1–148. [Google Scholar]
- Lewis, B.E. The Dynamic Nature and Biophysical Characterization of Isu1, Fe-S Cluster Assembly Scaffold Protein in Saccharomyces Cerevisiae, and Its Significance to Human Disease. Ph.D. Thesis, Wayne State University, Detroit, MI, USA, 2019. [Google Scholar]
- Becker, E.M.; Greer, J.M.; Ponka, P.; Richardson, D.R. Erythroid differentiation and protoporphyrin IX down-regulate frataxin expression in Friend cells: Characterization of frataxin expression compared to molecules involved in iron metabolism and hemoglobinization. Blood 2002, 99, 3813–3822. [Google Scholar] [CrossRef]
- Shimberg, G.D.; Michalek, J.L.; Oluyadi, A.A.; Rodrigues, A.V.; Zucconi, B.E.; Neu, H.M.; Ghosh, S.; Sureschandra, K.; Wilson, G.M.; Stemmler, T.L.; et al. Cleavage and polyadenylation specificity factor 30: An RNA-binding zinc-finger protein with an unexpected 2Fe-2S cluster. Proc. Natl. Acad. Sci. USA 2016, 113, 4700–4705. [Google Scholar] [CrossRef]
- Shimomura, Y.; Wada, K.; Fukuyama, K.; Takahashi, Y. The asymmetric trimeric architecture of [2Fe-2S] IscU: Implications for its scaffolding during iron-sulfur cluster biosynthesis. J. Mol. Biol. 2008, 383, 133–143. [Google Scholar] [CrossRef]
- Markley, J.L.; Kim, J.H.; Dai, Z.; Bothe, J.R.; Cai, K.; Frederick, R.O.; Tonelli, M. Metamorphic protein IscU alternates conformations in the course of its role as the scaffold protein for iron-sulfur cluster biosynthesis and delivery. FEBS Lett. 2013, 587, 1172–1179. [Google Scholar] [CrossRef]
- Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. ISCU(M108I) and ISCU(D39V) Differ from Wild-Type ISCU in Their Failure To Form Cysteine Desulfurase Complexes Containing Both Frataxin and Ferredoxin. Biochemistry 2018, 57, 1491–1500. [Google Scholar] [CrossRef]
- Manicki, M.; Majewska, J.; Ciesielski, S.; Schilke, B.; Blenska, A.; Kominek, J.; Marszalek, J.; Craig, E.A.; Dutkiewicz, R. Overlapping Binding Sites of the Frataxin Homologue Assembly Factor and the Heat Shock Protein 70 Transfer Factor on teh Isu Iron-Sulfur Cluster Scaffold Protein. J. Biol. Chem. 2014, 289, 30268–30278. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gakh, O.; Smith, D.Y.t.; Isaya, G. Oligomeric yeast frataxin drives assembly of core machinery for mitochondrial iron-sulfur cluster synthesis. J. Biol. Chem. 2009, 284, 21971–21980. [Google Scholar] [CrossRef] [PubMed]
- McCranor, B.J.; Bozym, R.A.; Vitolo, M.I.; Fierke, C.A.; Bambrick, L.; Polster, B.M.; Fiskum, G.; Thompson, R.B. Quantitative imaging of mitochondrial and cytosolic free zinc levels in an in vitro model of ischemia/reperfusion. J. Bioenerg. Biomembr. 2012, 44, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Analyzing free zinc(II) ion concentrations in cell biology with fluorescent chelating molecules. Metallomics 2015, 7, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Irving, H.M.N.H.; Williams, R.J.P. The stability of transition-metal complexes. J. Chem. Soc. 1953, 75, 3192–3210. [Google Scholar] [CrossRef]
- Holmes-Hampton, G.P.; Miao, R.; Garber Morales, J.; Guo, Y.; Munck, E.; Lindahl, P.A. A nonheme high-spin ferrous pool in mitochondria isolated from fermenting Saccharomyces cerevisiae. Biochemistry 2010, 49, 4227–4234. [Google Scholar] [CrossRef] [PubMed]
- Kunichika, K.; Nakamura, R.; Fujishiro, T.; Takahashi, Y. The Structure of the Dimeric State of IscU Harboring Two Adjacent [2Fe-2S] Clusters Provides Mechanistic Insights into Cluster Conversion to [4Fe-4S]. Biochemistry 2021. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Human | Fly | Yeast | |||
|---|---|---|---|---|---|---|
| Symbol | MW (kDa) | Symbol | MW (kDa) | Symbol | MW (kDa) | |
| Cysteine desulfurase | NFS1 | 50.2 | dNfs1 | 51.1 | Nfs1 | 54.5 |
| LYR Motif-containing protein 4 | ISD11 | 10.7 | dIsd11 | 11 | Isd11 | 11.2 |
| Acyl carrier protein | ACP/NDUFAB | 17.4 | dAcp | 17.2 | Acp1 | 13.9 |
| Iron–sulfur cluster assembly scaffold | ISCU | 15.3 | dIscU | 16.7 | Isu1 | 17.8 |
| Frataxin | FXN | 23.1 | dFh | 20.9 | Yfh1 | 19.5 |
| Function | Residue | Mutation | Location on Protein | Organism | Hs Equivalent | Reference |
|---|---|---|---|---|---|---|
| Iron Binding | E92 | E92A | α1 | Hs | E92 | [55] |
| E96 | E96A | α1 | Hs | E96 | [55] | |
| E100 | E100A | α1 | Hs | E100 | [55] | |
| E101 | E101A | α1 | Hs | E101 | [55] | |
| D104 | D104A | α1 | Hs | D104 | [55] | |
| D86 | D86A | α1 | Sc | E108 | [58] | |
| E108 | E108A | α1 | Hs | E108 | [17,42] | |
| E111 | E111A | α1 | Hs | E111 | [55,59] | |
| D112 | D112A | α1 | Hs | D112 | [60] | |
| E90 | E90A | α1 | Sc | D112 | [58] | |
| E93 | E93A | α1 | Sc | D115 | [58] | |
| D115 | D115A | α1-β1 Loop | Hs | D115 | [60] | |
| E121 | E121A | α1-β1 Loop | Hs | E121 | [60] | |
| D122 | D122A/Y | β1 | Hs | D122 | [56,57,60,61] | |
| D124 | D124A | β1 | Hs | D124 | [60] | |
| D101 | D101A | β1 | Sc | D124 | [58] | |
| E103 | E103A | β1 | Sc | S126 | [58] | |
| NFS Binding | D104 | *1 | α1 | Hs | D104 | [17] |
| E108 | *1 | α1 | Hs | E108 | [19] | |
| E111 | *1 | α1 | Hs | E111 | [19] | |
| E121-Y123 | *2 | α1-β1 Loop | Hs | E121-Y123 | [19] | |
| D124 | D124A/K | β1 | Hs | D124 | [19,62] | |
| G128 | *2 | β1 | Hs | G128 | [19] | |
| V131 | *2 | β2 | Hs | V131 | [19] | |
| N146 | N146K | β3 | Hs | N146 | [19,56] | |
| N151 | N151A | β3-β4 Loop | Hs | N151 | [19] | |
| W155 | W155R | β4 | Hs | W155 | [19,56,61] | |
| Y175 | *2 | β6 | Hs | Y175 | [19] | |
| H177 | *2 | β6-α2 Loop | Hs | H177 | [19] | |
| ISCU Binding | T142 | *2 | β3 | Hs | T142 | [19] |
| V120 | *1 | β3 | Sc | V144 | [28] | |
| V144 | *2 | β3 | Hs | V144 | [19] | |
| N146 | *2 | β3 | Hs | N146 | [19] | |
| N122 | N122A/K | β3 | Sc | N146 | [27] | |
| Q124 | Q124A | β3 | Sc | Q148 | [27] | |
| T149 | *1 | β3-β4 Loop | Hs | T149 | [17] | |
| P150 | *2 | β3-β4 Loop | Hs | P150 | [19] | |
| N151 | N151A | β3-β4 Loop | Hs | N151 | [19] | |
| N127 | *1 | β3-β4 Loop | Sc | N151 | [28] | |
| Q153 | *1 | β4 | Hs | Q153 | [17] | |
| Q129 | Q129A | β3-β4 Loop | Sc | Q153 | [63] | |
| W155 | W155R/A/F | β4 | Hs | W155 | [61,64] | |
| W131 | W131A/F | β4 | Sc | W155 | [63] | |
| S157 | *1 | β4 | Hs | S157 | [17] | |
| P163 | P163G | β5 | Hs | P163 | [19] | |
| R165 | R165C | β5 | Hs | R165 | [19] | |
| R141 | R141A | β5 | Sc | R165 | [63] | |
| Stability | L106 | L106S | α1 | Hs | L106 | [65] |
| D86 | D86A | α1 | Sc | E108 | [66] | |
| F109 | F109L | α1 | Hs | F109 | [67] | |
| E93 | E93A | α1 | Sc | D115 | [66] | |
| D122 | D122Y | α1-β1 Loop | Hs | D122 | [66] | |
| Y123 | Y123S | α1-β1 Loop | Hs | Y123 | [67] | |
| D101 | 101A | β1 | Sc | D124 | [66] | |
| E103 | 103A | β1 | Sc | S126 | [66] | |
| G130 | G130Y | β1 | Hs | G130 | [66] | |
| T110A | T110A | β2 | Sc | V134 | [63] | |
| G137 | G137V | β2- β3 Loop | Hs | G137 | [68] | |
| T118A | T118A | β3 | Sc | T142 | [63] | |
| V120 | V120A | β3 | Sc | V144 | [33] | |
| Q129A | Q129A | β3-β4 Loop | Sc | Q153 | [63] | |
| I130 | I130A | β4 | Sc | I154 | [63] | |
| I154 | I154F | β4 | Hs | I154 | [66] | |
| W155 | W155R | β4 | Hs | W155 | [66] | |
| W131 | W131A/F | β4 | Sc | W155 | [63] | |
| L132 | L132A | β4 | Sc | L156 | [63] | |
| S161 | S161I | β4- β5 Loop | Hs | S161 | [67] | |
| W173 | W173G | β6 | Hs | W173 | [69] | |
| S181 | S181F | α2 | Hs | S181 | [67] | |
| L182 | L182F | α2 | Hs | L182 | [70] | |
| L185-L186 | *3 | α2 | Hs | L185-L186 | [70] | |
| L190 | *3 | α2 | Hs | L190 | [70] | |
| L194 | *3 | α2 | Hs | L194 | [70] | |
| T196-K197 | Truncation | C-Termini | Hs | T196-K197 | [70] | |
| L198 | L198R/A/C | C-Termini | Hs | L198 | [71] | |
| D199 | Truncation | C-Termini | Hs | D199 | [70] | |
| L200 | L200C | C-Termini | Hs | L200 | [71] | |
| S201-A210 | Truncation | C-Termini | Hs | S201-A210 | [70] |
| Function | Residue | Mutation | Location on Protein | Organism | H sapien Equivalent | Reference |
|---|---|---|---|---|---|---|
| Iron Binding | C69 | C69A/S | β1–β2 Loop | Hs | C69 | [74,75] |
| C95 | C95A/S | α2 | Hs | C95 | [74,75] | |
| H137 | *1 | α4 | Hs | H137 | [74,75] | |
| C138 | C138A/S | α4 | Hs | C138 | [74,75] | |
| Active Site | C69 | C69A/S | β1–β2 Loop | Hs | C69 | [24] |
| D71 | D71A, D71V | β2 | Sc, Hs | D71 | [76] | |
| C95 | C95A/S | α2 | Hs | C95 | [24] | |
| H137 | *1 | α4 | Hs | H137 | [24] | |
| C138 | C138A/S | α4 | Hs | C138 | [24] | |
| NFS Binding | Y35 | Y3F/H/W | N-terminus | Ec | Y35 | [77] |
| L63 | L63A | β1 | Sc | L63 | [78] | |
| A66 | *3 | β1–β2 Loop | Hs | A66 | [16,18] | |
| P67 | *3 | β1–β2 Loop | Hs | P67 | [16,18] | |
| A68 | *3 | β1–β2 Loop | Hs | A68 | [16,18] | |
| C69 | C69A/S | β1–β2 Loop | Hs | C69 | [16] | |
| D71 | D71A | β2 | Sc | D71 | [16,17] | |
| V72 | V72A | β2 | Sc | V72 | [78] | |
| F94 | F94A | β3-α2 Loop | Sc | F93 | [78] | |
| C95 | C95A/S | α2 | Hs | C95 | [16] | |
| H137 | *1 | α4 | Hs | H137 | [16] | |
| C138 | C138A/S | α4 | Hs | C138 | [16] | |
| M138 | M138I | α4 | Sc | M140 | [48] | |
| FXN Binding | A66 | *2 | β1–β2 Loop | Hs | A66 | [19] |
| P67 | *2 | β1–β2 Loop | Hs | P67 | [19] | |
| A68 | *2 | β1–β2 Loop | Hs | A68 | [19] | |
| C69 | *2 | β1–β2 Loop | Hs | C69 | [19] | |
| G70 | *2 | β1–β2 Loop | Hs | G70 | [19] | |
| D71 | *2 | β1–β2 Loop | Hs | D71 | [19] | |
| L131 | *3 | α4 | Hs | L131 | [19] | |
| P132 | *3 | α4 | Hs | P132 | [19] | |
| P133 | *3 | α4 | Hs | P133 | [19] | |
| V134 | *3 | α4 | Hs | V134 | [19] | |
| K135 | *3 | α4 | Hs | K135 | [19] | |
| L136 | *2 | α4 | Hs | L136 | [19] | |
| H137 | *2 | α4 | Hs | H137 | [19] | |
| C138 | *2 | α4 | Hs | C138 | [19] | |
| S139 | *2 | α4 | Hs | S139 | [19] | |
| M140 | *2 | α4 | HS | M140 | [16,17] | |
| Stability | C69 | C69A/S, *1 | β1–β2 Loop | Hs | C69 | [79] |
| D71 | D71A/V, *1 | β2 | Sc | D71 | [79] | |
| C95 | C95A/S, *1 | α2 | Hs | C95 | [79] | |
| W108 | *1 | α2 | Hs | W108 | [32] | |
| H137 | *1 | α4 | Hs | H137 | [79] | |
| E144 | E144A | α4 | Sc | E143 | [80] | |
| E144Δ | Truncation | α4 | Sc | E143 | [81] | |
| D145 | D145A | α4 | Sc | D144 | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campbell, C.J.; Pall, A.E.; Naik, A.R.; Thompson, L.N.; Stemmler, T.L. Molecular Details of the Frataxin–Scaffold Interaction during Mitochondrial Fe–S Cluster Assembly. Int. J. Mol. Sci. 2021, 22, 6006. https://doi.org/10.3390/ijms22116006
Campbell CJ, Pall AE, Naik AR, Thompson LN, Stemmler TL. Molecular Details of the Frataxin–Scaffold Interaction during Mitochondrial Fe–S Cluster Assembly. International Journal of Molecular Sciences. 2021; 22(11):6006. https://doi.org/10.3390/ijms22116006
Chicago/Turabian StyleCampbell, Courtney J., Ashley E. Pall, Akshata R. Naik, Lindsey N. Thompson, and Timothy L. Stemmler. 2021. "Molecular Details of the Frataxin–Scaffold Interaction during Mitochondrial Fe–S Cluster Assembly" International Journal of Molecular Sciences 22, no. 11: 6006. https://doi.org/10.3390/ijms22116006
APA StyleCampbell, C. J., Pall, A. E., Naik, A. R., Thompson, L. N., & Stemmler, T. L. (2021). Molecular Details of the Frataxin–Scaffold Interaction during Mitochondrial Fe–S Cluster Assembly. International Journal of Molecular Sciences, 22(11), 6006. https://doi.org/10.3390/ijms22116006