
An Engineered sgsh Mutant Zebrafish Recapitulates Molecular and Behavioural Pathobiology of Sanfilippo Syndrome A/MPS IIIA

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results
2.1. Generation of the sgshΔex5−6 Zebrafish Mutant by CRISPR/Cas9-Mediated Dual-gRNA Targeted Mutagenesis
2.2. sgshΔex5−6 Mutant Zebrafish Exhibit Total Absence of Sgsh Activity and Accumulate Heparan Sulfate Partial Degradation Products
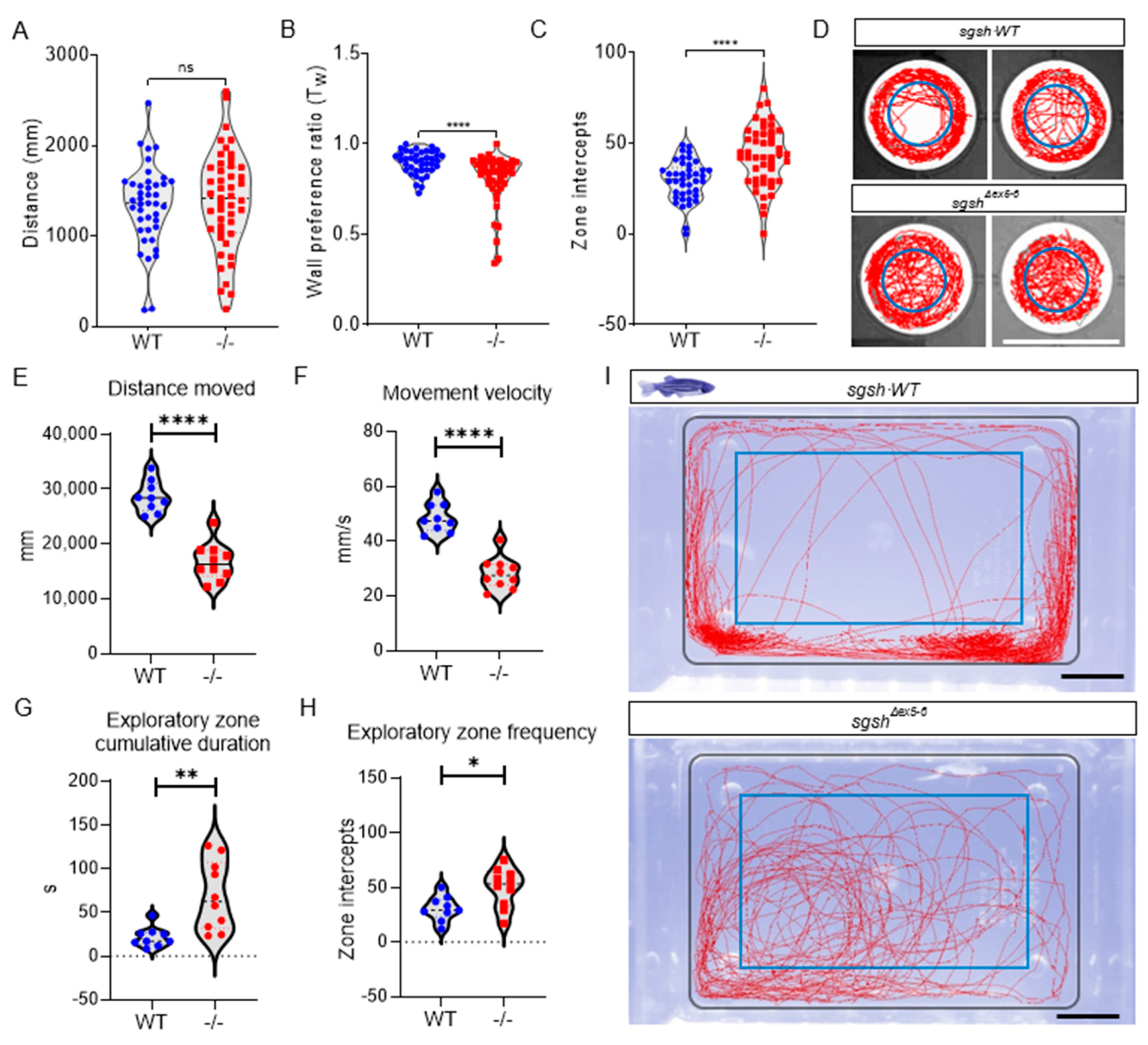
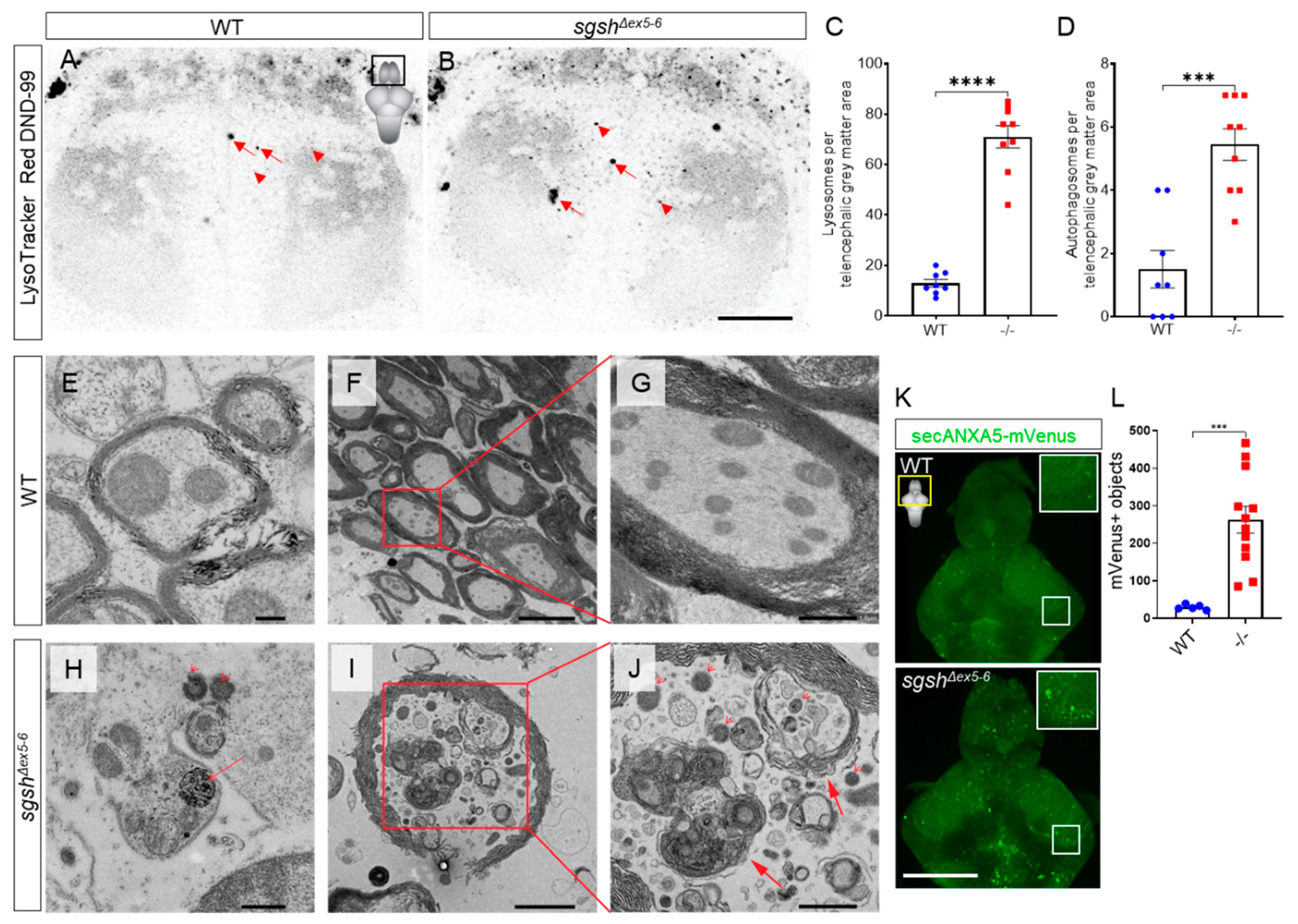
2.3. Behavioural Perturbations and Cellular Pathology in the CNS of Homozygous sgshΔex5−6 Zebrafish Recapitulates Features of Human MPS IIIA
2.4. Early-Onset and Lifelong Neuroinflammation Is a Hallmark of sgshΔex5−6 Zebrafish Mutant Pathology
2.5. Pharmacological Blockade of Il-1β Maturation Ameliorates Early-Developmental Microglial Activation in sgshΔex5−6 Larvae
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Generation of sgshΔex5−6 Zebrafish
4.3. Reverse Transcription Polymerase Chain Reaction (RT-PCR) of Wild Type and Mutant sgsh Transcript
4.4. In-Silico Modelling of sgshΔex5−6 Monomeric Protein Structure
4.5. Sgsh Enzymatic Activity Assay
4.6. Larval and Adult Behavioural Analysis
4.7. Caspase-3/7 Activity Assay
4.8. Larval Ac-YVAD-cmk Treatment
4.9. LysoTracker Staining of Zebrafish Larvae
4.10. Tissue Processing for Section Immunohistochemistry
4.11. Immunohistochemistry
4.12. Wholemount Immunochemistry
4.13. Tissue Processing for Transmission Electron Microscopy
4.14. Mass Spectrometry
4.15. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, W.; Xu, Y.; Liu, J.; Ho, M. Epitope mapping by a Wnt-blocking antibody: Evidence of the Wnt binding domain in heparan sulfate. Sci. Rep. 2016, 6, 26245. [Google Scholar] [CrossRef]
- Fuster, M.M.; Wang, L. Endothelial Heparan Sulfate in Angiogenesis. Prog. Mol. Biol. Transl. Sci. 2010, 93, 179–212. [Google Scholar]
- Beckouche, N.; Bignon, M.; Lelarge, V.; Mathivet, T.; Pichol-Thievend, C.; Berndt, S.; Hardouin, J.; Garand, M.; Ardidie-Robouant, C.; Barret, A.; et al. The interaction of heparan sulfate proteoglycans with endothelial transglutaminase-2 limits VEGF165-induced angiogenesis. Sci. Signal. 2015, 8, ra70. [Google Scholar] [CrossRef]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Heparan sulfate and heparan sulfate proteoglycans in cancer initiation and progression. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef]
- Cleary, M.; Wraith, J. Management of mucopolysaccharidosis type III. Arch. Dis. Child. 1993, 69, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Marchal, J.P.; Grootenhuis, M.; Colland, V.; Wijburg, F.A. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet J. Rare Dis. 2011, 6, 1–6. [Google Scholar] [CrossRef]
- Valstar, M.J.; Neijs, S.; Bruggenwirth, H.T.; Olmer, R.; Ruijter, G.J.G.; Wevers, R.A.; Van Diggelen, O.P.; Poorthuis, B.J.; Halley, D.J.; Wijburg, F.A. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann. Neurol. 2010, 68, 876–887. [Google Scholar] [CrossRef]
- Meyer, A.; Kossow, K.; Gal, A.; Ullrich, K.; Braulke, T.; Muschol, N.; Mühlhausen, C. Scoring Evaluation of the Natural Course of Mucopolysaccharidosis Type IIIA (Sanfilippo Syndrome Type A). Pediatrics 2007, 120, 1255. [Google Scholar] [CrossRef]
- Héron, B.; Mikaeloff, Y.; Froissart, R.; Caridade, G.; Maire, I.; Caillaud, C.; Levade, T.; Chabrol, B.; Feillet, F.; Ogier, H.; et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am. J. Med. Genet. Part A 2011, 155, 58–68. [Google Scholar] [CrossRef]
- Van De Kamp, J.J.P.; Niermeijer, M.F.; Von Figura, K.; Giesberts, M.A.H. Genetic heterogeneity and clinical variability in the Sanfilippo syndrome (types A, B, and C). Clin. Genet. 2008, 20, 152–160. [Google Scholar] [CrossRef]
- Zelei, T.; Csetneki, K.; Vokó, Z.; Siffel, C. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J. Rare Dis. 2018, 13, 53. [Google Scholar] [CrossRef]
- Lavery, C.; Hendriksz, C.J.; Jones, S.A. Mortality in patients with Sanfilippo syndrome. Orphanet J. Rare Dis. 2017, 12, 1–7. [Google Scholar] [CrossRef]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain pathology in mucopolysaccharidoses (MPS) patients with neurological forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef]
- Wijburg, F.A.; Węgrzyn, G.; Burton, B.K.; Tylki-Szymańska, A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013, 102, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Kresse, H. Mucopolysaccharidosis III A (Sanfilippo A disease): Deficiency of a heparin sulfamidase in skin fibroblasts and leucocytes. Biochem. Biophys. Res. Commun. 1973, 54, 1111–1118. [Google Scholar] [CrossRef]
- Kowalewski, B.; Lamanna, W.C.; Lawrence, R.; Damme, M.; Stroobants, S.; Padva, M.; Kalus, I.; Frese, M.-A.; Lübke, T.; Lüllmann-Rauch, R.; et al. Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10310–10315. [Google Scholar] [CrossRef]
- Kowalewski, B.; Heimann, P.; Ortkras, T.; Lüllmann-Rauch, R.; Sawada, T.; Walkley, S.U.; Dierks, T.; Damme, M. Ataxia is the major neuropathological finding in arylsulfatase G-deficient mice: Similarities and dissimilarities to Sanfilippo disease (mucopolysaccharidosis type III). Hum. Mol. Genet. 2014, 24, 1856–1868. [Google Scholar] [CrossRef]
- Khateb, S.; Kowalewski, B.; Bedoni, N.; Damme, M.; Pollack, N.; Saada, A.; Obolensky, A.; Ben-Yosef, T.; Gross, M.; Dierks, T.; et al. A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet. Med. 2018, 20, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Abad-Morales, V.; Navarro, R.; Burés-Jelstrup, A.; Pomares, E. Identification of a novel homozygous ARSG mutation as the second cause of Usher syndrome type 4. Am. J. Ophthalmol. Case Rep. 2020, 19, 100736. [Google Scholar] [CrossRef]
- Fowler, N.H.; El-Rashedy, M.I.; Chishti, E.A.; Van der Kooi, C.W.; Maldonado, R.S. Multimodal imaging and genetic findings in a case of ARSG-related atypical Usher syndrome. Ophthalmic Genet. 2021, 1–6. [Google Scholar] [CrossRef]
- Peter, V.G.; Quinodoz, M.; Sadio, S.; Held, S.; Rodrigues, M.; Soares, M.; Sousa, A.B.; Santos, L.C.; Damme, M.; Rivolta, C. New clinical and molecular evidence linking mutations in ARSG to Usher syndrome type IV. Hum. Mutat. 2021, 42, 261–271. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Nijmeijer, S.C.M.; Ijlst, L.; Brinke, H.T.; Van Vlies, N.; Wijburg, F.A. Prediction of phenotypic severity in mucopolysaccharidosis type IIIA. Ann. Neurol. 2017, 82, 686–696. [Google Scholar] [CrossRef]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Günther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.-L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nat. Cell Biol. 2019, 568, 193–197. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Sidhu, N.S.; Schreiber, K.; Pröpper, K.; Becker, S.; Usón, I.; Sheldrick, G.M.; Gärtner, J.; Krätzner, R.; Steinfeld, R. Structure of sulfamidase provides insight into the molecular pathology of mucopolysaccharidosis IIIA. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Karpova, E.; Voznyi, Y.V.; Keulemans, J.; Hoogeveen, A.; Winchester, B.; Tsvetkova, I.; Van Diggelen, O. A fluorimetric enzyme assay for the diagnosis of sanfilippo disease type A (MPS IIIA). J. Inherit. Metab. Dis. 1996, 19, 278–285. [Google Scholar] [CrossRef]
- Whyte, L.S.; Hopwood, J.J.; Hemsley, K.M.; Lau, A.A. Variables influencing fluorimetric N-sulfoglucosamine sulfohydrolase (SGSH) activity measurement in brain homogenates. Mol. Genet. Metab. Rep. 2015, 5, 60–62. [Google Scholar] [CrossRef]
- He, Q.Q.; Trim, P.J.; Lau, A.A.; King, B.M.; Hopwood, J.J.; Hemsley, K.M.; Snel, M.F.; Ferro, V. Synthetic Disaccharide Standards Enable Quantitative Analysis of Stored Heparan Sulfate in MPS IIIA Murine Brain Regions. ACS Chem. Neurosci. 2019, 10, 3847–3858. [Google Scholar] [CrossRef]
- Henry, J.; Rodriguez, A.; Wlodkowic, D. Impact of digital video analytics on accuracy of chemobehavioural phenotyping in aquatic toxicology. PeerJ 2019, 7, e7367. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Gebhardt, M.; Stewart, A.M.; Cachat, J.M.; Brimmer, M.; Chawla, J.S.; Craddock, C.; Kyzar, E.J.; Roth, A.; Landsman, S.; et al. Towards a Comprehensive Catalog of Zebrafish Behavior 1.0 and Beyond. Zebrafish 2013, 10, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.G.; Nestrasil, I.; Ahmed, A.; Wey, A.; Rudser, K.R.; Delaney, K.A.; Rumsey, R.K.; Haslett, P.A.; Whitley, C.B.; Potegal, M. Quantifying behaviors of children with Sanfilippo syndrome: The Sanfilippo Behavior Rating Scale. Mol. Genet. Metab. 2015, 114, 594–598. [Google Scholar] [CrossRef]
- Langford-Smith, A.; Langford-Smith, K.J.; Jones, S.A.; Wynn, R.F.; Wraith, J.; Wilkinson, F.L.; Bigger, B.W. Female mucopolysaccharidosis IIIA mice exhibit hyperactivity and a reduced sense of danger in the open field test. PLoS ONE 2011, 6, e25717. [Google Scholar] [CrossRef]
- Chikte, S.; Panchal, N.; Warnes, G. Use of LysoTracker dyes: A flow cytometric study of autophagy. Cytom. Part A 2014, 85, 169–178. [Google Scholar] [CrossRef]
- Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J. Clin. Med. 2020, 9, 344. [Google Scholar] [CrossRef]
- Arfi, A.; Richard, M.; Gandolphe, C.; Bonnefont-Rousselot, D.; Thérond, P.; Scherman, D. Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: The positive effect of long-term aspirin treatment. Mol. Genet. Metab. 2011, 103, 18–25. [Google Scholar] [CrossRef]
- Morsch, M.; Radford, R.; Lee, A.; Don, E.K.; Badrock, A.P.; Hall, T.E.; Cole, N.J.; Chung, R. In vivo characterization of microglial engulfment of dying neurons in the zebrafish spinal cord. Front. Cell. Neurosci. 2015, 9, 321. [Google Scholar] [CrossRef]
- Blewett, M. Lipid autoreactivity in multiple sclerosis. Med. Hypotheses 2010, 74, 433–442. [Google Scholar] [CrossRef]
- Fraldi, A.; Hemsley, K.; Crawley, A.; Lombardi, A.; Lau, A.; Sutherland, L.; Auricchio, A.; Ballabio, A.; Hopwood, J.J. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum. Mol. Genet. 2007, 16, 2693–2702. [Google Scholar] [CrossRef]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in Mouse Models of Mucopolysaccharidosis Type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef] [PubMed]
- Ausseil, J.; Desmaris, N.; Bigou, S.; Attali, R.; Corbineau, S.; Vitry, S.; Parent, M.; Cheillan, D.; Fuller, M.; Maire, I.; et al. Early Neurodegeneration Progresses Independently of Microglial Activation by Heparan Sulfate in the Brain of Mucopolysaccharidosis IIIB Mice. PLoS ONE 2008, 3, e2296. [Google Scholar] [CrossRef]
- Martins, C.; Hůlková, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, C.A.; Scudder, S.L.; Lin, Y.; Dozier, L.E.; Phan, D.; Allen, N.J.; Patrick, G.N.; Esko, J.D. Neurodevelopmental Changes in Excitatory Synaptic Structure and Function in the Cerebral Cortex of Sanfilippo Syndrome IIIA Mice. Sci. Rep. 2017, 7, 46576. [Google Scholar] [CrossRef]
- Bernardos, R.L.; Raymond, P.A. GFAP transgenic zebrafish. Gene Expr. Patterns 2006, 6, 1007–1013. [Google Scholar] [CrossRef]
- Lindsey, B.W.; Hall, Z.J.; Heuzé, A.; Joly, J.-S.; Tropepe, V.; Kaslin, J. The role of neuro-epithelial-like and radial-glial stem and progenitor cells in development, plasticity, and repair. Prog. Neurobiol. 2018, 170, 99–114. [Google Scholar] [CrossRef]
- Liu, J.; Fraser, S.D.; Faloon, P.W.; Rollins, E.L.; Berg, J.V.; Starovic-Subota, O.; Laliberte, A.L.; Chen, J.-N.; Serluca, F.C.; Childs, S.J. A βPix–Pak2a signaling pathway regulates cerebral vascular stability in zebrafish. Proc. Natl. Acad. Sci. USA 2007, 104, 13990–13995. [Google Scholar] [CrossRef] [PubMed]
- Keightley, M.-C.; Wang, C.-H.; Pazhakh, V.; Lieschke, G.J. Delineating the roles of neutrophils and macrophages in zebrafish regeneration models. Int. J. Biochem. Cell Biol. 2014, 56, 92–106. [Google Scholar] [CrossRef]
- Kyritsis, N.; Kizil, C.; Zocher, S.; Kroehne, V.; Kaslin, J.; Freudenreich, D.; Iltzsche, A.; Brand, M. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 2012, 338, 1353–1356. [Google Scholar] [CrossRef]
- Redd, M.J.; Kelly, G.; Dunn, G.; Way, M.; Martin, P. Imaging macrophage chemotaxis in vivo: Studies of microtubule function in zebrafish wound inflammation. Cell Motil. Cytoskeleton 2006, 63, 415–422. [Google Scholar] [CrossRef]
- Kroehne, V.; Freudenreich, D.; Hans, S.; Kaslin, J.; Brand, M. Regeneration of the adult zebrafish brain from neurogenic radial glia-type progenitors. Development 2011, 138, 4831–4841. [Google Scholar] [CrossRef] [PubMed]
- Kaslin, J.; Kroehne, V.; Ganz, J.; Hans, S.; Brand, M. Distinct roles of neuroepithelial-like and radial glia-like progenitor cells in cerebellar regeneration. Development 2017, 144, 1462–1471. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Wu, S.; Nguyen, L.T.M.; Pan, H.; Hassan, S.; Dai, Y.; Xu, J.; Wen, Z. Two phenotypically and functionally distinct microglial populations in adult zebrafish. Sci. Adv. 2020, 6, eabd1160. [Google Scholar] [CrossRef]
- Keating, D.; Winter, M.; Hemsley, K.; MacKenzie, K.; Teo, E.; Hopwood, J.; Brooks, D.; Parkinson-Lawrence, E. Exocytosis is impaired in mucopolysaccharidosis IIIA mouse chromaffin cells. Neuroscience 2012, 227, 110–118. [Google Scholar] [CrossRef]
- Machaczka, M.; Klimkowska, M.; Regenthal, S.; Hägglund, H. Gaucher disease with foamy transformed macrophages and erythrophagocytic activity. J. Inherit. Metab. Dis. 2010, 34, 233–235. [Google Scholar] [CrossRef]
- Aerts, J.M.; Kuo, C.-L.; Lelieveld, L.T.; Boer, D.E.; van der Lienden, M.J.; Overkleeft, H.S.; Artola, M. Glycosphingolipids and lysosomal storage disorders as illustrated by gaucher disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. [Google Scholar] [CrossRef]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 1–25. [Google Scholar] [CrossRef]
- Alam, M.S.; Getz, M.; Safeukui, I.; Yi, S.; Tamez, P.; Shin, J.; Velázquez, P.; Haldar, K. Genomic Expression Analyses Reveal Lysosomal, Innate Immunity Proteins, as Disease Correlates in Murine Models of a Lysosomal Storage Disorder. PLoS ONE 2012, 7, e48273. [Google Scholar] [CrossRef]
- Lee, J.-P.; Jeyakumar, M.; Gonzalez, R.; Takahashi, H.; Lee, P.-J.; Baek, R.C.; Clark, D.; Rose, H.; Fu, G.; Clarke, J.; et al. Stem cells act through multiple mechanisms to benefit mice with neurodegenerative metabolic disease. Nat. Med. 2007, 13, 439–447. [Google Scholar] [CrossRef]
- Reddy, A.S.; Kim, J.H.; Hawkins-Salsbury, J.A.; Macauley, S.L.; Tracy, E.T.; Vogler, C.A.; Han, X.; Song, S.-K.; Wozniak, D.F.; Fowler, S.C.; et al. Bone Marrow Transplantation Augments the Effect of Brain- and Spinal Cord-Directed Adeno-Associated Virus 2/5 Gene Therapy by Altering Inflammation in the Murine Model of Globoid-Cell Leukodystrophy. J. Neurosci. 2011, 31, 9945–9957. [Google Scholar] [CrossRef]
- Parker, H.; Ellison, S.M.; Holley, R.J.; O’Leary, C.; Liao, A.; Asadi, J.; Glover, E.; Ghosh, A.; Jones, S.; Wilkinson, F.L.; et al. Haematopoietic stem cell gene therapy with IL-1Ra rescues cognitive loss in mucopolysaccharidosis IIIA. EMBO Mol. Med. 2020, 12, e11185. [Google Scholar] [CrossRef] [PubMed]
- Sergijenko, A.; Langford-Smith, A.; Liao, A.Y.; Pickford, C.; McDermott, J.; Nowinski, G.; Langford-Smith, K.J.; Merry, C.L.; Jones, S.A.; Wraith, J.E.; et al. Myeloid/Microglial Driven Autologous Hematopoietic Stem Cell Gene Therapy Corrects a Neuronopathic Lysosomal Disease. Mol. Ther. 2013, 21, 1938–1949. [Google Scholar] [CrossRef]
- Holley, R.J.; Ellison, S.M.; Fil, D.; O’Leary, C.; McDermott, J.; Senthivel, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; D’Souza, Z.; Parker, H.; et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain 2017, 141, 99–116. [Google Scholar] [CrossRef]
- Kysil, E.V.; Meshalkina, D.; Frick, E.E.; Echevarria, D.J.; Rosemberg, D.B.; Maximino, C.; Lima, M.G.; Abreu, M.S.; Giacomini, A.C.; Barcellos, L.J.G.; et al. Comparative Analyses of Zebrafish Anxiety-Like Behavior Using Conflict-Based Novelty Tests. Zebrafish 2017, 14, 197–208. [Google Scholar] [CrossRef]
- Peng, X.; Lin, J.; Zhu, Y.; Liu, X.; Zhang, Y.; Ji, Y.; Yang, X.; Zhang, Y.; Guo, N.; Li, Q. Anxiety-related behavioral responses of pentylenetetrazole-treated zebrafish larvae to light-dark transitions. Pharmacol. Biochem. Behav. 2016, 145, 55–65. [Google Scholar] [CrossRef]
- Walpitagama, M.; Carve, M.; Douek, A.M.; Trestrail, C.; Bai, Y.; Kaslin, J.; Wlodkowic, D. Additives migrating from 3D-printed plastic induce developmental toxicity and neuro-behavioural alterations in early life zebrafish (Danio rerio). Aquat. Toxicol. 2019, 213, 105227. [Google Scholar] [CrossRef]
- Nakamura, T.; Gehrke, A.R.; Lemberg, J.; Szymaszek, J.; Shubin, T.N. Digits and fin rays share common developmental histories. Nat. Cell Biol. 2016, 537, 225–228. [Google Scholar] [CrossRef]
- Lele, Z.; Krone, P. The zebrafish as a model system in developmental, toxicological and transgenic research. Biotechnol. Adv. 1996, 14, 57–72. [Google Scholar] [CrossRef]
- Lindsey, B.W.; Aitken, G.E.; Tang, J.K.; Khabooshan, M.; Douek, A.M.; Vandestadt, C.; Kaslin, J. Midbrain tectal stem cells display diverse regenerative capacities in zebrafish. Sci. Rep. 2019, 9, 1–20. [Google Scholar] [CrossRef]
- Vandestadt, C.; Vanwalleghem, G.C.; Castillo, H.A.; Li, M.; Schulze, K.; Khabooshan, M.; Don, E.; Anko, M.-L.; Scott, E.K.; Kaslin, J. Early migration of precursor neurons initiates cellular and functional regeneration after spinal cord injury in zebrafish. bioRxiv 2019, 539940. [Google Scholar]
- Ratnayake, D.; Nguyen, P.D.; Rossello, F.J.; Wimmer, V.C.; Tan, J.L.; Galvis, L.A.; Julier, Z.; Wood, A.J.; Boudier, T.; Isiaku, A.I.; et al. Macrophages provide a transient muscle stem cell niche via NAMPT secretion. Nat. Cell Biol. 2021, 591, 281–287. [Google Scholar] [CrossRef]
- Sztal, T.E.; Stainier, D.Y.R. Transcriptional adaptation: A mechanism underlying genetic robustness. Development 2020, 147, 15. [Google Scholar] [CrossRef]
- Fischer, A.; Carmichael, K.P.; Munnell, J.F.; Jhabvala, P.; Thompson, J.N.; Matalon, R.; Jezyk, P.F.; Wang, P.; Giger, U. Sulfamidase Deficiency in a Family of Dachshunds: A Canine Model of Mucopolysaccharidosis IIIA (Sanfilippo A). Pediatr. Res. 1998, 44, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Yogalingam, G.; Pollard, T.; Gliddon, B.; Jolly, R.D.; Hopwood, J.J. Identification of a mutation causing mucopolysaccharidosis type IIIA in New Zealand Huntaway dogs. Genomics 2002, 79, 150–153. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Gliddon, B.; Beccari, T.; Hopwood, J.J.; Stanley, P. A novel missense mutation in lysosomal sulfamidase is the basis of MPS III A in a spontaneous mouse mutant. Glycobiology 2001, 11, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.A.; King, B.M.; Thorsen, C.L.; Hassiotis, S.; Beard, H.; Trim, P.J.; Whyte, L.S.; Tamang, S.J.; Duplock, S.K.; Snel, M.F.; et al. A novel conditional Sgsh knockout mouse model recapitulates phenotypic and neuropathic deficits of Sanfilippo syndrome. J. Inherit. Metab. Dis. 2017, 40, 715–724. [Google Scholar] [CrossRef]
- Webber, D.L.; Choo, A.; Hewson, L.J.; Trim, P.J.; Snel, M.F.; Hopwood, J.J.; Richards, R.I.; Hemsley, K.M.; O’Keefe, L.V. Neuronal-specific impairment of heparan sulfate degradation in Drosophila reveals pathogenic mechanisms for Mucopolysaccharidosis type IIIA. Exp. Neurol. 2018, 303, 38–47. [Google Scholar] [CrossRef]
- Crawley, A.C.; Gliddon, B.L.; Auclair, D.; Brodie, S.L.; Hirte, C.; King, B.M.; Fuller, M.; Hemsley, K.M.; Hopwood, J.J. Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res. 2006, 1104, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.A.; Tamang, S.J.; Hemsley, K.M. MPS-IIIA mice acquire autistic behaviours with age. J. Inherit. Metab. Dis. 2018, 41, 669–677. [Google Scholar] [CrossRef]
- Hemsley, K.M.; Hopwood, J.J. Development of motor deficits in a murine model of mucopolysaccharidosis type IIIA (MPS-IIIA). Behav. Brain Res. 2005, 158, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Hemsley, K.M.; King, B.; Hopwood, J.J. Injection of recombinant human sulfamidase into the CSF via the cerebellomedullary cistern in MPS IIIA mice. Mol. Genet. Metab. 2007, 90, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.A.; Crawley, A.C.; Hopwood, J.J.; Hemsley, K.M. Open field locomotor activity and anxiety-related behaviors in mucopolysaccharidosis type IIIA mice. Behav. Brain Res. 2008, 191, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.A.; Hannouche, H.; Rozaklis, T.; Hassiotis, S.; Hopwood, J.J.; Hemsley, K.M. Allogeneic stem cell transplantation does not improve neurological deficits in mucopolysaccharidosis type IIIA mice. Exp. Neurol. 2010, 225, 445–454. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.; Byers, S.; Anson, D.S. Correction of mucopolysaccharidosis type IIIA somatic and central nervous system pathology by lentiviral-mediated gene transfer. J. Genet. Med. 2010, 12, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.; Meikle, P.; Hopwood, J.; Fuller, M. Distribution of heparan sulfate oligosaccharides in murine mucopolysaccharidosis type IIIA. Metabolites 2014, 4, 1088–1100. [Google Scholar] [CrossRef]
- Gesslbauer, B.; Derler, R.; Handwerker, C.; Seles, E.; Kungl, A.J. Exploring the glycosaminoglycan–protein interaction network by glycan-mediated pull-down proteomics. Electrophoresis 2016, 37, 1437–1447. [Google Scholar] [CrossRef]
- Loo, B.-M.; Salmivirta, M. Heparin/Heparan Sulfate Domains in Binding and Signaling of Fibroblast Growth Factor 8b. J. Biol. Chem. 2002, 277, 32616–32623. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Z.; Lin, X.; Beenken, A.; Eliseenkova, A.V.; Mohammadi, M.; Linhardt, R.J. Compositional Analysis of Heparin/Heparan Sulfate Interacting with Fibroblast Growth Factor Fibroblast Growth Factor Receptor Complexes. Biochemistry 2009, 48, 8379–8386. [Google Scholar] [CrossRef]
- Yu, S.R.; Burkhardt, M.; Nowak, M.; Ries, J.; Petrášek, Z.; Scholpp, S.; Schwille, P.; Brand, M. Fgf8 morphogen gradient forms by a source-sink mechanism with freely diffusing molecules. Nat. Cell Biol. 2009, 461, 533–536. [Google Scholar] [CrossRef]
- Hopwood, J.J. Prenatal diagnosis of Sanfilippo syndrome. Prenat. Diagn. 2005, 25, 148–150. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J. Lysosomal storage disorders: Emerging therapeutic options require early diagnosis. Eur. J. Nucl. Med. Mol. Imaging 2003, 162, S34–S37. [Google Scholar] [CrossRef] [PubMed]
- Beard, H.; Hassiotis, S.; Gai, W.-P.; Parkinson-Lawrence, E.; Hopwood, J.J.; Hemsley, K.M. Axonal dystrophy in the brain of mice with Sanfilippo syndrome. Exp. Neurol. 2017, 295, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Caspase-independent cell death: Leaving the set without the final cut. Oncogene 2008, 27, 6452–6461. [Google Scholar] [CrossRef]
- Novoa, B.; Bowman, T.; Zon, L.; Figueras, A. LPS response and tolerance in the zebrafish (Danio rerio). Fish Shellfish. Immunol. 2009, 26, 326–331. [Google Scholar] [CrossRef]
- Sullivan, C.; Charette, J.; Catchen, J.; Lage, C.R.; Giasson, G.; Postlethwait, J.H.; Millard, P.J.; Kim, C.H. The gene history of zebrafish tlr4a and tlr4b is predictive of their divergent functions. J. Immunol. 2009, 183, 5896–5908. [Google Scholar] [CrossRef]
- Sepulcre, M.P.; Alcaraz-Pérez, F.; López-Muñoz, A.; Roca, F.J.; Meseguer, J.; Cayuela, M.L.; Mulero, V. Evolution of lipopolysaccharide (LPS) recognition and signaling: Fish TLR4 does not recognize LPS and negatively regulates NF-κB activation. J. Immunol. 2009, 182, 1836–1845. [Google Scholar] [CrossRef]
- Brennan, T.V.; Lin, L.; Huang, X.; Cardona, D.; Li, Z.; Dredge, K.; Chao, N.J.; Yang, Y. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood 2012, 120, 2899–2908. [Google Scholar] [CrossRef]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish. Available online: http://zfin.org/zf_info/zfbook/zfbk.html (accessed on 1 April 2021).
- Schnörr, S.; Steenbergen, P.; Richardson, M.; Champagne, D. Measuring thigmotaxis in larval zebrafish. Behav. Brain Res. 2012, 228, 367–374. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (IDT Identifier) | Sequence (PAM Not Included for crRNAs) |
|---|---|
| sgsh crRNA 1 (CD.Cas9.JJFS2685.AM) | 5′-TCCGGACACTCCTGCAGCGA-3′ |
| sgsh crRNA 2 (CD.Cas9.JJFS2685.AB) | 5′-CCAGGCCTACGTCAGTCTAC-3′ |
| sgsh genotyping F primer | 5′-GGATGTCTTTATCCCGAACATAAC-3′ |
| sgsh genotyping R primer | 5′-ATTTACTCACCCTTGGACCATTTA-3′ |
| sgsh RT-PCR exon 1 F primer | 5′-TGATGTAGGAGGATGCAGAAGTAG-3′ |
| sgsh RT-PCR exon 7 R primer | 5′-AGCCAGATTCACCTTCTCCA-3′ |
| eef1a1l1 RT-PCR F primer | 5′-GAAGACAACCCCAAGGCTCTC-3′ |
| eef1a1l1 RT-PCR R primer | 5′-CCACCGATTTTCTTCTCAACG-3′ |
| Time | % A | % B | Flow (mL/min) |
|---|---|---|---|
| 0.00 | 80 | 20 | 0.350 |
| 3.50 | 60 | 40 | 0.350 |
| 3.51 | 1 | 99 | 0.350 |
| 4.50 | 80 | 20 | 0.350 |
| 5.50 | 80 | 20 | 0.350 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douek, A.M.; Amiri Khabooshan, M.; Henry, J.; Stamatis, S.-A.; Kreuder, F.; Ramm, G.; Änkö, M.-L.; Wlodkowic, D.; Kaslin, J. An Engineered sgsh Mutant Zebrafish Recapitulates Molecular and Behavioural Pathobiology of Sanfilippo Syndrome A/MPS IIIA. Int. J. Mol. Sci. 2021, 22, 5948. https://doi.org/10.3390/ijms22115948
Douek AM, Amiri Khabooshan M, Henry J, Stamatis S-A, Kreuder F, Ramm G, Änkö M-L, Wlodkowic D, Kaslin J. An Engineered sgsh Mutant Zebrafish Recapitulates Molecular and Behavioural Pathobiology of Sanfilippo Syndrome A/MPS IIIA. International Journal of Molecular Sciences. 2021; 22(11):5948. https://doi.org/10.3390/ijms22115948
Chicago/Turabian StyleDouek, Alon M., Mitra Amiri Khabooshan, Jason Henry, Sebastian-Alexander Stamatis, Florian Kreuder, Georg Ramm, Minna-Liisa Änkö, Donald Wlodkowic, and Jan Kaslin. 2021. "An Engineered sgsh Mutant Zebrafish Recapitulates Molecular and Behavioural Pathobiology of Sanfilippo Syndrome A/MPS IIIA" International Journal of Molecular Sciences 22, no. 11: 5948. https://doi.org/10.3390/ijms22115948
APA StyleDouek, A. M., Amiri Khabooshan, M., Henry, J., Stamatis, S.-A., Kreuder, F., Ramm, G., Änkö, M.-L., Wlodkowic, D., & Kaslin, J. (2021). An Engineered sgsh Mutant Zebrafish Recapitulates Molecular and Behavioural Pathobiology of Sanfilippo Syndrome A/MPS IIIA. International Journal of Molecular Sciences, 22(11), 5948. https://doi.org/10.3390/ijms22115948