
Anti-Inflammatory Effect of Auranofin on Palmitic Acid and LPS-Induced Inflammatory Response by Modulating TLR4 and NOX4-Mediated NF-κB Signaling Pathway in RAW264.7 Macrophages

 , , , ,
, , , ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. PA Amplifies LPS-Stimulated Release of NO and Inflammatory Cytokines in RAW264.7 Macrophages
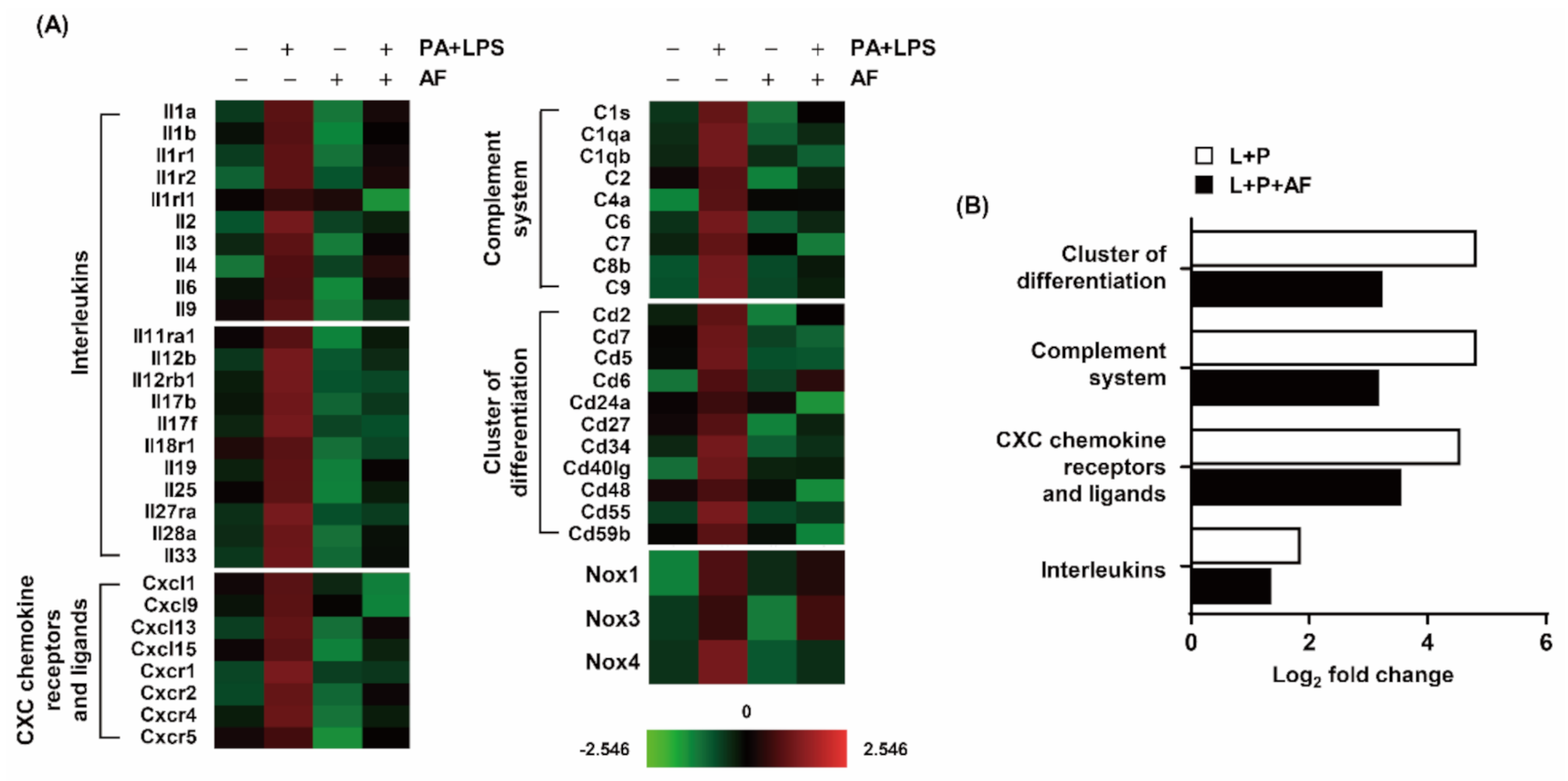
2.2. Auranofin Suppresses PA and LPS-Induced Expression Profiles of Pro-Inflammatory Genes
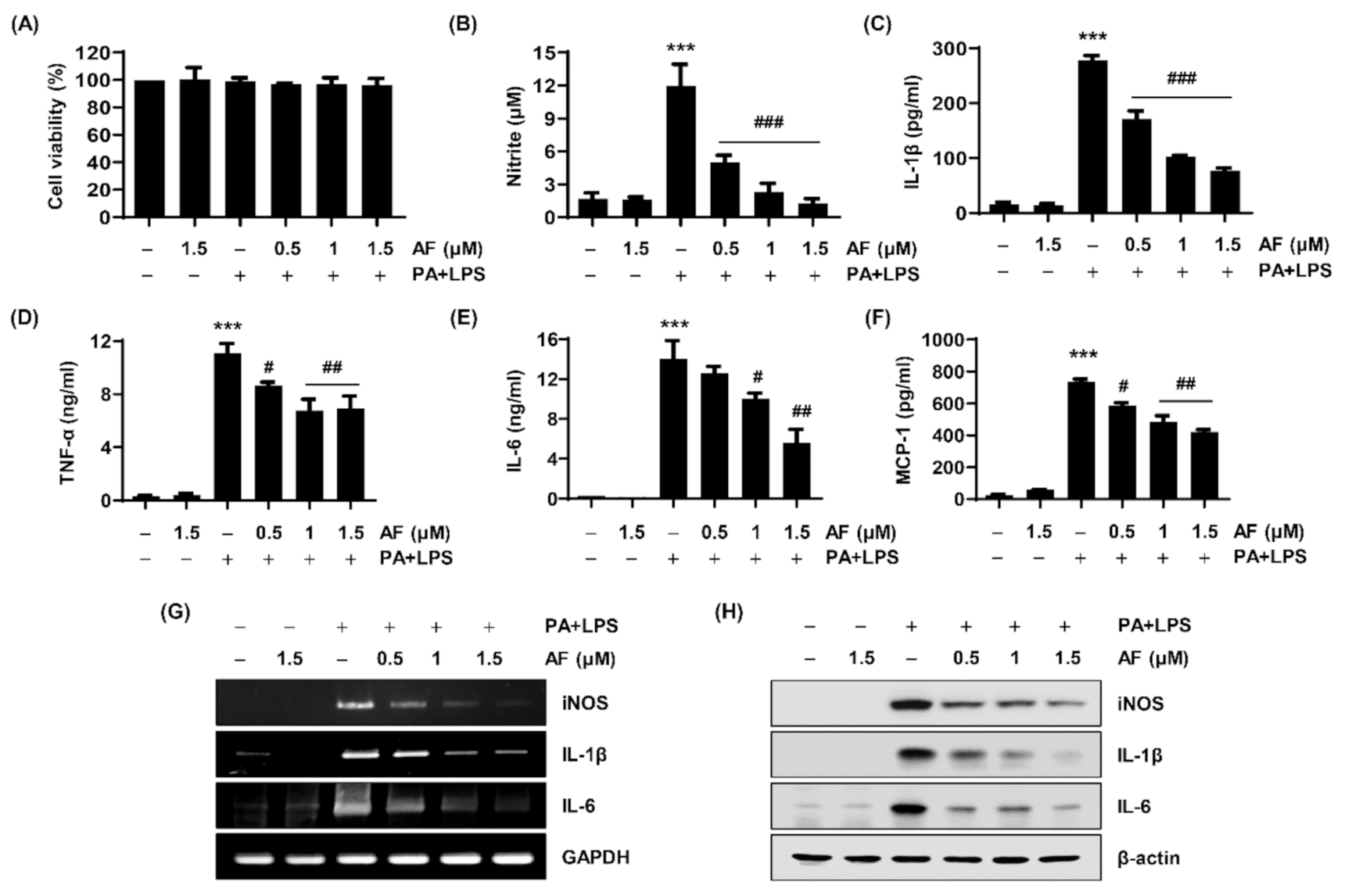
2.3. Auranofin Attenuates the Production of NO, Cytokine Secretion, and the Expression of Their Regulatory Genes
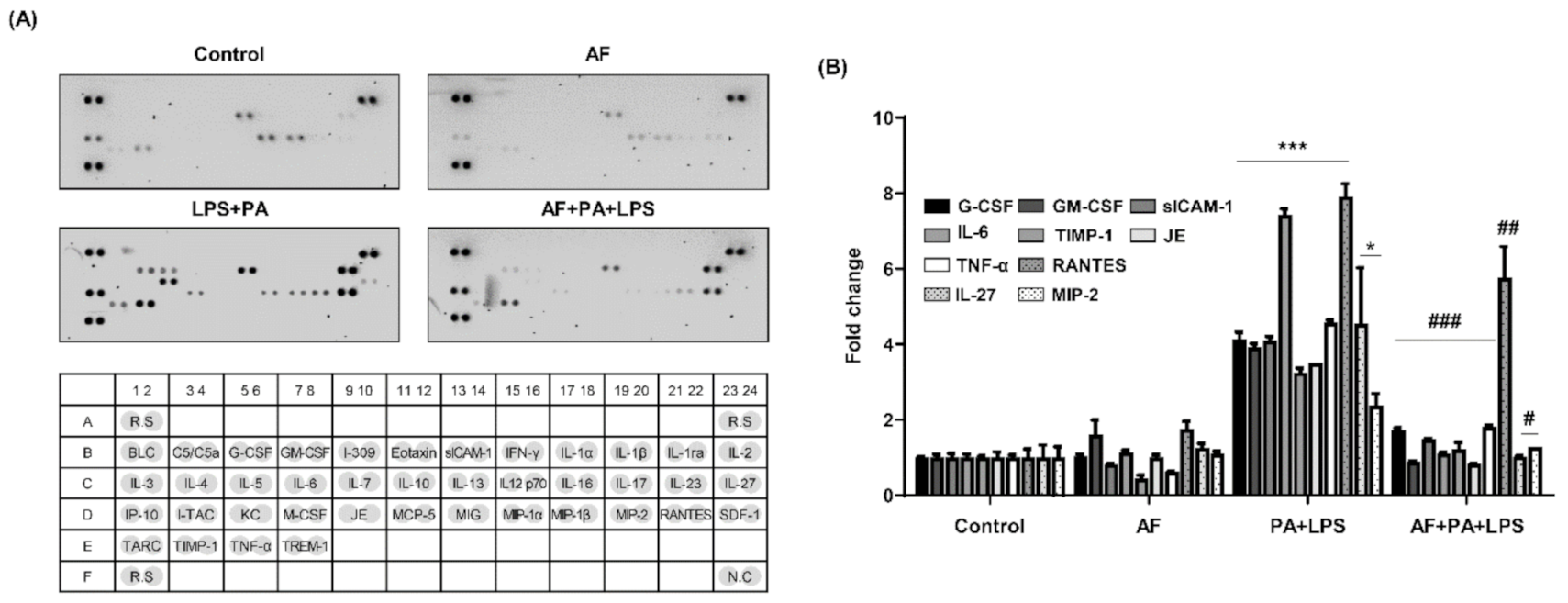
2.4. Auranofin Decreases the PA and LPS-Induced Releases of Cytokines and Chemokines
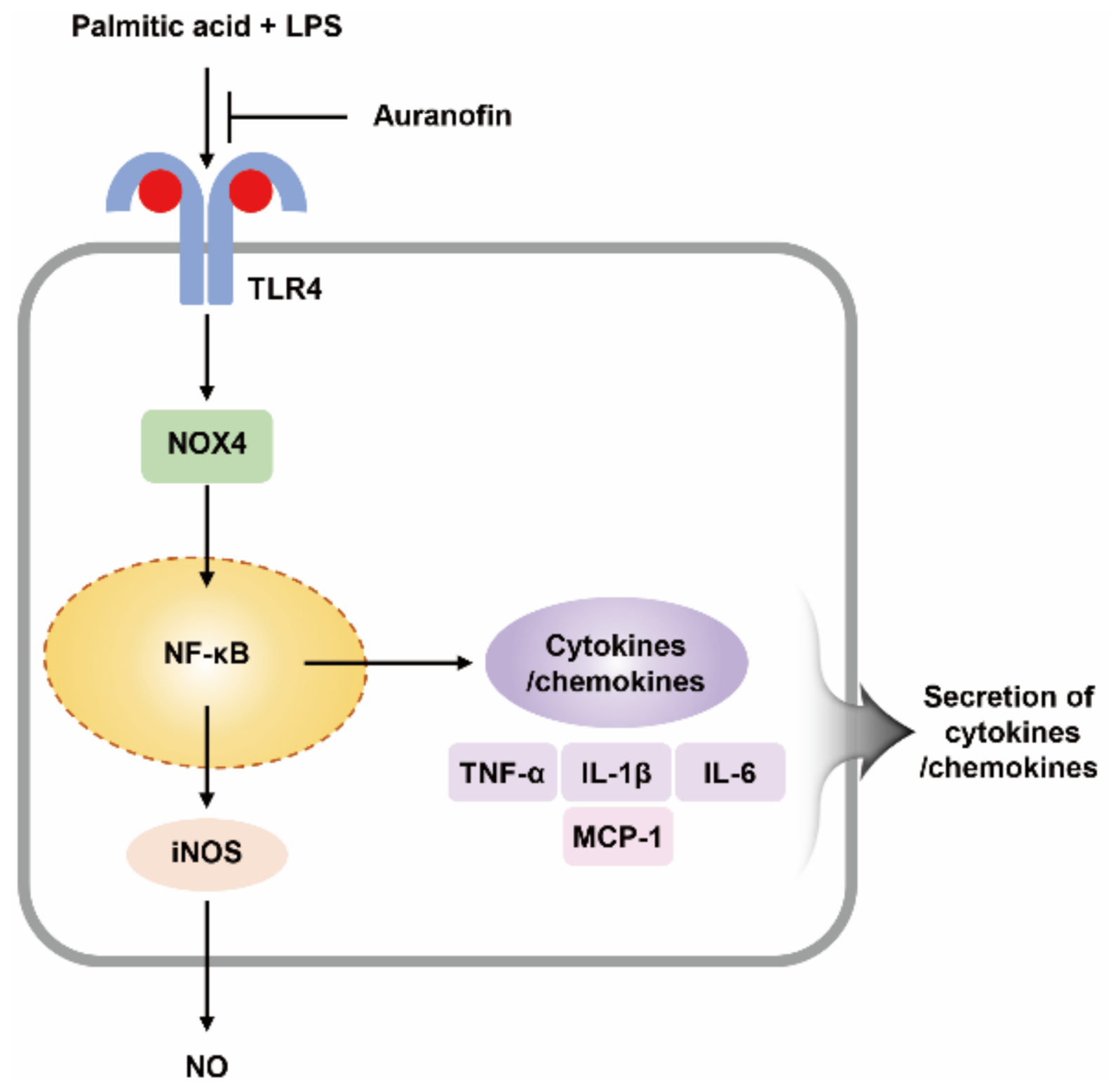
2.5. PA and LPS Increases NOX4 Expression, and Auranofin Reduces the NOX4-Mediated Inflammatory Response
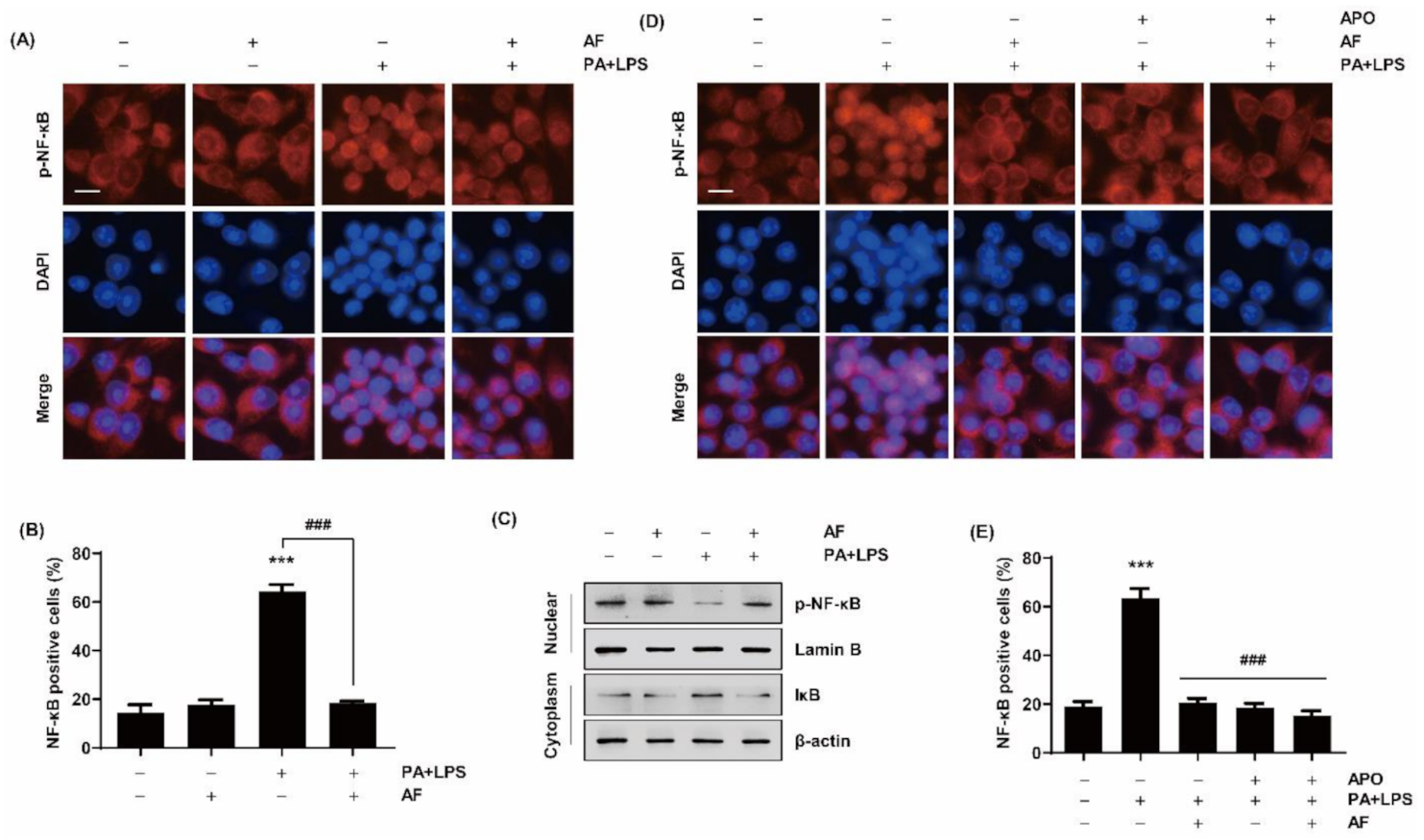
2.6. Auranofin Inhibits the Translocation of NF-κB to the Nucleus by PA and LPS
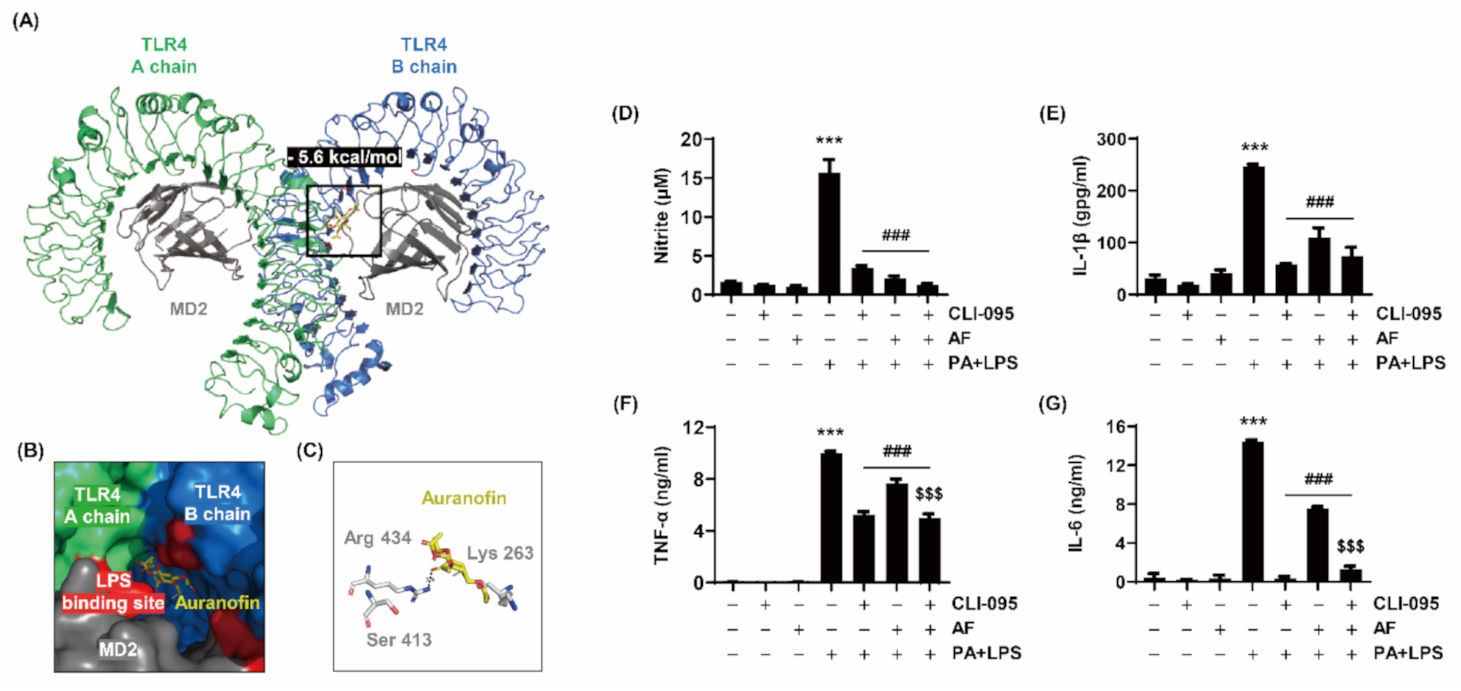
2.7. Auranofin Inhibits PA and LPS-Stimulated Inflammation by Interacting with the TLR4/MD2 Complex
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Treatment of Chemicals
4.4. MTT Assay
4.5. Assessment of Nitrite Production
4.6. Measurement of Cytokine Level
4.7. Analysis of Gene Expression in the Immune Response
4.8. RT-PCR Analysis
4.9. Western Blot Analysis
4.10. Cytokine Array Analysis
4.11. Immunofluorescence
4.12. Molecular Docking Prediction for TLR4 and Auranofin Interaction
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Stavitsky, A.B. The innate immune response to infection, toxins and trauma evolved into networks of interactive, defensive, reparative, regulatory, injurious and pathogenic pathways. Mol. Immunol. 2007, 44, 2787–2799. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef]
- Satou, R.; Penrose, H.; Navar, L.G. Inflammation as a Regulator of the Renin-Angiotensin System and Blood Pressure. Curr. Hypertens. Rep. 2018, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Skiba, D.S.; Touyz, R.M.; Harrison, D.G. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc. Res. 2017, 113, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; D’Ovidio, C.; Conti, C.; Gallenga, C.E.; Lauritano, D.; Caraffa, A.; Kritas, S.K.; Ronconi, G. Progression in migraine: Role of mast cells and pro-inflammatory and anti-inflammatory cytokines. Eur. J. Pharmacol. 2019, 844, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Pålsson-McDermott, E.M.; O’Neill, L.A. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef]
- Sachithanandan, N.; Graham, K.L.; Galic, S.; Honeyman, J.E.; Fynch, S.L.; Hewitt, K.A.; Steinberg, G.R.; Kay, T.W. Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes 2011, 60, 2023–2031. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, X.; Deng, B.; Xiao, J.; Jin, J.; Huang, Z. Lipopolysaccharide and palmitic acid synergistically induced MCP-1 production via MAPK-meditated TLR4 signaling pathway in RAW264.7 cells. Lipids Health Dis. 2019, 18, 71. [Google Scholar] [CrossRef]
- Jin, J.; Lu, Z.; Li, Y.; Cowart, L.A.; Lopes-Virella, M.F.; Huang, Y. Docosahexaenoic acid antagonizes the boosting effect of palmitic acid on LPS inflammatory signaling by inhibiting gene transcription and ceramide synthesis. PLoS ONE 2018, 13, e0193343. [Google Scholar] [CrossRef]
- Essakalli, M.; Atouf, O.; Bennani, N.; Benseffaj, N.; Ouadghiri, S.; Brick, C. Toll-like récepteurs [Toll-like receptors]. Pathol. Biol. 2009, 57, 430–438. [Google Scholar] [CrossRef]
- Yoon, H.J.; Chay, K.O.; Yang, S.Y. Native low density lipoprotein increases the production of both nitric oxide and reactive oxygen species in the human umbilical vein endothelial cells. Genes Genom. 2019, 41, 373–379. [Google Scholar] [CrossRef]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef]
- Cochet, F.; Peri, F. The Role of Carbohydrates in the Lipopolysaccharide (LPS)/Toll-Like Receptor 4 (TLR4) Signalling. Int. J. Mol. Sci. 2017, 18, 2318. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef]
- Du, S.; Miao, J.; Lu, X.; Shi, L.; Sun, J.; Xu, E.; Wang, X.; Zhao, M.; Chen, H.; Wang, F.; et al. NADPH oxidase 4 is correlated with gastric cancer progression and predicts a poor prognosis. Am. J. Transl. Res. 2019, 11, 3518–3530. [Google Scholar]
- Suzuki, S.; Pitchakarn, P.; Sato, S.; Shirai, T.; Takahashi, S. Apocynin, an NADPH oxidase inhibitor, suppresses progression of prostate cancer via Rac1 dephosphorylation. Exp. Toxicol. Pathol. 2013, 65, 1035–1041. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Bertran, E.; Sancho, P.; López-Luque, J.; Fernando, J.; Sánchez, A.; Fernández, M.; Navarro, E.; Fabregat, I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014, 69, 338–347. [Google Scholar] [CrossRef]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting edge: Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef]
- Hu, H.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, Z.; Liu, K.; Liu, X.; Sun, H. Catalpol Inhibits Homocysteine-induced Oxidation and Inflammation via Inhibiting Nox4/NF-κB and GRP78/PERK Pathways in Human Aorta Endothelial Cells. Inflammation 2019, 42, 64–80. [Google Scholar] [CrossRef]
- Liu, X.; Qin, Z.; Liu, C.; Song, M.; Luo, X.; Zhao, H.; Qian, D.; Chen, J.; Huang, L. Nox4 and soluble epoxide hydrolase synergistically mediate homocysteine-induced inflammation in vascular smooth muscle cells. Vascul. Pharmacol. 2019, 120, 106544. [Google Scholar] [CrossRef]
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef]
- Yu, M.H.; Li, X.; Li, Q.; Mo, S.J.; Ni, Y.; Han, F.; Wang, Y.B.; Tu, Y.X. SAA1 increases NOX4/ROS production to promote LPS-induced inflammation in vascular smooth muscle cells through activating p38MAPK/NF-κB pathway. BMC Mol. Cell Biol. 2019, 20, 15. [Google Scholar] [CrossRef]
- Williams, C.R.; Lu, X.; Sutliff, R.L.; Hart, C.M. Rosiglitazone attenuates NF-κB-mediated Nox4 upregulation in hyperglycemia-activated endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C213–C223. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, X.; Feng, X.; Zhao, Y.; Song, M.; Wang, C.; Fan, H. Dexmedetomidine alleviates lipopolysaccharide-induced acute kidney injury by inhibiting the NLRP3 inflammasome activation via regulating the TLR4/NOX4/NF-κB pathway. J. Cell Biochem. 2019, 120, 18509–18523. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R&D 2015, 15, 13–20. [Google Scholar]
- Harbut, M.B.; Vilchèze, C.; Luo, X.; Hensler, M.E.; Guo, H.; Yang, B.; Chatterjee, A.K.; Nizet, V.; Jacobs, W.R., Jr.; Schultz, P.G.; et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 4453–4458. [Google Scholar] [CrossRef]
- Yamada, R.; Sano, H.; Hla, T.; Hashiramoto, A.; Fukui, W.; Miyazaki, S.; Kohno, M.; Tsubouchi, Y.; Kusaka, Y.; Kondo, M. Auranofin inhibits interleukin-1beta-induced transcript of cyclooxygenase-2 on cultured human synoviocytes. Eur. J. Pharmacol. 1999, 385, 71–79. [Google Scholar] [CrossRef]
- Jeon, K.I.; Jeong, J.Y.; Jue, D.M. Thiol-reactive metal compounds inhibit NF-kappa B activation by blocking I kappa B kinase. J. Immunol. 2000, 164, 5981–5989. [Google Scholar] [CrossRef]
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef]
- Zhang, X.; Xue, C.; Xu, Q.; Zhang, Y.; Li, H.; Li, F.; Liu, Y.; Guo, C. Caprylic acid suppresses inflammation via TLR4/NF-κB signaling and improves atherosclerosis in ApoE-deficient mice. Nutr. Metab. 2019, 16, 40. [Google Scholar] [CrossRef]
- Sun, X.; Hao, H.; Han, Q.; Song, X.; Liu, J.; Dong, L.; Han, W.; Mu, Y. Human umbilical cord-derived mesenchymal stem cells ameliorate insulin resistance by suppressing NLRP3 inflammasome-mediated inflammation in type 2 diabetes rats. Stem Cell Res. Ther. 2017, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, A.; Sagawa, M.; Muto, A.; Uchida, H.; Ikeda, Y.; Kizaki, M. The gold compound auranofin induces apoptosis of human multiple myeloma cells through both down-regulation of STAT3 and inhibition of NF-κB activity. Leuk. Res. 2011, 35, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. 2019, 67, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Isakov, E.; Weisman-Shomer, P.; Benhar, M. Suppression of the pro-inflammatory NLRP3/interleukin-1β pathway in macrophages by the thioredoxin reductase inhibitor auranofin. Biochim. Biophys. Acta 2014, 1840, 3153–3161. [Google Scholar] [CrossRef]
- Park, H.Y.; Kim, G.Y.; Hyun, J.W.; Hwang, H.J.; Kim, N.D.; Kim, B.W.; Choi, Y.H. 7,8-Dihydroxyflavone exhibits anti-inflammatory properties by downregulating the NF-κB and MAPK signaling pathways in lipopolysaccharide-treated RAW264.7 cells. Int. J. Mol. Med. 2012, 29, 1146–1152. [Google Scholar]
- Yamashita, M.; Ashino, S.; Oshima, Y.; Kawamura, S.; Ohuchi, K.; Takayanagi, M. Inhibition of TPA-induced NF-kappaB nuclear translocation and production of NO and PGE2 by the anti-rheumatic gold compounds. J. Pharm. Pharmacol. 2003, 55, 245–251. [Google Scholar] [CrossRef]
- Hu, M.; Zhang, Z.; Liu, B.; Zhang, S.; Chai, R.; Chen, X.; Kong, T.; Zhang, F.; Zhang, J.; Liu, S.; et al. Deubiquitinase Inhibitor Auranofin Attenuated Cardiac Hypertrophy by Blocking NF-κB Activation. Cell Physiol. Biochem. 2018, 45, 2421–2430. [Google Scholar] [CrossRef]
- Yang, G.; Lee, S.J.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Zouboulis, C.C.; Han, S.H.; Ma, K.H.; Jang, J.K.; Lee, J.Y. Repurposing Auranofin, an Anti-Rheumatic Gold Compound, to Treat Acne Vulgaris by Targeting the NLRP3 Inflammasome. Biomol. Ther. 2020, 28, 437–442. [Google Scholar] [CrossRef]
- Meijles, D.N.; Pagano, P.J. Nox and Inflammation in the Vascular Adventitia. Hypertension 2016, 67, 14–19. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Csányi, G.; Taylor, W.R.; Pagano, P.J. NOX and inflammation in the vascular adventitia. Free Radic. Biol. Med. 2009, 47, 1254–1266. [Google Scholar] [CrossRef]
- Ohto, U.; Fukase, K.; Miyake, K.; Shimizu, T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. USA 2012, 109, 7421–7426. [Google Scholar] [CrossRef]
- Gayeon You, G.; Kim, Y.; Lee, J.H.; Song, J.; Mok, H. Exosome-modified PLGA microspheres for improved internalization into dendritic cells and macrophages. Biotechnol. Bioprocess Eng. 2020, 25, 521–527. [Google Scholar]
- Choi, Y.H. Trans-cinnamaldehyde protects C2C12 myoblasts from DNA damage, mitochondrial dysfunction and apoptosis caused by oxidative stress through inhibiting ROS production. Genes Genom. 2021, 43, 303–312. [Google Scholar] [CrossRef]
- Chae, B.S. Effect of low-dose corticosterone pretreatment on the production of inflammatory mediators in super-low-dose LPS-primed immune cells. Toxicol. Res. 2020, 37, 47–57. [Google Scholar] [CrossRef]
- Ji, S.Y.; Cha, H.J.; Molagoda, I.M.N.; Kim, M.Y.; Kim, S.Y.; Hwangbo, H.; Lee, H.; Kim, G.Y.; Kim, D.H.; Hyun, J.W.; et al. Suppression of lipopolysaccharide-induced inflammatory and oxidative response by 5-aminolevulinic acid in RAW 264.7 macrophages and zebrafish larvae. Biomol. Ther. 2021. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, S.J.; Kim, J.E.; Kang, M.J.; Bae, S.J.; Choi, Y.J.; Gong, J.E.; Kim, K.S.; Jung, Y.S.; Cho, J.Y.; et al. Comparison of response to LPS-induced sepsis in three DBA/2 stocks derived from different sources. Lab. Anim. Res. 2021, 37, 2. [Google Scholar] [CrossRef]
- Kim, H.O.; Shin, K.R.; Jang, B.-C.; Kim, Y.C. Action mechanism of anti-wrinkle effect of Rhamnus yoshinoi methanol extract in human dermal fibroblast and keratinocyte cell lines. Toxicol. Res. 2021, 36, 69–77. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Compound | Binding Affinity (kcal/mol) | Binding Site | Distance (Å) |
|---|---|---|---|---|
| Toll-like receptor 4 (PDB ID 3VQ2) | Auranofin (CID 70788951) | –5.6 | N (Arg 434) | 2.9 |
| Gene | Primers | Tm (°C) | |
|---|---|---|---|
| iNOS | F | CGT GTT TAC CAT GAG GCT GA | 55 |
| R | GCT TCA GGT TCC TGA TCC AA | ||
| IL-1 β | F | AAT CTC GCA GCA GCA CAT CA | 55 |
| R | AGC CCA TAC TTT AGG AAG AC | ||
| IL-6 | F | CTG GTG ACA ACC ACG GCC TTC CCT A | 65 |
| R | ATG CTT AGG CAT AAAC GCA CTA CCT T | ||
| GAPDH | F | GAA GAG TGG GAG TTG CTG TT | 55 |
| R | GGA GAA ACC TGC CAA GTA TG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwangbo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Lee, H.; Kim, G.-Y.; Kim, S.; Cheong, J.; Choi, Y.H. Anti-Inflammatory Effect of Auranofin on Palmitic Acid and LPS-Induced Inflammatory Response by Modulating TLR4 and NOX4-Mediated NF-κB Signaling Pathway in RAW264.7 Macrophages. Int. J. Mol. Sci. 2021, 22, 5920. https://doi.org/10.3390/ijms22115920
Hwangbo H, Ji SY, Kim MY, Kim SY, Lee H, Kim G-Y, Kim S, Cheong J, Choi YH. Anti-Inflammatory Effect of Auranofin on Palmitic Acid and LPS-Induced Inflammatory Response by Modulating TLR4 and NOX4-Mediated NF-κB Signaling Pathway in RAW264.7 Macrophages. International Journal of Molecular Sciences. 2021; 22(11):5920. https://doi.org/10.3390/ijms22115920
Chicago/Turabian StyleHwangbo, Hyun, Seon Yeong Ji, Min Yeong Kim, So Young Kim, Hyesook Lee, Gi-Young Kim, Suhkmann Kim, JaeHun Cheong, and Yung Hyun Choi. 2021. "Anti-Inflammatory Effect of Auranofin on Palmitic Acid and LPS-Induced Inflammatory Response by Modulating TLR4 and NOX4-Mediated NF-κB Signaling Pathway in RAW264.7 Macrophages" International Journal of Molecular Sciences 22, no. 11: 5920. https://doi.org/10.3390/ijms22115920
APA StyleHwangbo, H., Ji, S. Y., Kim, M. Y., Kim, S. Y., Lee, H., Kim, G.-Y., Kim, S., Cheong, J., & Choi, Y. H. (2021). Anti-Inflammatory Effect of Auranofin on Palmitic Acid and LPS-Induced Inflammatory Response by Modulating TLR4 and NOX4-Mediated NF-κB Signaling Pathway in RAW264.7 Macrophages. International Journal of Molecular Sciences, 22(11), 5920. https://doi.org/10.3390/ijms22115920