
Serotonin Promotes Serum Albumin Interaction with the Monomeric Amyloid β Peptide

 ,
,  and
and 
Abstract
1. Introduction
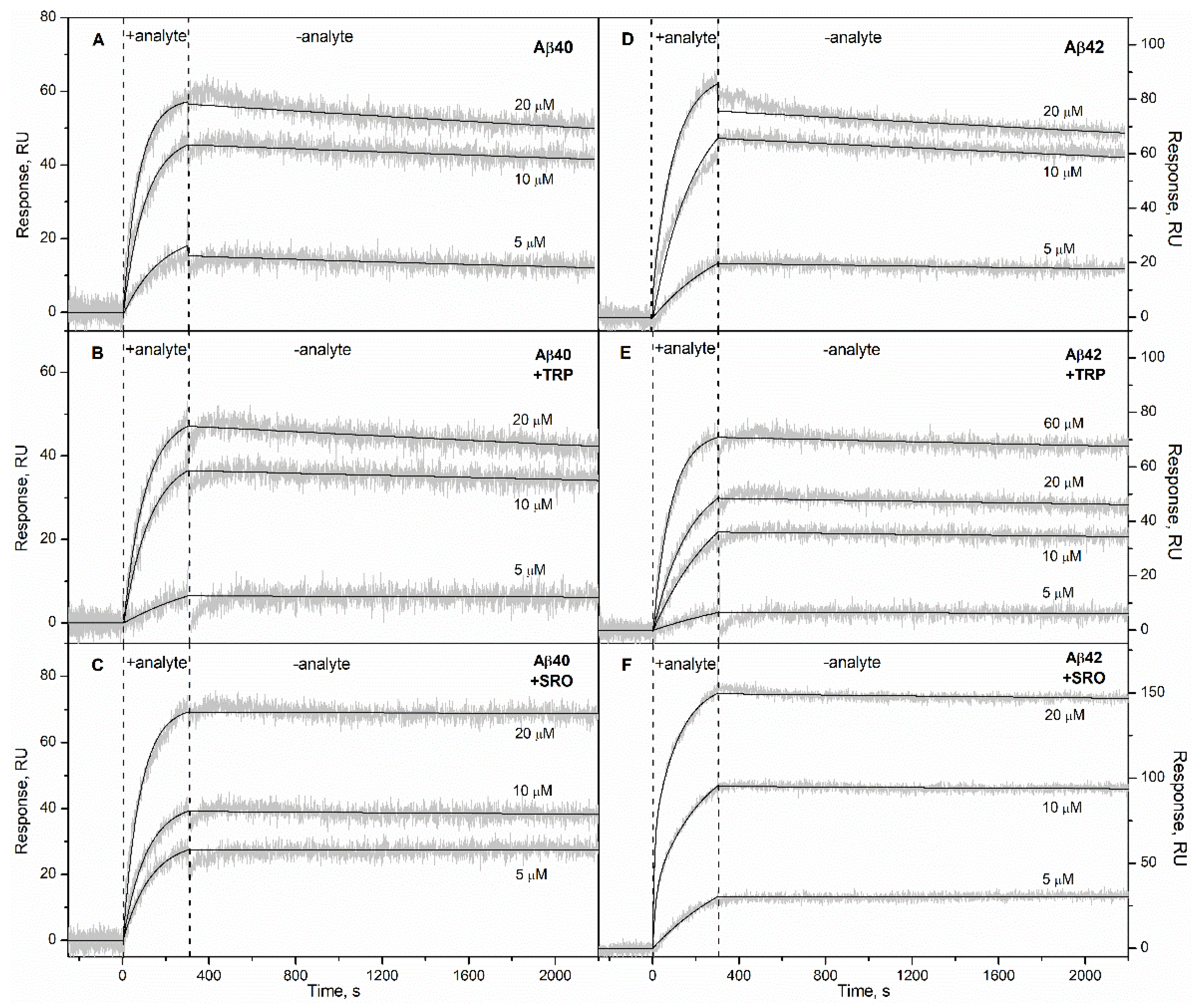
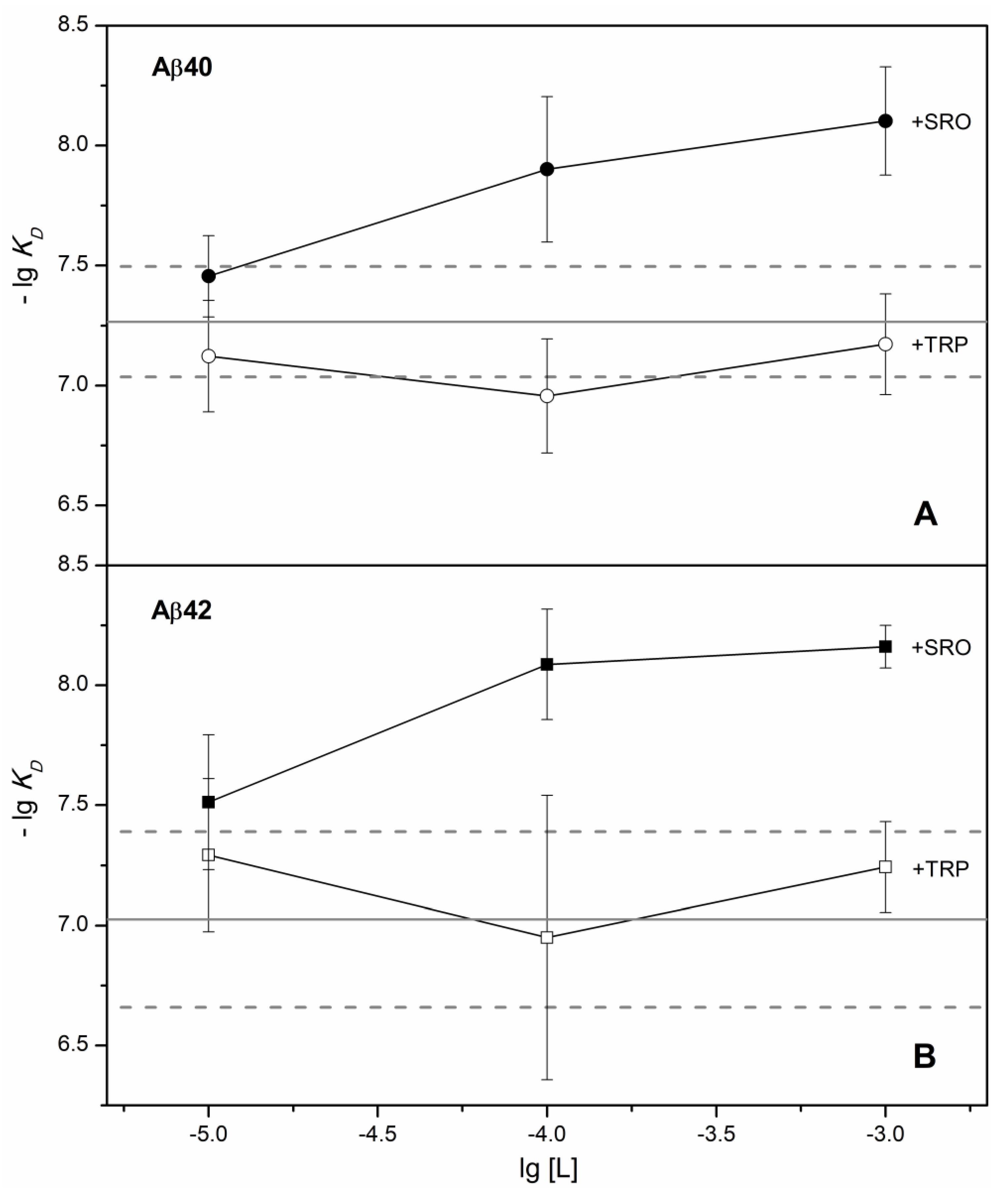
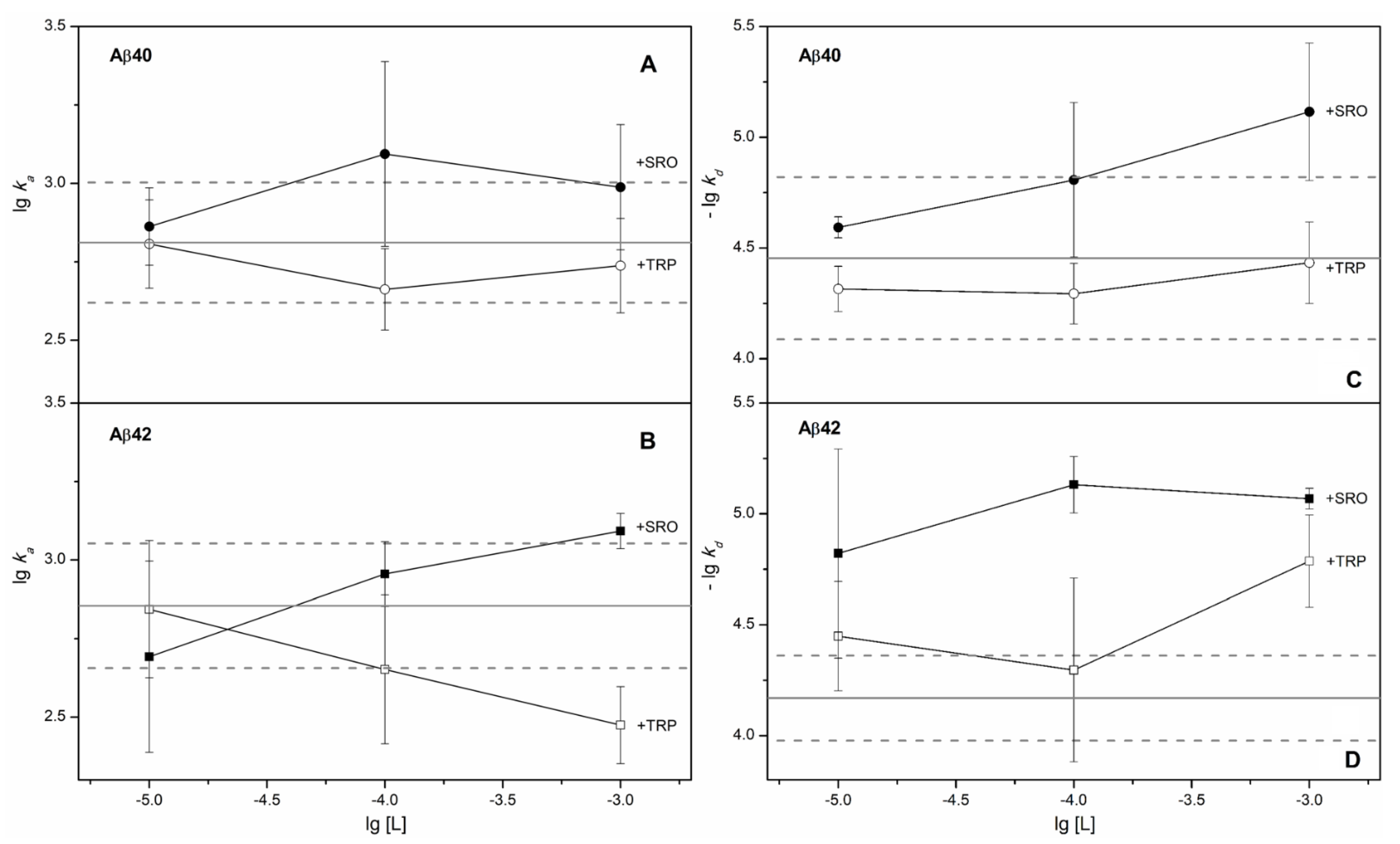
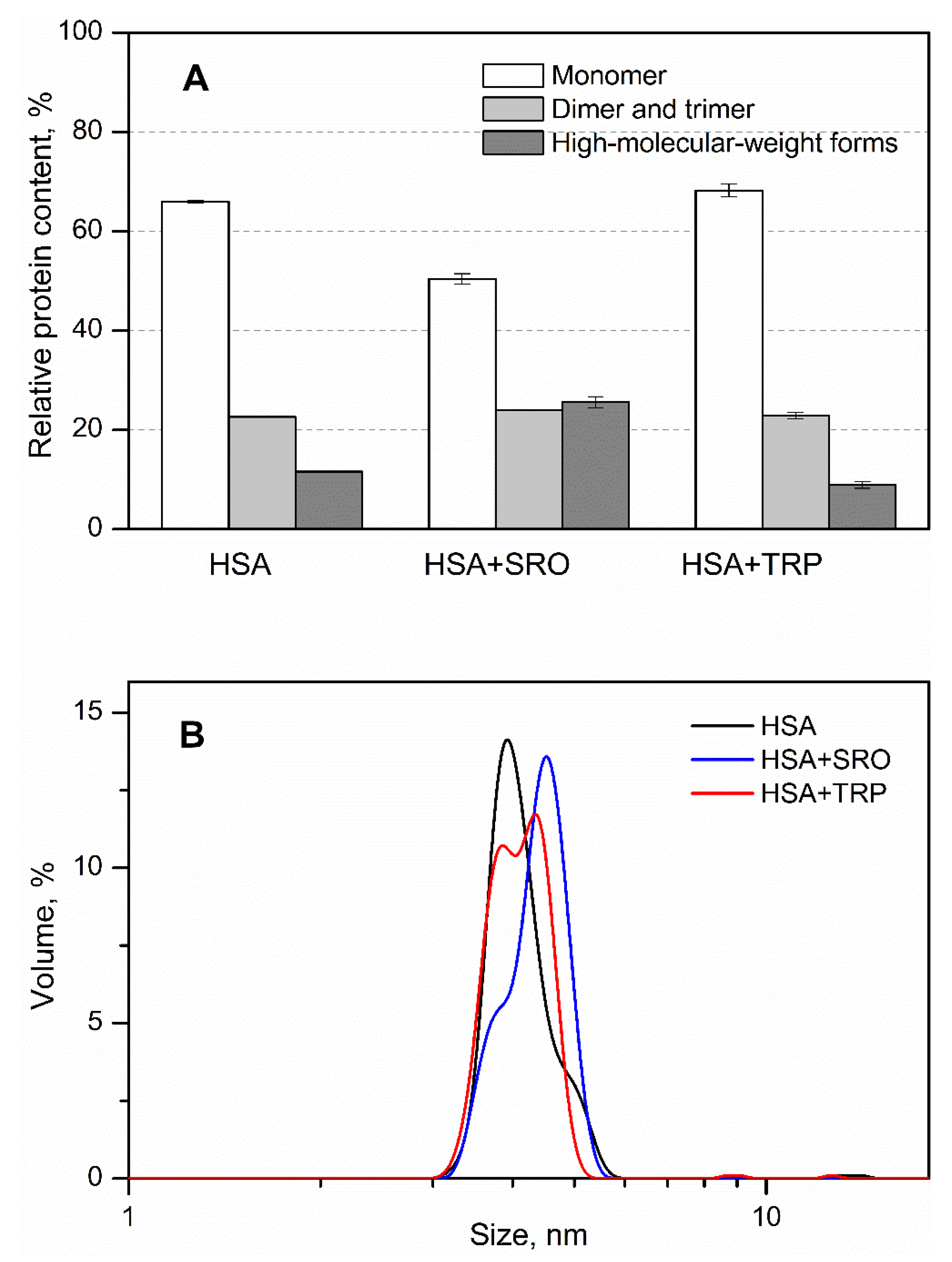
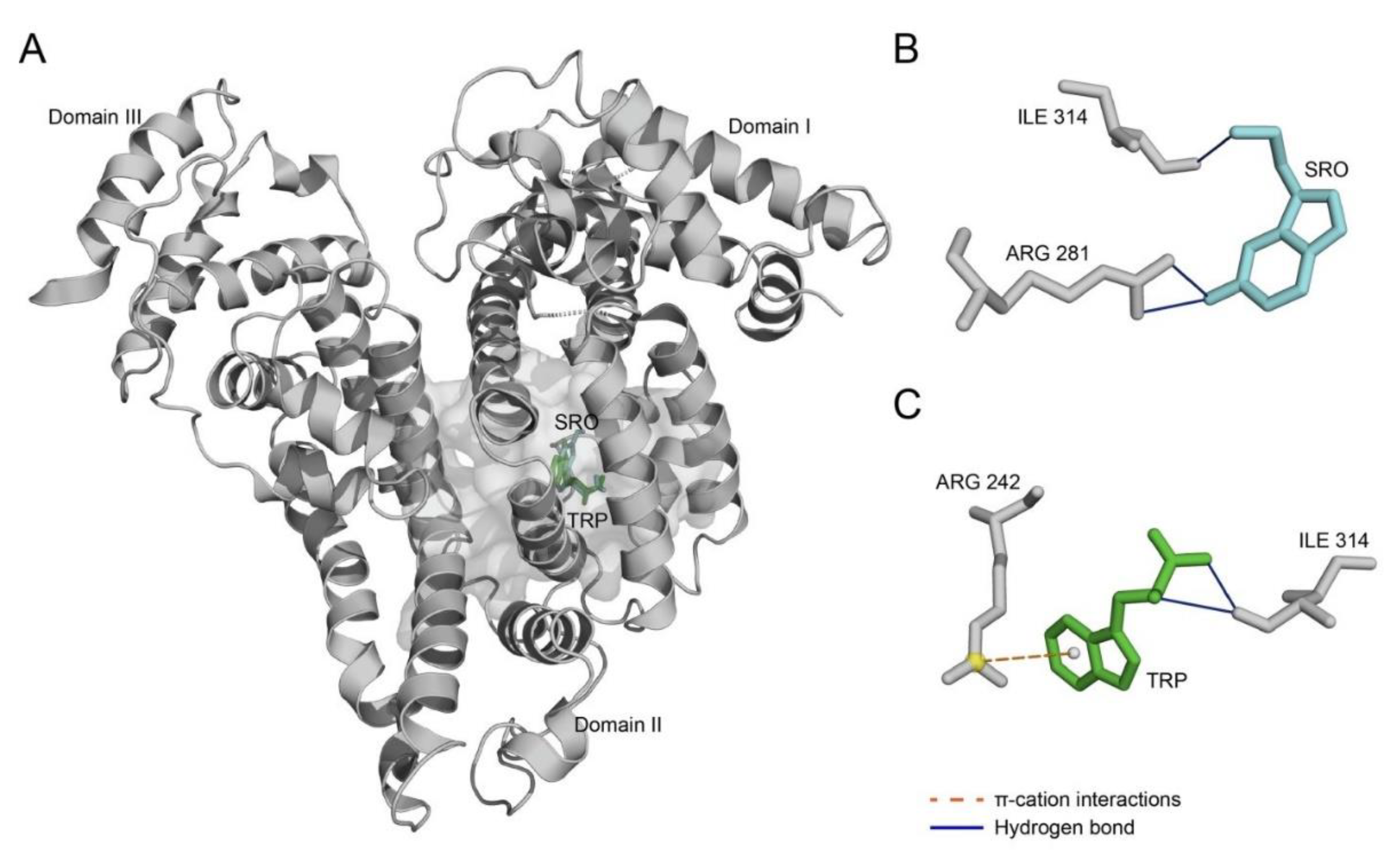
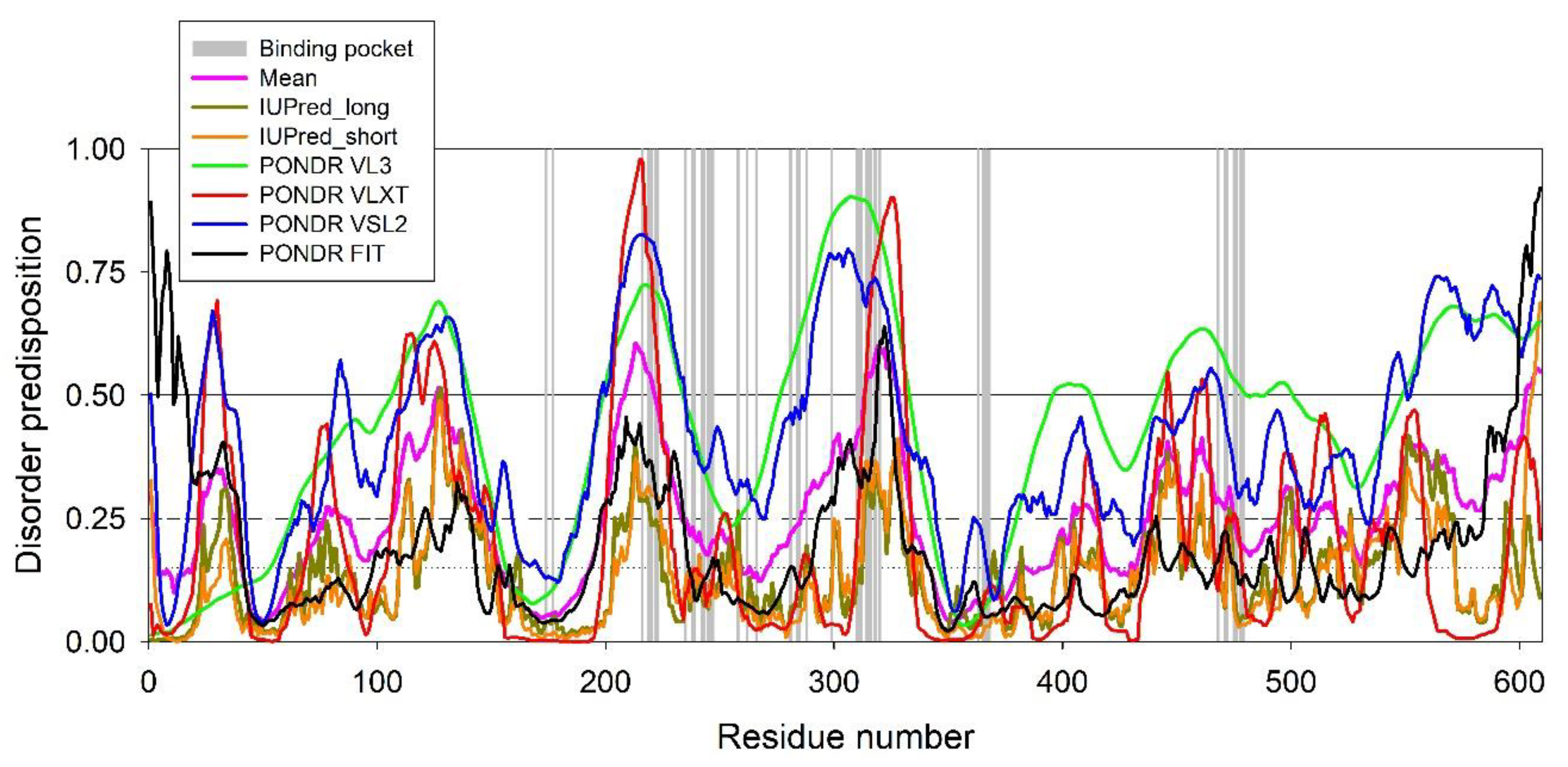
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Expression and Purification of Recombinant Human Aβ40
4.3. Pretreatment of Aβ Samples for SPR Experiments
4.4. Surface Plasmon Resonance Studies
4.5. Chemical Crosslinking of Proteins
4.6. Dynamic Light Scattering Studies
4.7. Prediction of Ligand-Binding Sites in HSA
4.8. Evaluation of the Per-Residue Intrinsic Disorder Predisposition of HSA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-ME | 2-mercaptoethanol |
| Aβ | amyloid β peptide |
| Aβ40/Aβ42/Aβ20 | amyloid β peptide, residues 1-40/42/20 |
| AD | Alzheimer’s disease |
| CSF | cerebrospinal fluid |
| DLS | dynamic light scattering |
| DMSO | dimethyl sulfoxide |
| DS | disorder score |
| EDTA | ethylenediaminetetraacetic acid |
| HEPES | 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid |
| HSA | human serum albumin |
| PAGE | polyacrylamide gel electrophoresis |
| PDB | Protein Data Bank |
| RU | resonance unit |
| SDS | sodium dodecyl sulfate |
| SPR | surface plasmon resonance |
| SRO | serotonin, 5-hydroxytryptamine, 5-HT |
| SSRI | selective serotonin reuptake inhibitor |
| TFA | trifluoroacetic acid |
| Tris | tris(hydroxymethyl)aminomethane |
| TRP | L-tryptophan |
| Usp2-cc | ubiquitin carboxyl-terminal hydrolase 2, catalytic core |
References
- Litus, E.A.; Permyakov, S.E.; Uversky, V.N.; Permyakov, E.A. Intrinsically disordered regions in serum albumin: What are they for? Cell Biochem. Biophys. 2017, 76, 39–57. [Google Scholar] [CrossRef]
- Al-Harthi, S.; Lachowicz, J.I.; Nowakowski, M.E.; Jaremko, M.; Jaremko, L. Towards the functional high-resolution coordination chemistry of blood plasma human serum albumin. J. Inorg. Biochem. 2019, 198, 110716. [Google Scholar] [CrossRef]
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Biere, A.L.; Ostaszewski, B.; Stimson, E.R.; Hyman, B.T.; Maggio, J.E.; Selkoe, D.J. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J. Biol. Chem. 1996, 271, 32916–32922. [Google Scholar] [CrossRef]
- Bohrmann, B.; Tjernberg, L.; Kuner, P.; Poli, S.; Levet-Trafit, B.; Naslund, J.; Richards, G.; Huber, W.; Dobeli, H.; Nordstedt, C. Endogenous proteins controlling amyloid beta-peptide polymerization. Possible implications for beta-amyloid formation in the central nervous system and in peripheral tissues. J. Biol. Chem. 1999, 274, 15990–15995. [Google Scholar] [CrossRef] [PubMed]
- Stanyon, H.F.; Viles, J.H. Human serum albumin can regulate amyloid-β peptide fiber growth in the brain interstitium: Implications for Alzheimer disease. J. Biol. Chem. 2012, 287, 28163–28168. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, F.; Xu, L.; Jia, L. HSA targets multiple Aβ42 species and inhibits the seeding-mediated aggregation and cytotoxicity of Aβ42 aggregates. RSC Adv. 2016, 6, 71165–71175. [Google Scholar] [CrossRef]
- Ezra, A.; Rabinovich-Nikitin, I.; Rabinovich-Toidman, P.; Solomon, B. Chapter 11—Multifunctional effects of human serum albumin toward neuroprotection in Alzheimer disease. In Neuroprotection in Alzheimer’s Disease; Gozes, I., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 217–238. [Google Scholar]
- Boada, M.; Ortiz, P.; Anaya, F.; Hernandez, I.; Munoz, J.; Nunez, L.; Olazaran, J.; Roca, I.; Cuberas, G.; Tarraga, L.; et al. Amyloid-targeted therapeutics in Alzheimer’s disease: Use of human albumin in plasma exchange as a novel approach for Abeta mobilization. Drug News Perspect. 2009, 22, 325–339. [Google Scholar] [CrossRef]
- Boada, M.; Anaya, F.; Ortiz, P.; Olazaran, J.; Shua-Haim, J.R.; Obisesan, T.O.; Hernandez, I.; Munoz, J.; Buendia, M.; Alegret, M.; et al. Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid-beta concentrations and cognition outcomes in Alzheimer’s disease patients: A multicenter, randomized, controlled clinical trial. J. Alzheimers Dis. 2017, 56, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Boada, M.; Lopez, O.; Nunez, L.; Szczepiorkowski, Z.M.; Torres, M.; Grifols, C.; Paez, A. Plasma exchange for Alzheimer’s disease management by albumin replacement (AMBAR) trial: Study design and progress. Alzheimer’s Dement. 2019, 5, 61–69. [Google Scholar] [CrossRef]
- Litus, E.A.; Kazakov, A.S.; Sokolov, A.S.; Nemashkalova, E.L.; Galushko, E.I.; Dzhus, U.F.; Marchenkov, V.V.; Galzitskaya, O.V.; Permyakov, E.A.; Permyakov, S.E. The binding of monomeric amyloid β peptide to serum albumin is affected by major plasma unsaturated fatty acids. Biochem. Biophys. Res. Commun. 2019, 510, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.M.; Ma, Q.-L.; Teter, B.; Jones, M.; Frautschy, S.A. [P3–124]: Dietary linoleic acid differentially influences brain fads activities increasing an N-6 metabolite that inhibits inflammation and promotes amyloid-β clearance. Alzheimer’s Dement. 2017, 13, P982. [Google Scholar] [CrossRef]
- Bode, D.C.; Stanyon, H.F.; Hirani, T.; Baker, M.D.; Nield, J.; Viles, J.H. Serum albumin’s protective inhibition of amyloid-β fiber formation is suppressed by cholesterol, fatty acids and warfarin. J. Mol. Biol. 2018, 430, 919–934. [Google Scholar] [CrossRef]
- Smith, G.S.; Barrett, F.S.; Joo, J.H.; Nassery, N.; Savonenko, A.; Sodums, D.J.; Marano, C.M.; Munro, C.A.; Brandt, J.; Kraut, M.A.; et al. Molecular imaging of serotonin degeneration in mild cognitive impairment. Neurobiol. Dis. 2017, 105, 33–41. [Google Scholar] [CrossRef]
- Whiley, L.; Chappell, K.E.; D’Hondt, E.; Lewis, M.R.; Jiménez, B.; Snowden, S.G.; Soininen, H.; Kłoszewska, I.; Mecocci, P.; Tsolaki, M.; et al. Metabolic phenotyping reveals a reduction in the bioavailability of serotonin and kynurenine pathway metabolites in both the urine and serum of individuals living with Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Yabut, J.M.; Crane, J.D.; Green, A.E.; Keating, D.J.; Khan, W.I.; Steinberg, G.R. Emerging roles for serotonin in regulating metabolism: New implications for an ancient molecule. Endocr. Rev. 2019, 40, 1092–1107. [Google Scholar] [CrossRef]
- Ferrero, H.; Solas, M.; Francis, P.T.; Ramirez, M.J. Serotonin 5-HT(6) receptor antagonists in Alzheimer’s disease: Therapeutic rationale and current development status. CNS Drugs 2017, 31, 19–32. [Google Scholar] [CrossRef]
- Travis, J.; Pannell, R. Selective removal of albumin from plasma by affinity chromatography. Clin. Chim. Acta 1973, 49, 49–52. [Google Scholar] [CrossRef]
- Yang, J.; Hage, D.S. Characterization of the binding and chiral separation of D- and L-tryptophan on a high-performance immobilized human serum albumin column. J. Chromatogr. 1993, 645, 241–250. [Google Scholar] [CrossRef]
- Sengupta, B.; Chaudhuri, S.; Banerjee, A.; Sengupta, P.K. Characterization of serotonin in protein and membrane mimetic environments: A spectroscopic study. Chem. Biodivers. 2004, 1, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Hornedo-Ortega, R.; da Costa, G.; Cerezo, A.B.; Troncoso, A.M.; Richard, T.; Garcia-Parrilla, M.C. In vitro effects of serotonin, melatonin, and other related indole compounds on amyloid-β kinetics and neuroprotection. Mol. Nutr. Food Res. 2018, 62, 1700383. [Google Scholar] [CrossRef] [PubMed]
- Nemashkalova, E.L.; Permyakov, E.A.; Permyakov, S.E.; Litus, E.A. Modulation of linoleic acid-binding properties of human serum albumin by divalent metal cations. Biometals 2017, 30, 341–353. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Modeling 2012, 52, 360–372. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Permyakov, S.E.; Ismailov, R.G.; Xue, B.; Denesyuk, A.I.; Uversky, V.N.; Permyakov, E.A. Intrinsic disorder in S100 proteins. Mol. Biosyst. 2011, 7, 2164–2180. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef]
- Milojevic, J.; Costa, M.; Ortiz, A.M.; Jorquera, J.I.; Melacini, G. In vitro amyloid-β binding and inhibition of amyloid-β self-association by therapeutic albumin. J. Alzheimer’s Dis. 2014, 38, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Milojevic, J.; Raditsis, A.; Melacini, G. Human serum albumin inhibits Abeta fibrillization through a “monomer-competitor” mechanism. Biophys. J. 2009, 97, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Rózga, M.; Kłoniecki, M.; Jabłonowska, A.; Dadlez, M.; Bal, W. The binding constant for amyloid Aβ40 peptide interaction with human serum albumin. Biochem. Biophys. Res. Commun. 2007, 364, 714–718. [Google Scholar] [CrossRef]
- Costa, M.; Ortiz, A.M.; Jorquera, J.I. Therapeutic albumin binding to remove amyloid-β. J. Alzheimer’s Dis. 2012, 29, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Nazarali, A.J.; Reynolds, G.P. Monoamine neurotransmitters and their metabolites in brain regions in Alzheimer’s disease: A postmortem study. Cell. Mol. Neurobiol. 1992, 12, 581–587. [Google Scholar] [CrossRef]
- Tietz, N.W.; Finley, P.R.; Pruden, E.; Amerson, A.B. Clinical Guide to Laboratory Tests, 2nd ed.; Saunders: Philadelphia, PA, USA, 1990; pp. 1–931. [Google Scholar]
- Bunin, M.A.; Wightman, R.M. Quantitative evaluation of 5-hydroxytryptamine (serotonin) neuronal release and uptake: An investigation of extrasynaptic transmission. J. Neurosci. 1998, 18, 4854–4860. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Disabato, B.M.; Restivo, J.L.; Verges, D.K.; Goebel, W.D.; Sathyan, A.; Hayreh, D.; D’Angelo, G.; Benzinger, T.; Yoon, H.; et al. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 14968–14973. [Google Scholar] [CrossRef]
- Vijayan, D.; Chandra, R. Amyloid beta hypothesis in Alzheimer’s disease: Major culprits and recent therapeutic strategies. Curr. Drug Targets 2020, 21, 148–166. [Google Scholar] [CrossRef] [PubMed]
- Catanzariti, A.-M.; Soboleva, T.A.; Jans, D.A.; Board, P.G.; Baker, R.T. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 2004, 13, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [PubMed]
- Zagorski, M.G.; Yang, J.; Shao, H.; Ma, K.; Zeng, H.; Hong, A. [13] Methodological and chemical factors affecting amyloid β peptide amyloidogenicity. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1999; Volume 309, pp. 189–204. [Google Scholar]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallographica 2002, 58, 899–907. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSA Ligand | [HSA Ligand] | Aβ40 | Aβ42 | ||||
|---|---|---|---|---|---|---|---|
| ka × 10−3, M−1s−1 | kd × 104, s−1 | KD × 107, M | ka × 10−3, M−1s−1 | kd × 104, s−1 | KD × 107, M | ||
| - | - | 0.7 ± 0.3 | 0.4 ± 0.3 | 0.6 ± 0.3 | 0.8 ± 0.3 | 0.7 ± 0.3 | 1.2 ± 0.7 |
| SRO | 10 µM | 0.7 ± 0.2 | 0.26 ± 0.03 | 0.4 ± 0.2 | 0.6 ± 0.4 | 0.20 ± 0.14 | 0.4 ± 0.2 |
| 100 µM | 1.5 ± 1.2 | 0.19 ± 0.12 | 0.15 ± 0.13 | 0.9 ± 0.2 | 0.08 ± 0.02 | 0.09 ± 0.04 | |
| 1 mM | 1.0 ± 0.5 | 0.09 ± 0.05 | 0.09 ± 0.05 | 1.2 ± 0.2 | 0.086 ± 0.009 | 0.070 ± 0.015 | |
| TRP | 10 µM | 0.7 ± 0.2 | 0.49 ± 0.12 | 0.8 ± 0.4 | 0.8 ± 0.4 | 0.4 ± 0.2 | 0.6 ± 0.4 |
| 100 µM | 0.47 ± 0.15 | 0.53 ± 0.17 | 1.2 ± 0.6 | 0.5 ± 0.3 | 0.7 ± 0.6 | 1.8 ± 1.8 | |
| 1 mM | 0.6 ± 0.2 | 0.4 ± 0.2 | 0.7 ± 0.4 | 0.29 ± 0.07 | 0.18 ± 0.07 | 0.6 ± 0.3 | |
| Ligand | Index | Residue # | Residue | Distance, Å |
|---|---|---|---|---|
| SRO | Hydrogen bonds | |||
| 1 | 281 | Arg | 3.16 | |
| 2 | 281 | Arg | 3.18 | |
| 3 | 314 | Ile | 2.22 | |
| TRP | Hydrogen bonds | |||
| 1 | 314 | Ile | 2.99 | |
| 2 | 314 | Ile | 3.21 | |
| π–cation interactions | ||||
| 1 | 242 | Arg | 5.57 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litus, E.A.; Kazakov, A.S.; Deryusheva, E.I.; Nemashkalova, E.L.; Shevelyova, M.P.; Nazipova, A.A.; Permyakova, M.E.; Raznikova, E.V.; Uversky, V.N.; Permyakov, S.E. Serotonin Promotes Serum Albumin Interaction with the Monomeric Amyloid β Peptide. Int. J. Mol. Sci. 2021, 22, 5896. https://doi.org/10.3390/ijms22115896
Litus EA, Kazakov AS, Deryusheva EI, Nemashkalova EL, Shevelyova MP, Nazipova AA, Permyakova ME, Raznikova EV, Uversky VN, Permyakov SE. Serotonin Promotes Serum Albumin Interaction with the Monomeric Amyloid β Peptide. International Journal of Molecular Sciences. 2021; 22(11):5896. https://doi.org/10.3390/ijms22115896
Chicago/Turabian StyleLitus, Ekaterina A., Alexey S. Kazakov, Evgenia I. Deryusheva, Ekaterina L. Nemashkalova, Marina P. Shevelyova, Aliya A. Nazipova, Maria E. Permyakova, Elena V. Raznikova, Vladimir N. Uversky, and Sergei E. Permyakov. 2021. "Serotonin Promotes Serum Albumin Interaction with the Monomeric Amyloid β Peptide" International Journal of Molecular Sciences 22, no. 11: 5896. https://doi.org/10.3390/ijms22115896
APA StyleLitus, E. A., Kazakov, A. S., Deryusheva, E. I., Nemashkalova, E. L., Shevelyova, M. P., Nazipova, A. A., Permyakova, M. E., Raznikova, E. V., Uversky, V. N., & Permyakov, S. E. (2021). Serotonin Promotes Serum Albumin Interaction with the Monomeric Amyloid β Peptide. International Journal of Molecular Sciences, 22(11), 5896. https://doi.org/10.3390/ijms22115896