
Integration of the Microbiome, Metabolome and Transcriptomics Data Identified Novel Metabolic Pathway Regulation in Colorectal Cancer


 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results
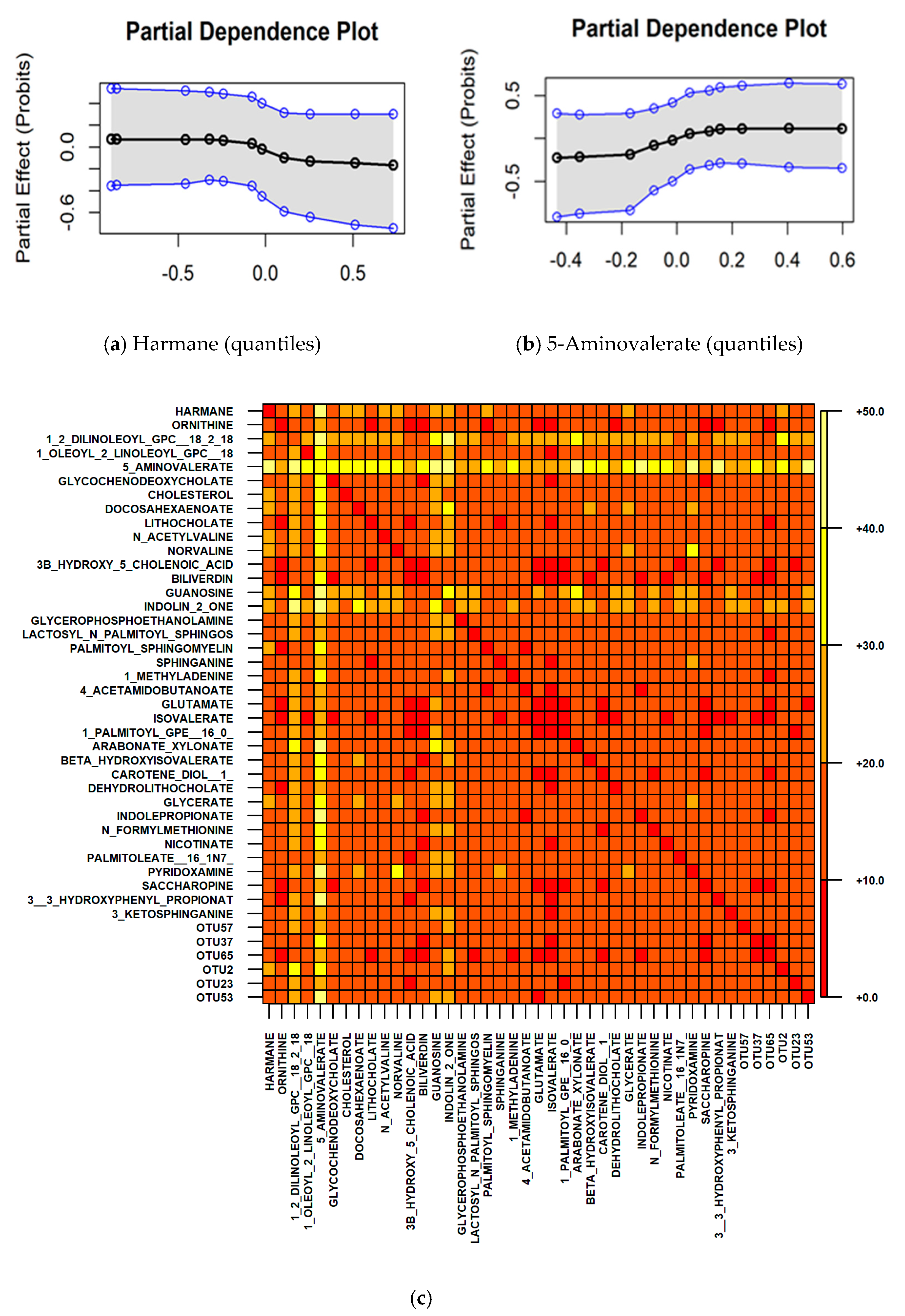
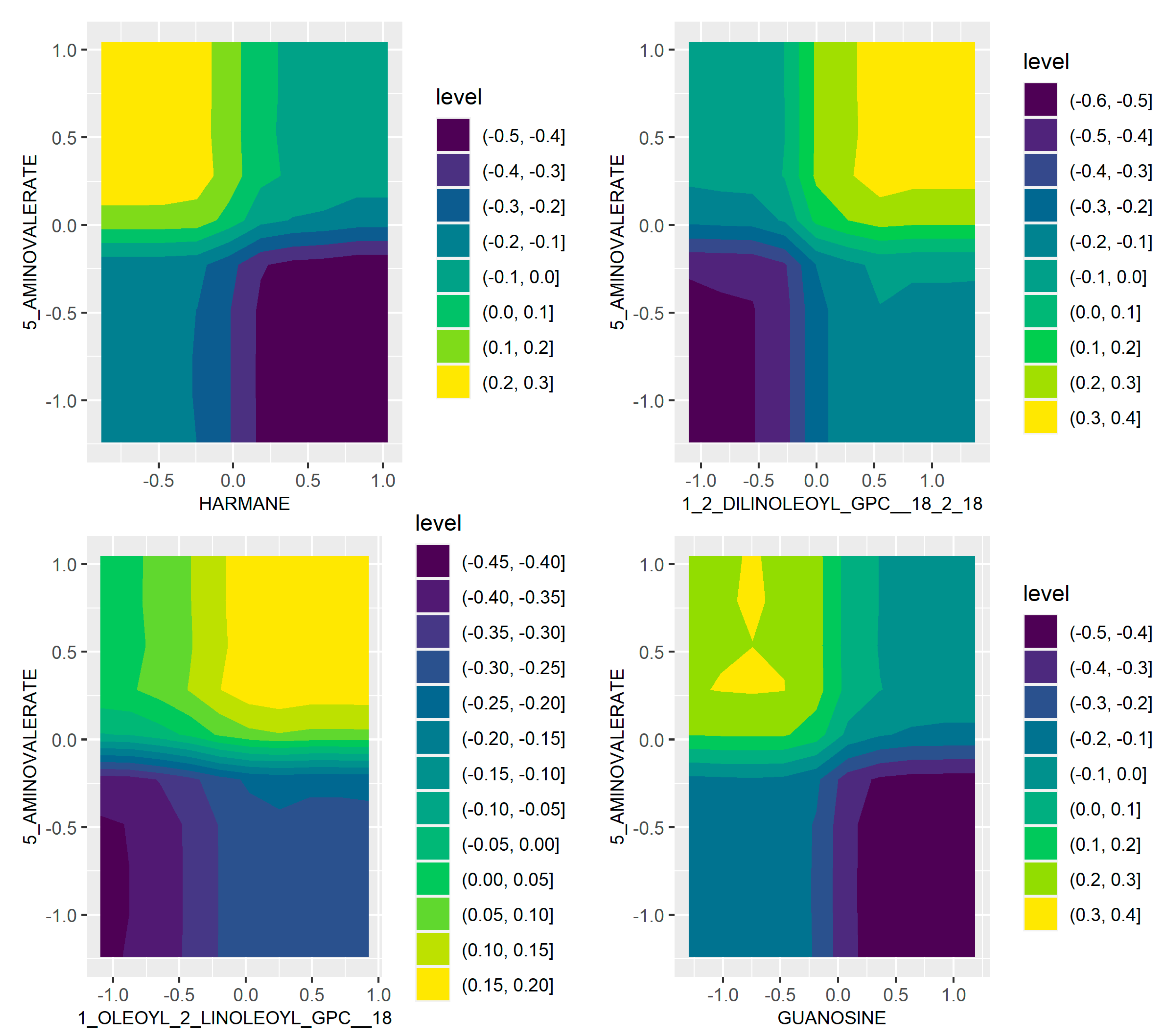
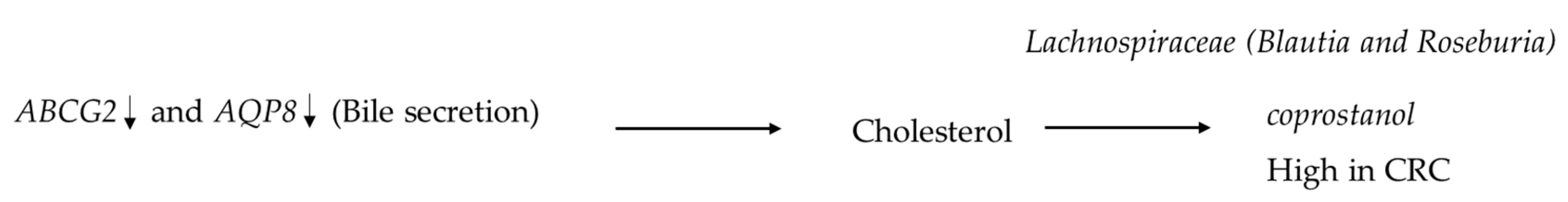
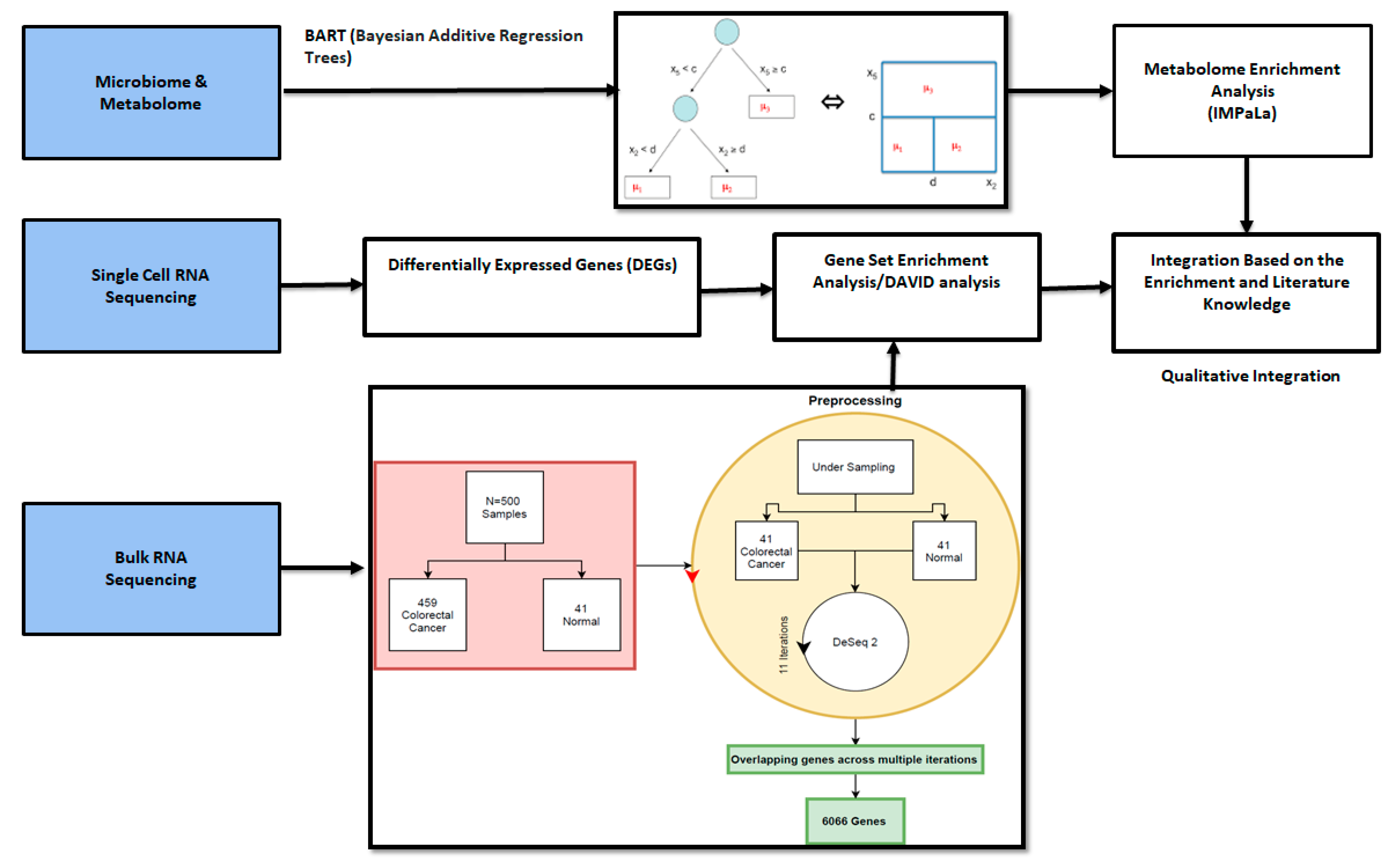
2.1. Microbiome and Metabolome Analysis Identified Novel Interactions
2.2. Metabolite Enrichment Analysis
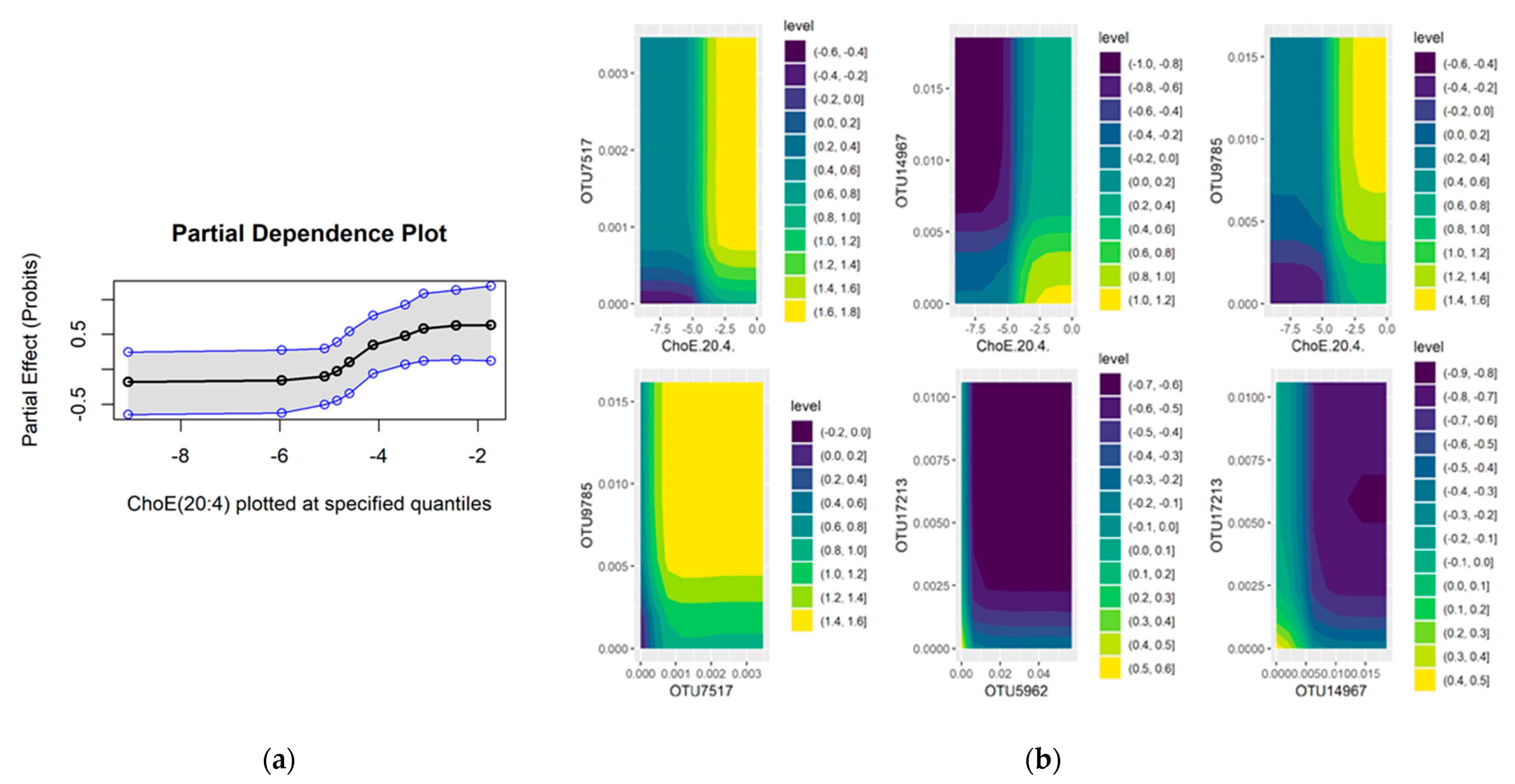
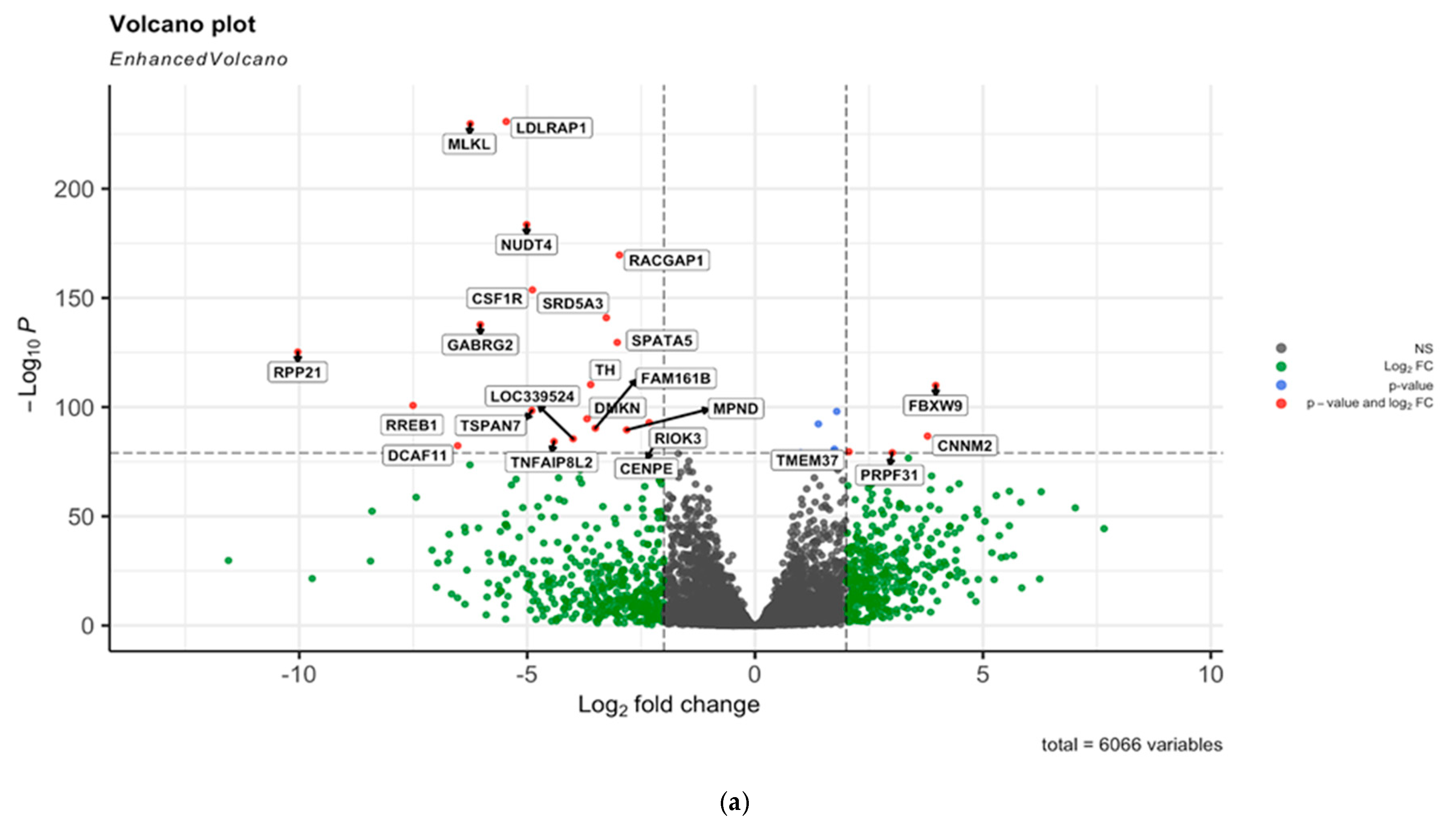
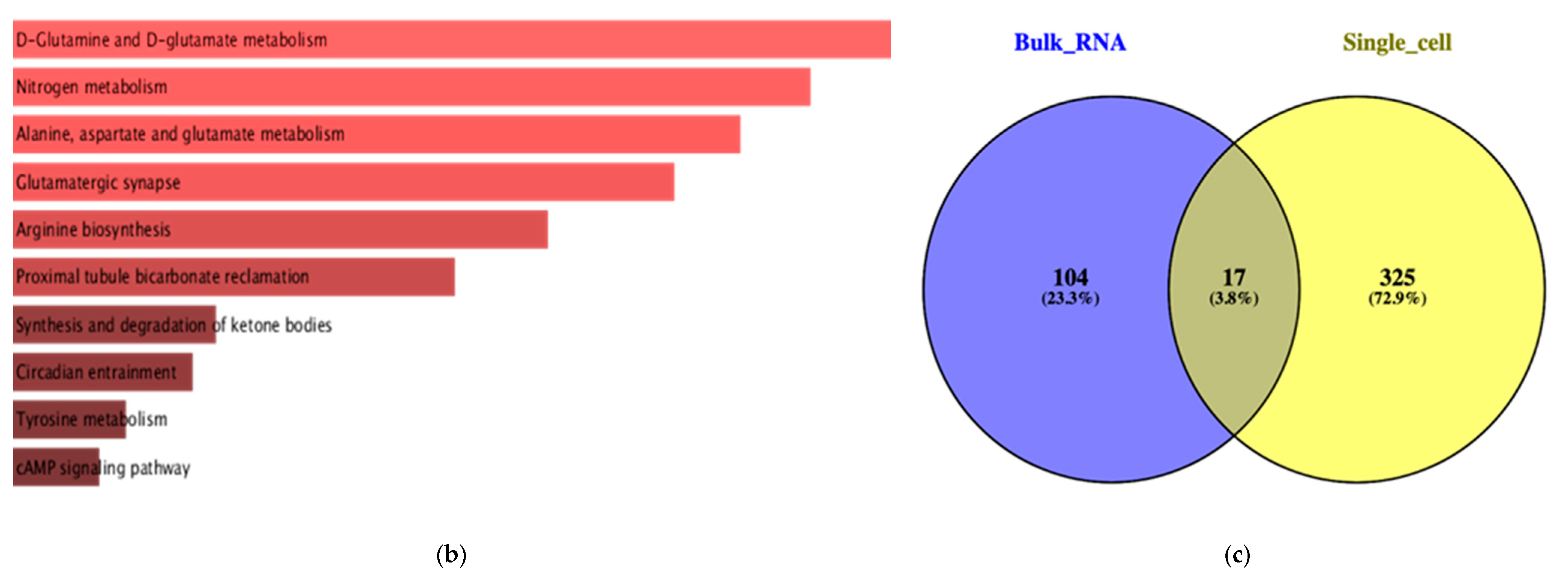
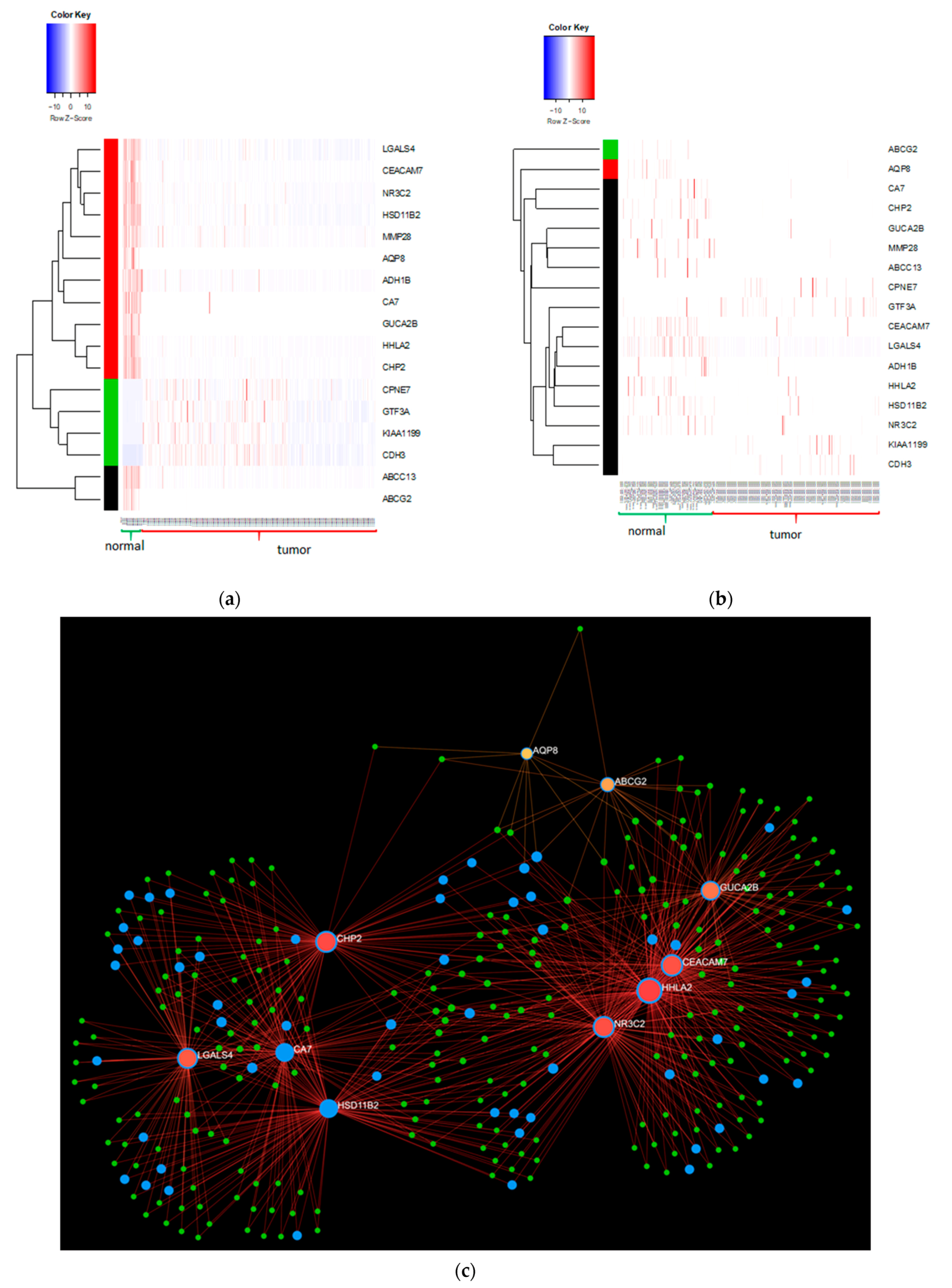
2.3. Bulk RNA Seq Analysis
2.4. Single Cell RNA Seq Analysis
2.5. Qualitative Integration
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of Colorectal Cancer: Incidence, Mortality, Survival, and Risk Factors. Przeglad Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Winawer, S. Colorectal Cancer Screening and Surveillance: Clinical Guidelines and Rationale?Update Based on New Evidence. Gastroenterology 2003, 124, 544–560. [Google Scholar] [CrossRef]
- Keku, T.O.; Dulal, S.; Deveaux, A.; Jovov, B.; Han, X. The Gastrointestinal Microbiota and Colorectal Cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G351–G363. [Google Scholar] [CrossRef]
- Ternes, D.; Karta, J.; Tsenkova, M.; Wilmes, P.; Haan, S.; Letellier, E. Microbiome in Colorectal Cancer: How to Get from Meta-Omics to Mechanism? Trends Microbiol. 2020, 28, 401–423. [Google Scholar] [CrossRef]
- Yang, Y.; Misra, B.B.; Liang, L.; Bi, D.; Weng, W.; Wu, W.; Cai, S.; Qin, H.; Goel, A.; Li, X.; et al. Integrated Microbiome and Metabolome Analysis Reveals a Novel Interplay between Commensal Bacteria and Metabolites in Colorectal Cancer. Theranostics 2019, 9, 4101–4114. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Nookaew, I.; Sommer, N.; Fogelstrand, P.; Bäckhed, F. Site-Specific Programming of the Host Epithelial Transcriptome by the Gut Microbiota. Genome Biol. 2015, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ye, J.; Fang, D.; Lv, L.; Wu, W.; Shi, D.; Li, Y.; Yang, L.; Bian, X.; Wu, J.; et al. Multi-Omic Profiling Reveals Associations between the Gut Mucosal Microbiome, the Metabolome, and Host DNA Methylation Associated Gene Expression in Patients with Colorectal Cancer. BMC Microbiol. 2020, 20, 83. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Vogtmann, E.; Ahlquist, D.A.; Devens, M.E.; Kisiel, J.B.; Taylor, W.R.; White, B.A.; Hale, V.L.; Sung, J.; Chia, N.; et al. Fecal Metabolomic Signatures in Colorectal Adenoma Patients Are Associated with Gut Microbiota and Early Events of Colorectal Cancer Pathogenesis. mBio 2020, 11, e03186-19. [Google Scholar] [CrossRef]
- Tong, D.; Tian, Y.; Zhou, T.; Ye, Q.; Li, J.; Ding, K.; Li, J. Improving Prediction Performance of Colon Cancer Prognosis Based on the Integration of Clinical and Multi-Omics Data. BMC Med. Inform. Decis. Mak. 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Yu, J. Gut Microbiota in Colorectal Cancer: Mechanisms of Action and Clinical Applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef] [PubMed]
- Clos-Garcia, M.; Garcia, K.; Alonso, C.; Iruarrizaga-Lejarreta, M.; D’Amato, M.; Crespo, A.; Iglesias, A.; Cubiella, J.; Bujanda, L.; Falcón-Pérez, J.M. Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer. Cancers 2020, 12, 1142. [Google Scholar] [CrossRef] [PubMed]
- Integrated Pathway-Level Analysis of Transcriptomics and Metabolomics Data with IMPaLA | Bioinformatics | Oxford Academic. Available online: https://academic.oup.com/bioinformatics/article/27/20/2917/202905 (accessed on 13 January 2021).
- Consensus-Phenotype Integration of Transcriptomic and Metabolomic Data Implies a Role for Metabolism in the Chemosensitivity of Tumour Cells. Available online: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1001113 (accessed on 13 January 2021).
- GDC Data Portal Exploration. Available online: https://portal.gdc.cancer.gov/ (accessed on 13 January 2021).
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification Using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference Component Analysis of Single-Cell Transcriptomes Elucidates Cellular Heterogeneity in Human Colorectal Tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Zhang, G.-L.; Pan, L.-L.; Huang, T.; Wang, J.-H. The Transcriptome Difference between Colorectal Tumor and Normal Tissues Revealed by Single-Cell Sequencing. J. Cancer 2019, 10, 5883–5890. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A Visual Analytics Platform for Comprehensive Gene Expression Profiling and Meta-Analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed]
- Crous-Bou, M.; Rennert, G.; Cuadras, D.; Salazar, R.; Cordero, D.; Saltz Rennert, H.; Lejbkowicz, F.; Kopelovich, L.; Monroe Lipkin, S.; Bernard Gruber, S.; et al. Polymorphisms in Alcohol Metabolism Genes ADH1B and ALDH2, Alcohol Consumption and Colorectal Cancer. PLoS ONE 2013, 8, e80158. [Google Scholar] [CrossRef] [PubMed]
- Sabates-Bellver, J.; Van der Flier, L.G.; de Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome Profile of Human Colorectal Adenomas. Mol. Cancer Res. MCR 2007, 5, 1263–1275. [Google Scholar] [CrossRef]
- Evensen, N.A.; Li, Y.; Kuscu, C.; Liu, J.; Cathcart, J.; Banach, A.; Zhang, Q.; Li, E.; Joshi, S.; Yang, J.; et al. Hypoxia Promotes Colon Cancer Dissemination through Up-Regulation of Cell Migration-Inducing Protein (CEMIP). Oncotarget 2015, 6, 20723–20739. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, L.C.; Pedersen, S.K.; Dunne, R.; Brown, G.S.; Pimlott, L.; Gaur, S.; McEvoy, A.; Thomas, M.; Wattchow, D.; Molloy, P.L.; et al. Discovery and Validation of Molecular Biomarkers for Colorectal Adenomas and Cancer with Application to Blood Testing. PLoS ONE 2012, 7, e29059. [Google Scholar] [CrossRef]
- Tiwari, A.; Schneider, M.; Fiorino, A.; Haider, R.; Okoniewski, M.J.; Roschitzki, B.; Uzozie, A.; Menigatti, M.; Jiricny, J.; Marra, G. Early Insights into the Function of KIAA1199, a Markedly Overexpressed Protein in Human Colorectal Tumors. PLoS ONE 2013, 8, e69473. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, L.; Shen, Q.; Lv, Q.; Jin, M.; Ma, H.; Nie, X.; Zheng, X.; Huang, S.; Zhou, P.; et al. Down-Regulation of KIAA1199/CEMIP by MiR-216a Suppresses Tumor Invasion and Metastasis in Colorectal Cancer. Int. J. Cancer 2017, 140, 2298–2309. [Google Scholar] [CrossRef]
- Fink, S.P.; Myeroff, L.L.; Kariv, R.; Platzer, P.; Xin, B.; Mikkola, D.; Lawrence, E.; Morris, N.; Nosrati, A.; Willson, J.K.V.; et al. Induction of KIAA1199/CEMIP Is Associated with Colon Cancer Phenotype and Poor Patient Survival. Oncotarget 2015, 6, 30500–30515. [Google Scholar] [CrossRef] [PubMed]
- Hibi, K.; Kitamura, Y.; Mizukami, H.; Goto, T.; Sakuraba, K.; Sakata, M.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; et al. Frequent CDH3 Demethylation in Advanced Gastric Carcinoma. Anticancer Res. 2009, 29, 3945–3947. [Google Scholar]
- Kumara, H.M.C.S.; Bellini, G.A.; Caballero, O.L.; Herath, S.A.C.; Su, T.; Ahmed, A.; Njoh, L.; Cekic, V.; Whelan, R.L. P-Cadherin (CDH3) Is Overexpressed in Colorectal Tumors and Has Potential as a Serum Marker for Colorectal Cancer Monitoring. Oncoscience 2017, 4, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-Z.; Hu, L.; Cai, J.; Chen, H.-Y.; Zhang, Y.; Feng, D.; Qi, C.-Y.; Zhai, Y.-X.; Gong, H.; Fu, H.; et al. Prognostic Value of Carbonic Anhydrase VII Expression in Colorectal Carcinoma. BMC Cancer 2015, 15, 209. [Google Scholar] [CrossRef]
- Hong, Y.; Liew, S.C.; Thean, L.F.; Tang, C.L.; Cheah, P.Y. Human Colorectal Cancer Initiation Is Bidirectional, and Cell Growth, Metabolic Genes and Transporter Genes Are Early Drivers of Tumorigenesis. Cancer Lett. 2018, 431, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, Y.; Wang, Z.; Lou, R.; Wu, J.; Feng, J. Integrated Analysis of Oncogenic Networks in Colorectal Cancer Identifies GUCA2A as a Molecular Marker. Biochem. Res. Int. 2019, 2019, 6469420. [Google Scholar] [CrossRef] [PubMed]
- Feodorova, Y.; Tashkova, D.; Koev, I.; Todorov, A.; Kostov, G.; Simitchiev, K.; Belovejdov, V.; Dimov, R.; Sarafian, V. Novel Insights into Transcriptional Dysregulation in Colorectal Cancer. Neoplasma 2018, 65, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.H.; Reverter, A. A Boolean-Based Systems Biology Approach to Predict Novel Genes Associated with Cancer: Application to Colorectal Cancer. BMC Syst. Biol. 2011, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, P.; Hlavac, V.; Mohelnikova-Duchonova, B.; Liska, V.; Pesta, M.; Soucek, P. Downregulation of ABC Transporters in Non-Neoplastic Tissues Confers Better Prognosis for Pancreatic and Colorectal Cancer Patients. J. Cancer 2017, 8, 1959–1971. [Google Scholar] [CrossRef]
- Lin, P.-C.; Lin, H.-H.; Lin, J.-K.; Lin, C.-C.; Yang, S.-H.; Li, A.F.-Y.; Chen, W.-S.; Chang, S.-C. Expression of ABCG2 Associated with Tumor Response in Metastatic Colorectal Cancer Patients Receiving First-Line FOLFOX Therapy--Preliminary Evidence. Int. J. Biol. Markers 2013, 28, 182–186. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, J.M.; Roh, Y.J.; Kim, I.-W.; Hasan, T.; Choi, M.-G. Enhanced Efficacy of Photodynamic Therapy by Inhibiting ABCG2 in Colon Cancers. BMC Cancer 2015, 15, 504. [Google Scholar] [CrossRef] [PubMed]
- Andersen, V.; Svenningsen, K.; Knudsen, L.A.; Hansen, A.K.; Holmskov, U.; Stensballe, A.; Vogel, U. Novel Understanding of ABC Transporters ABCB1/MDR/P-Glycoprotein, ABCC2/MRP2, and ABCG2/BCRP in Colorectal Pathophysiology. World J. Gastroenterol. 2015, 21, 11862–11876. [Google Scholar] [CrossRef]
- To, K.K.W.; Leung, W.W.; Ng, S.S.M. A Novel MiR-203-DNMT3b-ABCG2 Regulatory Pathway Predisposing Colorectal Cancer Development. Mol. Carcinog. 2017, 56, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Andersen, V.; Vogel, L.K.; Kopp, T.I.; Sæbø, M.; Nonboe, A.W.; Hamfjord, J.; Kure, E.H.; Vogel, U. High ABCC2 and Low ABCG2 Gene Expression Are Early Events in the Colorectal Adenoma-Carcinoma Sequence. PLoS ONE 2015, 10, e0119255. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, T.; Jin, Y.; Wei, J.; Yang, Y.; Zhang, H. ABCG2 Is Required for Self-Renewal and Chemoresistance of CD133-Positive Human Colorectal Cancer Cells. Tumour Biol. 2016, 37, 12889–12896. [Google Scholar] [CrossRef]
- Abstract 4455: Identification of Novel Oncogene, Copine-7 (CPNE7), in Colorectal Cancer—Cancer Research. Available online: https://cancerres.aacrjournals.org/content/77/13_Supplement/4455 (accessed on 13 January 2021).
- Zhu, Z.; Dong, W. Overexpression of HHLA2, a Member of the B7 Family, Is Associated with Worse Survival in Human Colorectal Carcinoma. OncoTargets Ther. 2018, 11, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Feng, H.; Cheng, X.; Liu, K.; Cai, D.; Zhao, R. Potential Therapeutic Targets of B7 Family in Colorectal Cancer. Front. Immunol. 2020, 11, 681. [Google Scholar] [CrossRef]
- Chan, C.H.F.; Cook, D.; Stanners, C.P. Increased Colon Tumor Susceptibility in Azoxymethane Treated CEABAC Transgenic Mice. Carcinogenesis 2006, 27, 1909–1916. [Google Scholar] [CrossRef] [PubMed]
- Messick, C.A.; Sanchez, J.; Dejulius, K.L.; Hammel, J.; Ishwaran, H.; Kalady, M.F. CEACAM-7: A Predictive Marker for Rectal Cancer Recurrence. Surgery 2010, 147, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Stenling, R.; Rubio, C.; Lindblom, A. Differential Expression of Aquaporin 8 in Human Colonic Epithelial Cells and Colorectal Tumors. BMC Physiol. 2001, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kwon, C.H.; Lee, S.J.; Park, J.; Shin, J.-Y.; Park, D.Y. Integrative Analysis of Oncogenic Fusion Genes and Their Functional Impact in Colorectal Cancer. Br. J. Cancer 2018, 119, 230–240. [Google Scholar] [CrossRef]
- Lorenc, Z.; Waniczek, D.; Lorenc-Podgórska, K.; Krawczyk, W.; Domagała, M.; Majewski, M.; Mazurek, U. Profile of Expression of Genes Encoding Matrix Metallopeptidase 9 (MMP9), Matrix Metallopeptidase 28 (MMP28) and TIMP Metallopeptidase Inhibitor 1 (TIMP1) in Colorectal Cancer: Assessment of the Role in Diagnosis and Prognostication. Med. Sci. Monit. 2017, 23, 1305–1311. [Google Scholar] [CrossRef]
- Xiao, R.; Li, C.; Chai, B. MiRNA-144 Suppresses Proliferation and Migration of Colorectal Cancer Cells through GSPT1. Biomed. Pharmacother. Biomed. Pharmacother. 2015, 74, 138–144. [Google Scholar] [CrossRef]
- Kim, S.W.; Park, K.C.; Jeon, S.M.; Ohn, T.B.; Kim, T.I.; Kim, W.H.; Cheon, J.H. Abrogation of Galectin-4 Expression Promotes Tumorigenesis in Colorectal Cancer. Cell. Oncol. Dordr. 2013, 36, 169–178. [Google Scholar] [CrossRef]
- Rao, U.S.; Rao, P.S. Surface-Bound Galectin-4 Regulates Gene Transcription and Secretion of Chemokines in Human Colorectal Cancer Cell Lines. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef]
- Ferlizza, E.; Solmi, R.; Miglio, R.; Nardi, E.; Mattei, G.; Sgarzi, M.; Lauriola, M. Colorectal Cancer Screening: Assessment of CEACAM6, LGALS4, TSPAN8 and COL1A2 as Blood Markers in Faecal Immunochemical Test Negative Subjects. J. Adv. Res. 2020, 24, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Rodia, M.T.; Solmi, R.; Pasini, F.; Nardi, E.; Mattei, G.; Ugolini, G.; Ricciardiello, L.; Strippoli, P.; Miglio, R.; Lauriola, M. LGALS4, CEACAM6, TSPAN8, and COL1A2: Blood Markers for Colorectal Cancer-Validation in a Cohort of Subjects With Positive Fecal Immunochemical Test Result. Clin. Colorectal Cancer 2018, 17, e217–e228. [Google Scholar] [CrossRef]
- Satelli, A.; Rao, P.S.; Thirumala, S.; Rao, U.S. Galectin-4 Functions as a Tumor Suppressor of Human Colorectal Cancer. Int. J. Cancer 2011, 129, 799–809. [Google Scholar] [CrossRef]
- Zbánková, S.; Bryndová, J.; Kment, M.; Pácha, J. Expression of 11beta-Hydroxysteroid Dehydrogenase Types 1 and 2 in Colorectal Cancer. Cancer Lett. 2004, 210, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, Q.-M.; Du, P.-C.; Ning, D.; Mo, J.; Zhu, H.-D.; Wang, C.; Ge, Q.-Y.; Cheng, Q.; Zhang, X.-W.; et al. Type-2 11β-Hydroxysteroid Dehydrogenase Promotes the Metastasis of Colorectal Cancer via the Fgfbp1-AKT Pathway. Am. J. Cancer Res. 2020, 10, 662–673. [Google Scholar] [PubMed]
- Andrieu, T.; Fustier, P.; Alikhani-Koupaei, R.; Ignatova, I.D.; Guettinger, A.; Frey, F.J.; Frey, B.M. Insulin, CCAAT/Enhancer-Binding Proteins and Lactate Regulate the Human 11β-Hydroxysteroid Dehydrogenase Type 2 Gene Expression in Colon Cancer Cell Lines. PLoS ONE 2014, 9, e105354. [Google Scholar] [CrossRef]
- Lian, W.; Jin, H.; Cao, J.; Zhang, X.; Zhu, T.; Zhao, S.; Wu, S.; Zou, K.; Zhang, X.; Zhang, M.; et al. Identification of Novel Biomarkers Affecting the Metastasis of Colorectal Cancer through Bioinformatics Analysis and Validation through QRT-PCR. Cancer Cell Int. 2020, 20, 105. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yu, H.-L.; Li, Q.-H.; Zhang, L.; Chen, Y.-X. MiR-4709 Overexpression Facilitates Cancer Proliferation and Invasion via Downregulating NR3C2 and Is an Unfavorable Prognosis Factor in Colon Adenocarcinoma. J. Biochem. Mol. Toxicol. 2019, 33, e22411. [Google Scholar] [CrossRef]
- Pesson, M.; Volant, A.; Uguen, A.; Trillet, K.; De La Grange, P.; Aubry, M.; Daoulas, M.; Robaszkiewicz, M.; Le Gac, G.; Morel, A.; et al. A Gene Expression and Pre-MRNA Splicing Signature That Marks the Adenoma-Adenocarcinoma Progression in Colorectal Cancer. PLoS ONE 2014, 9, e87761. [Google Scholar] [CrossRef]
- Bhalla, S.; Chaudhary, K.; Kumar, R.; Sehgal, M.; Kaur, H.; Sharma, S.; Raghava, G.P.S. Gene Expression-Based Biomarkers for Discriminating Early and Late Stage of Clear Cell Renal Cancer. Sci. Rep. 2017, 7, 44997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, H.; Wu, D.; Cao, D.; Ma, W. A Comprehensive Analysis of the Microbiota Composition and Gene Expression in Colorectal Cancer. BMC Microbiol. 2020, 20, 308. [Google Scholar] [CrossRef]
- Reddy, B.S.; Wynder, E.L. Metabolic Epidemiology of Colon Cancer. Fecal Bile Acids and Neutral Sterols in Colon Cancer Patients and Patients with Adenomatous Polyps. Cancer 1977, 39, 2533–2539. [Google Scholar] [CrossRef]
- Roomi, M.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Synergistic Effect of Combination of Lysine, Proline, Arginine, Ascorbic Acid, and Epigallocatechin Gallate on Colon Cancer Cell Line HCT 116. JANA 2004, 7, 40–43. [Google Scholar]
- Karimian, J.; Hadi, A.; Salehi-sahlabadi, A.; Kafeshani, M. The Effect of Arginine Intake on Colorectal Cancer: A Systematic Review of Literatures. Clin. Nutr. Res. 2019, 8, 209–218. [Google Scholar] [CrossRef]
- Shortt, C.; Hasselwander, O.; Meynier, A.; Nauta, A.; Fernández, E.N.; Putz, P.; Rowland, I.; Swann, J.; Türk, J.; Vermeiren, J.; et al. Systematic Review of the Effects of the Intestinal Microbiota on Selected Nutrients and Non-Nutrients. Eur. J. Nutr. 2018, 57, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Claus, S.P.; Tsang, T.M.; Wang, Y.; Cloarec, O.; Skordi, E.; Martin, F.-P.; Rezzi, S.; Ross, A.; Kochhar, S.; Holmes, E.; et al. Systemic Multicompartmental Effects of the Gut Microbiome on Mouse Metabolic Phenotypes. Mol. Syst. Biol. 2008, 4, 219. [Google Scholar] [CrossRef] [PubMed]
- Human Metabolome Database: Showing Metabocard for 5-Aminopentanoic Acid (HMDB0003355). Hmdb.ca. 2020. Available online: https://Hmdb.ca/Metabolites/HMDB0003355 (accessed on 30 November 2020).
- Martin, O.C.B.; Olier, M.; Ellero-Simatos, S.; Naud, N.; Dupuy, J.; Huc, L.; Taché, S.; Graillot, V.; Levêque, M.; Bézirard, V.; et al. Haem Iron Reshapes Colonic Luminal Environment: Impact on Mucosal Homeostasis and Microbiome through Aldehyde Formation. Microbiome 2019, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, B.F.; Meng, S.; Wu, J.T.; Archer, S.Y.; Hodin, R.A. The Effects of Short-Chain Fatty Acids on Human Colon Cancer Cell Phenotype Are Associated with Histone Hyperacetylation. J. Nutr. 2002, 132, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, I.R. Short Chain Fatty Acid Regulation of Signaling Genes Expressed by the Intestinal Epithelium. J. Nutr. 2004, 134, 2450S–2454S. [Google Scholar] [CrossRef]
- Acharjee, A.; Ament, Z.; West, J.A.; Stanley, E.; Griffin, J.L. Integration of Metabolomics, Lipidomics and Clinical Data Using a Machine Learning Method. BMC Bioinform. 2016, 17, 440. [Google Scholar] [CrossRef]
- Quraishi, M.N.; Acharjee, A.; Beggs, A.D.; Horniblow, R.; Tselepis, C.; Gkoutos, G.; Ghosh, S.; Rossiter, A.E.; Loman, N.; van Schaik, W.; et al. A Pilot Integrative Analysis of Colonic Gene Expression, Gut Microbiota, and Immune Infiltration in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Association of Disease with Bile Acid Pathways. J. Crohns Colitis 2020, 14, 935–947. [Google Scholar] [CrossRef]
- Martin, F.-P.J.; Wang, Y.; Sprenger, N.; Yap, I.K.S.; Lundstedt, T.; Lek, P.; Rezzi, S.; Ramadan, Z.; van Bladeren, P.; Fay, L.B.; et al. Probiotic Modulation of Symbiotic Gut Microbial–Host Metabolic Interactions in a Humanized Microbiome Mouse Model. Mol. Syst. Biol. 2008, 4, 157. [Google Scholar] [CrossRef]
- Gerl, M.J.; Vaz, W.L.C.; Domingues, N.; Klose, C.; Surma, M.A.; Sampaio, J.L.; Almeida, M.S.; Rodrigues, G.; Araújo-Gonçalves, P.; Ferreira, J.; et al. Cholesterol Is Inefficiently Converted to Cholesteryl Esters in the Blood of Cardiovascular Disease Patients. Sci. Rep. 2018, 8, 14764. [Google Scholar] [CrossRef]
- Bent, S.; Lawton, B.; Warren, T.; Widjaja, F.; Dang, K.; Fahey, J.W.; Cornblatt, B.; Kinchen, J.M.; Delucchi, K.; Hendren, R.L. Identification of Urinary Metabolites That Correlate with Clinical Improvements in Children with Autism Treated with Sulforaphane from Broccoli. Mol. Autism 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M. 2-Indolinone a Versatile Scaffold for Treatment of Cancer: A Patent Review (2008–2014). Expert Opin. Ther. Pat. 2016, 26, 149–173. [Google Scholar] [CrossRef]
- Kaminska, K.K.; Bertrand, H.C.; Tajima, H.; Stafford, W.C.; Cheng, Q.; Chen, W.; Wells, G.; Arner, E.S.J.; Chew, E.-H. Indolin-2-One Compounds Targeting Thioredoxin Reductase as Potential Anticancer Drug Leads. Oncotarget 2016, 7, 40233–40251. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, C.; Amawi, H.; Ashby, C.R.; Khare, V.M.; Jones, V.; Hari Narayana Moorthy, N.S.; Trivedi, P.; Tiwari, A.K. Novel 3-((2-Chloroquinolin-3-Yl)Methylene)Indolin-2-One Derivatives Produce Anticancer Efficacy in Ovarian Cancer in Vitro. Heliyon 2019, 5, e01603. [Google Scholar] [CrossRef]
- Louis, E.D.; Pellegrino, K.M.; Factor-Litvak, P.; Rios, E.; Jiang, W.; Henchcliffe, C.; Zheng, W. Cancer and Blood Concentrations of the Comutagen Harmane in Essential Tremor. Mov. Disord. 2008, 23, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, I.; Mastalier, B. Genetics of Colorectal Cancer. J. Med. Life 2014, 7, 507–511. [Google Scholar] [PubMed]
- Smith, G.; Carey, F.A.; Beattie, J.; Wilkie, M.J.V.; Lightfoot, T.J.; Coxhead, J.; Garner, R.C.; Steele, R.J.C.; Wolf, C.R. Mutations in APC, Kirsten-Ras, and P53—Alternative Genetic Pathways to Colorectal Cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 9433–9438. [Google Scholar] [CrossRef]
- Song, K.; Su, W.; Liu, Y.; Zhang, J.; Liang, Q.; Li, N.; Guan, Q.; He, J.; Bai, X.; Zhao, W.; et al. Identification of Genes with Universally Upregulated or Downregulated Expressions in Colorectal Cancer. J. Gastroenterol. Hepatol. 2019, 34, 880–889. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) RNA-Seq Meta-Analysis 2018. Available online: https://portal.gdc.cancer.gov/projects/TCGA-COAD (accessed on 30 November 2020).
- Kapelner, A.; Bleich, J. bartMachine: Machine Learning with Bayesian Additive Regression Trees. J. Stat. Softw. 2016, 70, 1–40. [Google Scholar] [CrossRef]
- Chipman, H.A.; George, E.I.; McCulloch, R.E. BART: Bayesian Additive Regression Trees. Ann. Appl. Stat. 2010, 4, 266–298. [Google Scholar] [CrossRef]
- RNA-Seq Workflow: Gene-Level Exploratory Analysis and Differential Expression. Available online: https://bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html (accessed on 13 January 2021).
- Sun, Y.; Wong, A.K.C.; Kamel, M.S. Classification of Imbalanced Data: A Review. Int. J. Pattern Recognit. Artif. Intell. 2009, 23, 687–719. [Google Scholar] [CrossRef]
- Yen, S.-J.; Lee, Y.-S. Under-Sampling Approaches for Improving Prediction of the Minority Class in an Imbalanced Dataset. In Intelligent Control and Automation, International Conference on Intelligent Computing, ICIC 2006 Kunming, China, 16–19 August 2006; Huang, D.-S., Li, K., Irwin, G.W., Eds.; Lecture Notes in Control and Information Sciences; Springer: Berlin/Heidelberg, Germany, 2006; pp. 731–740. ISBN 978-3-540-37256-1. [Google Scholar]
- R Core Team. Caret: Classification and Regression Training; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Normal vs. CRC (Bulk RNA Seq) | Normal vs. CRC (Single-Cell RNA Seq) Epithelial Cells | Reference |
|---|---|---|---|
| ADH1B | Downregulated | Downregulated | [21] |
| KIAA1199 (CEMIP) | Upregulated | Upregulated | [22,23,24,25,26,27] |
| CDH3 | Upregulated | Upregulated | [5,28,29] |
| CA7 | Downregulated | Downregulated | [30,31] |
| GUCA2B | Downregulated | Downregulated | [32,33,34] |
| ABCC13 | Downregulated | Downregulated | [35] |
| ABCG2 | Downregulated | Downregulated | [36,37,38,39,40,41] |
| CPNE7 | Upregulated | Upregulated | [42] |
| HHLA2 | Downregulated | Downregulated | [43,44] |
| CEACAM7 | Downregulated | Downregulated | [45,46] |
| AQP8 | Downregulated | Downregulated | [47] |
| GTF3A | Upregulated | Upregulated | [48] |
| MMP28 | Downregulated | Down regulated | [49,50] |
| LGALS4 | Downregulated | Downregulated | [51,52,53,54,55] |
| HSD11B2 | Downregulated | Downregulated | [56,57,58] |
| CHP2 | Downregulated | Downregulated | [59] |
| NR3C2 | Downregulated | Downregulated | [32,34,60,61,62] |
| Data Set | Features | Sample | Reference |
|---|---|---|---|
| Kim et al., 2020 | 16S rRNA and metabolomics | Normal (N = 102) vs. Colorectal cancer (N = 36) | [8] |
| Clos-Garcia et al., 2020 | 16S rRNA and metabolomics | Normal (N = 77) vs. Colorectal cancer (N = 99) | [11] |
| The Cancer Genome Atlas (TCGA) ColonAdenocarcinoma | RNA sequence | Normal (N = 41) vs. Colon Adenocarcinoma (N = 459) | [85] |
| Li et al., 2017 | Single-cell transcriptomes | Normal (N = 1591 cells) vs. Colorectal cancer (N = 1591 cells) | [17] |
| Zhang et al., 2019 | Single-cell transcriptomes | Normal (N = 160 cells) vs. Colorectal cancer (N = 272 cells) | [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisht, V.; Nash, K.; Xu, Y.; Agarwal, P.; Bosch, S.; Gkoutos, G.V.; Acharjee, A. Integration of the Microbiome, Metabolome and Transcriptomics Data Identified Novel Metabolic Pathway Regulation in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 5763. https://doi.org/10.3390/ijms22115763
Bisht V, Nash K, Xu Y, Agarwal P, Bosch S, Gkoutos GV, Acharjee A. Integration of the Microbiome, Metabolome and Transcriptomics Data Identified Novel Metabolic Pathway Regulation in Colorectal Cancer. International Journal of Molecular Sciences. 2021; 22(11):5763. https://doi.org/10.3390/ijms22115763
Chicago/Turabian StyleBisht, Vartika, Katrina Nash, Yuanwei Xu, Prasoon Agarwal, Sofie Bosch, Georgios V. Gkoutos, and Animesh Acharjee. 2021. "Integration of the Microbiome, Metabolome and Transcriptomics Data Identified Novel Metabolic Pathway Regulation in Colorectal Cancer" International Journal of Molecular Sciences 22, no. 11: 5763. https://doi.org/10.3390/ijms22115763
APA StyleBisht, V., Nash, K., Xu, Y., Agarwal, P., Bosch, S., Gkoutos, G. V., & Acharjee, A. (2021). Integration of the Microbiome, Metabolome and Transcriptomics Data Identified Novel Metabolic Pathway Regulation in Colorectal Cancer. International Journal of Molecular Sciences, 22(11), 5763. https://doi.org/10.3390/ijms22115763