
Protease Inhibition—An Established Strategy to Combat Infectious Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Protease Inhibition as a Strategy for the Treatment of Infectious Diseases
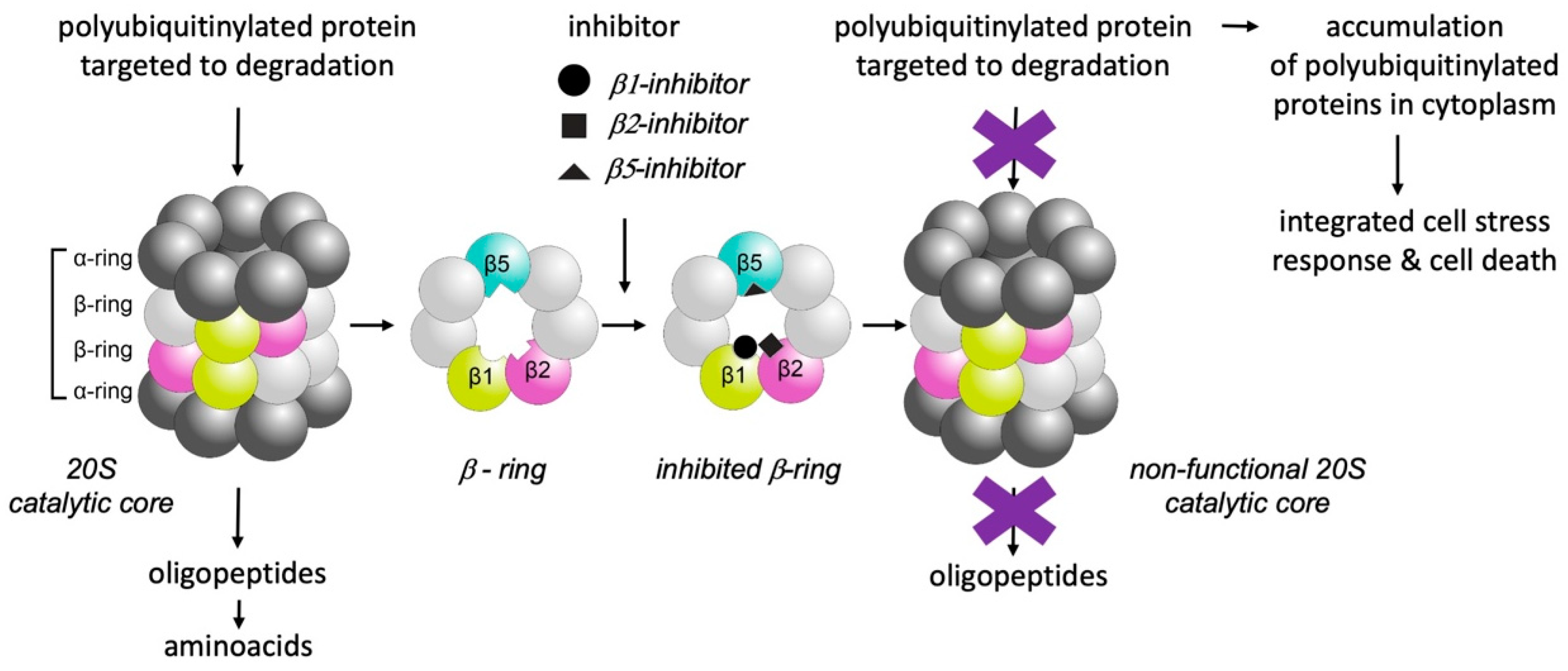
3. Selective Inhibition of Proteasomes
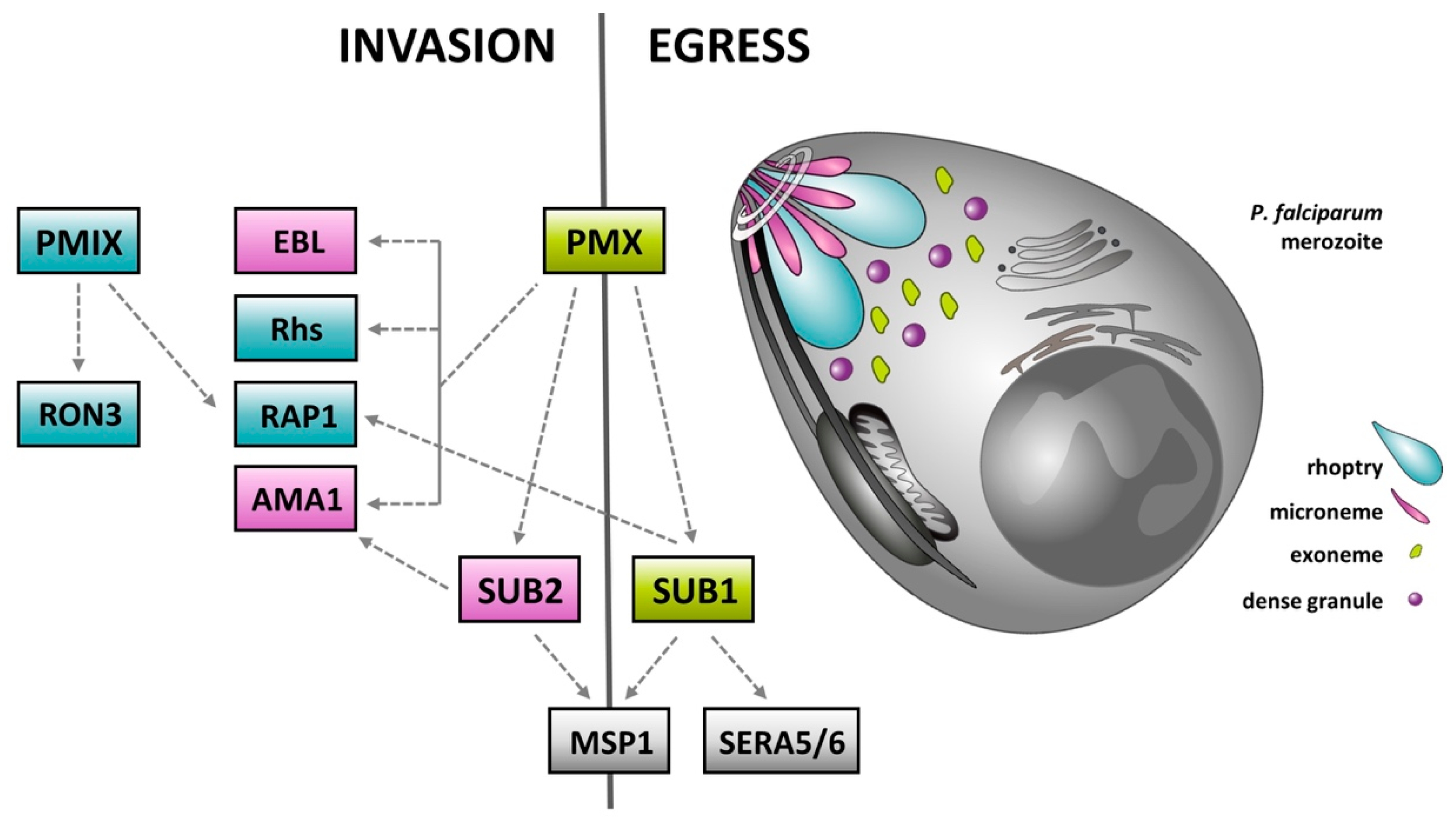
4. Plasmepsins—Rediscovered Molecular Targets for the Treatment of Malaria
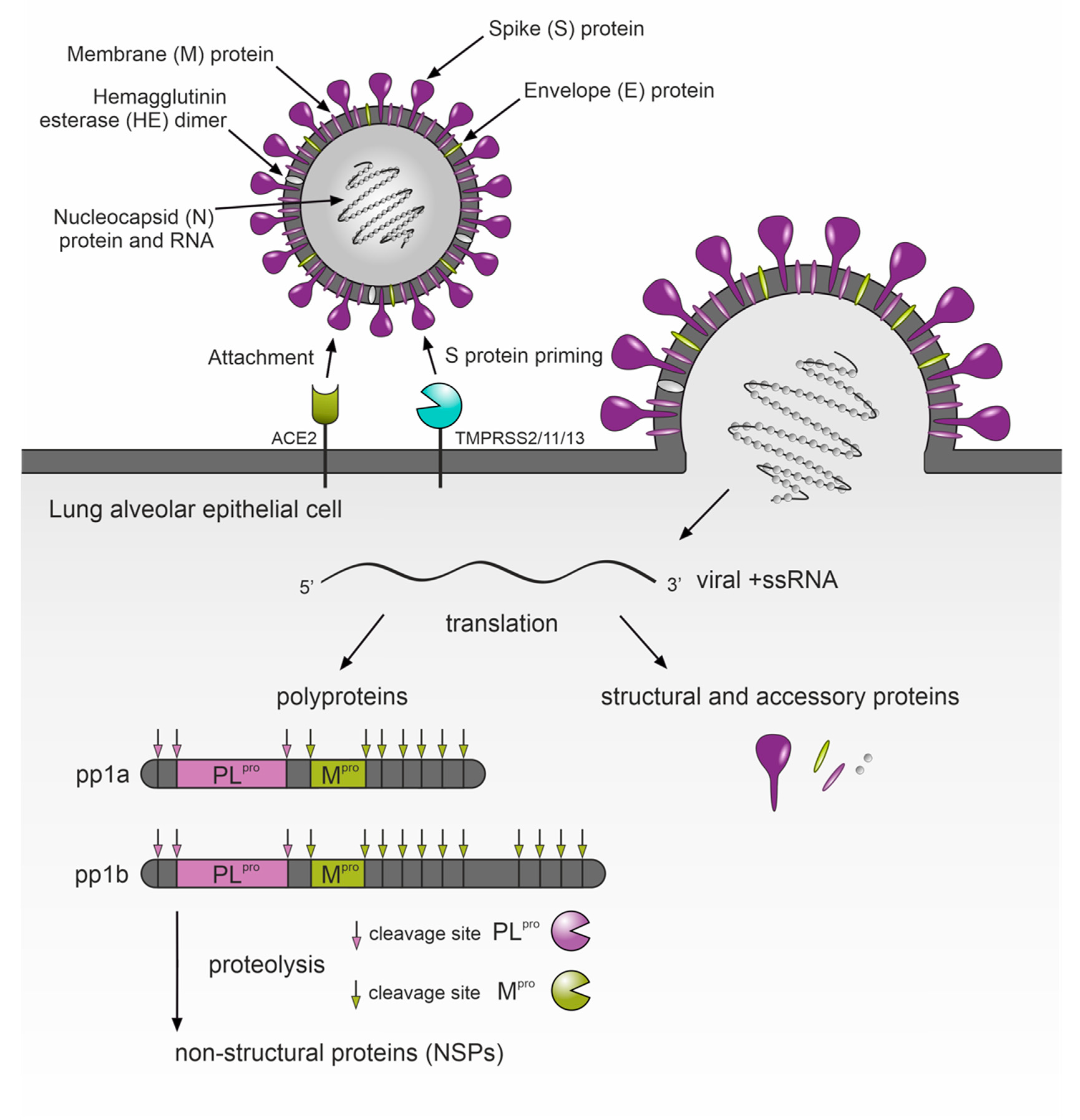
5. Protease Inhibitors as a Potential Therapy for COVID-19
6. “Tailored” Inhibitors for Better Bioavailability and Reduced Toxicity
7. Conclusions
Funding
Conflicts of Interest
References
- McKerrow, J.H. Designing Drugs for Parasitic Diseases of the Developing World. PLoS Med. 2005, 2, e210. [Google Scholar] [CrossRef]
- Despommier, D.D.; Griffin, D.O.; Gwadz, R.W.; Hotez, P.J.; Knirsch, C.A. Parasitic Diseases, 7th ed.; Parasites without Borders, Inc.: New York, NY, USA, 2019. [Google Scholar]
- Young, K.M.; Corrin, T.; Wilhelm, B.; Uhland, C.; Greig, J.; Mascarenhas, M.; Waddell, L.A. Zoonotic Babesia: A scoping review of the global evidence. PLoS ONE 2019, 14, e0226781. [Google Scholar] [CrossRef]
- Bond, J.S. Proteases: History, discovery, and roles in health and disease. J. Biol. Chem. 2019, 294, 1643–1651. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Bateman, A. How to use the MEROPS database and website to help understand peptidase specificity. Protein Sci. 2021, 30, 83–92. [Google Scholar] [CrossRef]
- Barrett, A.J.; Rawlings, N.D.; Salvesen, G.; Woessner, J.F. Handbook of Proteolytic Enzymes Introduction, 3rd ed.; Academic Press: Boston, MA, USA; London, UK, 2013. [Google Scholar]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Renslo, A.R.; McKerrow, J.H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H.; Caffrey, C.; Kelly, B.; Loke, P.; Sajid, M. PROTEASES IN PARASITIC DISEASES. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 497–536. [Google Scholar] [CrossRef]
- Kitchen, V.; Skinner, C.; Ariyoshi, K.; Weber, J.; Pinching, A.; Lane, E.; Duncan, I.; Burckhardt, J.; Burger, H.; Bragman, K. Safety and activity of saquinavir in HIV infection. Lancet 1995, 345, 952–955. [Google Scholar] [CrossRef]
- Kosalaraksa, P.; Ananworanich, J.; Puthanakit, T.; Pinyakorn, S.; Lumbiganon, P.; Chuanjaroen, T.; Chobkarjing, U.; Phanuphak, P.; Pancharoen, C.; Bunupuradah, T. Long-term Lopinavir/Ritonavir Monotherapy in HIV-infected Children. Pediatr. Infect. Dis. J. 2013, 32, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef]
- Sajid, M.; Robertson, S.A.; Brinen, L.S.; McKerrow, J.H. Cruzain: The path from target validation to the clinic. Adv. Exp. Med. Biol. 2011, 712, 100–115. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H. Update on drug development targeting parasite cysteine proteases. PLOS Negl. Trop. Dis. 2018, 12, e0005850. [Google Scholar] [CrossRef]
- Perez, J.; Kreinovich, V. Gartner’s hype cycle: A simple explanation. Int. J. Comput. Optim. 2018, 5, 1–4. [Google Scholar] [CrossRef]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.A.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G. Update on proteasome inhibitors in multiple myeloma. Clin. Adv. Hematol. Oncol. 2014, 12, 179–181. [Google Scholar]
- Bibo-Verdugo, B.; Jiang, Z.; Caffrey, C.R.; O’Donoghue, A.J. Targeting proteasomes in infectious organisms to combat disease. FEBS J. 2017, 284, 1503–1517. [Google Scholar] [CrossRef]
- Kreidenweiss, A.; Kremsner, P.G.; Mordmüller, B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar. J. 2008, 7, 187. [Google Scholar] [CrossRef]
- Gantt, S.M.; Myung, J.M.; Briones, M.R.S.; Li, W.D.; Corey, E.J.; Omura, S.; Nussenzweig, V.; Sinnis, P. Proteasome Inhibitors Block Development ofPlasmodium spp. Antimicrob. Agents Chemother. 1998, 42, 2731–2738. [Google Scholar] [CrossRef]
- Dekel, E.; Yaffe, D.; Rosenhek-Goldian, I.; Ben-Nissan, G.; Ofir-Birin, Y.; Morandi, M.I.; Ziv, T.; Sisquella, X.; Pimentel, M.A.; Nebl, T.; et al. 20S proteasomes secreted by the malaria parasite promote its growth. Nat. Commun. 2021, 12, 1–19. [Google Scholar] [CrossRef]
- Reynolds, J.M.; EL Bissati, K.; Brandenburg, J.; Günzl, A.; Ben Mamoun, C. Antimalarial activity of the anticancer and proteasome inhibitor bortezomib and its analog ZL3B. BMC Clin. Pharmacol. 2007, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Atul; Kolla, V.K.; Legac, J.; Singhal, N.; Navale, R.; Rosenthal, P.J.; Sijwali, P.S. Blocking Plasmodium falciparum Development via Dual Inhibition of Hemoglobin Degradation and the Ubiquitin Proteasome System by MG132. PLoS ONE 2013, 8, e73530. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ponder, E.L.; Verdoes, M.; Asbjornsdottir, K.H.; Deu, E.; Edgington-Mitchell, L.; Lee, J.T.; Kirk, C.J.; Demo, S.D.; Williamson, K.C.; et al. Validation of the Proteasome as a Therapeutic Target in Plasmodium Using an Epoxyketone Inhibitor with Parasite-Specific Toxicity. Chem. Biol. 2012, 19, 1535–1545. [Google Scholar] [CrossRef]
- Li, H.; Tsu, C.; Blackburn, C.; Li, G.; Hales, P.; Dick, L.; Bogyo, M. Identification of Potent and Selective Non-covalent Inhibitors of thePlasmodium falciparumProteasome. J. Am. Chem. Soc. 2014, 136, 13562–13565. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.C.; Gillett, D.L.; Spillman, N.J.; Tsu, C.; Luth, M.R.; Ottilie, S.; Duffy, S.; Gould, A.E.; Hales, P.; Seager, B.A.; et al. Target Validation and Identification of Novel Boronate Inhibitors of the Plasmodium falciparum Proteasome. J. Med. Chem. 2018, 61, 10053–10066. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.M.; Williamson, K.C. The proteasome as a target to combat malaria: Hits and misses. Transl. Res. 2018, 198, 40–47. [Google Scholar] [CrossRef]
- Li, H.; O’Donoghue, A.J.; van der Linden, H.L.W.A.; Xie, S.C.; Yoo, E.; Foe, I.T.; Tilley, L.; Craik, A.J.O.C.S.; Da Fonseca, P.C.A.; Bogyo, H.L.E.Y.I.T.F.M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nat. Cell Biol. 2016, 530, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Visone, J.; Ouellette, T.; Harris, J.C.; Wang, R.; Zhang, H.; Singh, P.K.; Ginn, J.; Sukenick, G.; Wong, T.-T.; et al. Improvement of Asparagine Ethylenediamines as Anti-malarial Plasmodium-Selective Proteasome Inhibitors. J. Med. Chem. 2019, 62, 6137–6145. [Google Scholar] [CrossRef]
- LaMonte, G.M.; Almaliti, J.; Bibo-Verdugo, B.; Keller, L.; Zou, B.Y.; Yang, J.; Antonova-Koch, Y.; Orjuela-Sanchez, P.; Boyle, C.A.; Vigil, E.; et al. Development of a Potent Inhibitor of the Plasmodium Proteasome with Reduced Mammalian Toxicity. J. Med. Chem. 2017, 60, 6721–6732. [Google Scholar] [CrossRef]
- Stokes, B.H.; Yoo, E.; Murithi, J.M.; Luth, M.R.; Afanasyev, P.; Da Fonseca, P.C.A.; Winzeler, E.A.; Ng, C.L.; Bogyo, M.; Fidock, D.A. Covalent Plasmodium falciparum-selective proteasome inhibitors exhibit a low propensity for generating resistance in vitro and synergize with multiple antimalarial agents. PLoS Pathog. 2019, 15, e1007722. [Google Scholar] [CrossRef]
- Shea, M.; Jäkle, U.; Liu, Q.; Berry, C.; Joiner, K.A.; Soldati-Favre, M.; Soldati-Favre, D. A Family of Aspartic Proteases and a Novel, Dynamic and Cell-Cycle-Dependent Protease Localization in the Secretory Pathway of Toxoplasma gondii. Traffic 2007, 8, 1018–1034. [Google Scholar] [CrossRef] [PubMed]
- Sojka, D.; Hartmann, D.; Bartošová-Sojková, P.; Dvořák, J. Parasite Cathepsin D-Like Peptidases and Their Relevance as Therapeutic Targets. Trends Parasitol. 2016, 32, 708–723. [Google Scholar] [CrossRef]
- Nasamu, A.S.; Polino, A.J.; Istvan, E.S.; Goldberg, D.E. Malaria parasite plasmepsins: More than just plain old degradative pepsins. J. Biol. Chem. 2020, 295, 8425–8441. [Google Scholar] [CrossRef] [PubMed]
- Meyers, M.J.; Goldberg, D.E. Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts. Curr. Top. Med. Chem. 2012, 12, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Gilson, P.R.; Chisholm, S.A.; Crabb, B.S.; de Koning-Ward, T. Host cell remodelling in malaria parasites: A new pool of potential drug targets. Int. J. Parasitol. 2017, 47, 119–127. [Google Scholar] [CrossRef]
- Klemba, M.; Goldberg, D.E. Characterization of plasmepsin V, a membrane-bound aspartic protease homolog in the endoplasmic reticulum of Plasmodium falciparum. Mol. Biochem. Parasitol. 2005, 143, 183–191. [Google Scholar] [CrossRef]
- Boddey, J.; Hodder, A.N.; Günther, S.; Gilson, P.R.; Patsiouras, H.; Kapp, E.A.; Pearce, J.A.; de Koning-Ward, T.; Simpson, R.J.; Crabb, B.S.; et al. An aspartyl protease directs malaria effector proteins to the host cell. Nat. Cell Biol. 2010, 463, 627–631. [Google Scholar] [CrossRef]
- Ho, C.-M.; Beck, J.R.; Lai, M.; Cui, Y.; Goldberg, D.E.; Egea, P.F.; Zhou, Z.H. Malaria parasite translocon structure and mechanism of effector export. Nat. Cell Biol. 2018, 561, 70–75. [Google Scholar] [CrossRef]
- Jennison, C.; Lucantoni, L.; O’Neill, M.T.; McConville, R.; Erickson, S.M.; Cowman, A.F.; Sleebs, B.E.; Avery, V.M.; Boddey, J.A. Inhibition of Plasmepsin V Activity Blocks Plasmodium falciparum Gametocytogenesis and Transmission to Mosquitoes. Cell Rep. 2019, 29, 3796–3806.e4. [Google Scholar] [CrossRef]
- Sleebs, B.E.; Gazdik, M.; O’Neill, M.T.; Rajasekaran, P.; Lopaticki, S.; Lackovic, K.; Lowes, K.; Smith, B.J.; Cowman, A.F.; Boddey, J.A. Transition State Mimetics of the Plasmodium Export Element are Potent Inhibitors of Plasmepsin V from P. falciparum and P. vivax. J. Med. Chem. 2014, 57, 7644–7662. [Google Scholar] [CrossRef]
- Hodder, A.N.; Sleebs, B.E.; Czabotar, P.E.; Gazdik, M.; Xu, Y.; O’Neill, M.T.; Lopaticki, S.; Nebl, T.; Triglia, T.; Smith, B.J.; et al. Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat. Struct. Mol. Biol. 2015, 22, 590–596. [Google Scholar] [CrossRef]
- Nguyen, W.; Hodder, A.N.; de Lezongard, R.B.; Czabotar, P.E.; Jarman, K.E.; O’Neill, M.T.; Thompson, J.K.; Sabroux, H.J.; Cowman, A.F.; Boddey, J.; et al. Enhanced antimalarial activity of plasmepsin V inhibitors by modification of the P 2 position of PEXEL peptidomimetics. Eur. J. Med. Chem. 2018, 154, 182–198. [Google Scholar] [CrossRef] [PubMed]
- Dogga, S.K.; Mukherjee, B.; Jacot, D.; Kockmann, T.; Molino, L.; Hammoudi, P.-M.; Hartkoorn, R.C.; Hehl, A.B.; Soldati-Favre, D. A druggable secretory protein maturase of Toxoplasma essential for invasion and egress. eLife 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Alaganan, A.; Singh, P.; Chitnis, C.E. Molecular mechanisms that mediate invasion and egress of malaria parasites from red blood cells. Curr. Opin. Hematol. 2017, 24, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Withers-Martinez, C.; Suarez, C.; Fulle, S.; Kher, S.; Penzo, M.; Ebejer, J.-P.; Koussis, K.; Hackett, F.; Jirgensons, A.; Finn, P.; et al. Plasmodium subtilisin-like protease 1 (SUB1): Insights into the active-site structure, specificity and function of a pan-malaria drug target. Int. J. Parasitol. 2012, 42, 597–612. [Google Scholar] [CrossRef]
- Collins, C.R.; Hackett, F.; Howell, S.A.; Snijders, A.P.; Russell, M.R.; Collinson, L.M.; Blackman, M.J. The malaria parasite sheddase SUB2 governs host red blood cell membrane sealing at invasion. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Favuzza, P.; Ruiz, M.D.L.; Thompson, J.K.; Triglia, T.; Ngo, A.; Steel, R.W.; Vavrek, M.; Christensen, J.; Healer, J.; Boyce, C.; et al. Dual Plasmepsin-Targeting Antimalarial Agents Disrupt Multiple Stages of the Malaria Parasite Life Cycle. Cell Host Microbe 2020, 27, 642–658.e12. [Google Scholar] [CrossRef]
- Pino, P.; Caldelari, R.; Mukherjee, B.; Vahokoski, J.; Klages, N.; Maco, B.; Collins, C.R.; Blackman, M.J.; Kursula, I.; Heussler, V.; et al. A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science 2017, 358, 522–528. [Google Scholar] [CrossRef]
- Nasamu, A.S.; Glushakova, S.; Russo, I.; Vaupel, B.; Oksman, A.; Kim, A.S.; Fremont, D.H.; Tolia, N.; Beck, J.R.; Meyers, M.J.; et al. Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science 2017, 358, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef]
- Cui, W.; Yang, K.; Yang, H. Recent Progress in the Drug Development Targeting SARS-CoV-2 Main Protease as Treatment for COVID-19. Front. Mol. Biosci. 2020, 7, 616341. [Google Scholar] [CrossRef]
- Han, Y.-S.; Chang, G.-G.; Juo, C.-G.; Lee, H.-J.; Yeh, S.-H.; Hsu, J.T.-A.; Chen, X. Papain-Like Protease 2 (PLP2) from Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV): Expression, Purification, Characterization, and Inhibition. Biochemistry 2005, 44, 10349–10359. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating Activity of SARS-CoV Papain-Like Protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti–COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef] [PubMed]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, T. COVID19 therapeutics: Expanding the antiviral arsenal. EBioMedicine 2021, 66, 103289. [Google Scholar] [CrossRef] [PubMed]
- Sakr, Y.; Bensasi, H.; Taha, A.; Bauer, M.; Khaeled, I. Camostat mesylate therapy in critically ill patients with COVID-19 pneumonia. Intensive Care Med. 2021, 12, 1–3. [Google Scholar] [CrossRef]
- Gunst, J.D.; Staerke, N.B.; Pahus, M.H.; Kristensen, L.H.; Bodilsen, J.; Lohse, N.; Dalgaard, L.S.; Brønnum, D.; Fröbert, O.; Hønge, B.; et al. Efficacy of the TMPRSS2 inhibitor camostat mesilate in patients hospitalized with Covid-19-a double-blind randomized controlled trial. EClinicalMedicine 2021, 100849. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, 00754-20. [Google Scholar] [CrossRef]
- Mahoney, M.; Damalanka, V.C.; Tartell, M.A.; Chung, D.H.; Lourenco, A.L.; Pwee, D.; Mayer Bridwell, A.E.; Hoffmann, M.; Voss, J.; Karmakar, P.; et al. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS-CoV viral entry and protect human epithelial lung cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, Z.; Chu, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS Res. Palliat. Care 2015, 7, 95–104. [Google Scholar] [CrossRef]
- Barber, J.; Sikakana, P.; Sadler, C.; Baud, D.; Valentin, J.-P.; Roberts, R. A target safety assessment of the potential toxicological risks of targeting plasmepsin IX/X for the treatment of malaria. Toxicol. Res. 2021, 10, 203–213. [Google Scholar] [CrossRef]
- Pereira, A.R.; Kale, A.J.; Fenley, A.T.; Byrum, T.; Debonsi, H.M.; Gilson, M.K.; Valeriote, F.A.; Moore, B.; Gerwick, W.H. The Carmaphycins: New Proteasome Inhibitors Exhibiting an α,β-Epoxyketone Warhead from a Marine Cyanobacterium. ChemBioChem 2012, 13, 810–817. [Google Scholar] [CrossRef]
- Kumar, T.M.; Rohini, K.; James, N.; Shanthi, V.; Ramanathan, K. Discovery of Potent Covid-19 Main Protease Inhibitors using Integrated Drug Repurposing Strategy. Biotechnol. Appl. Biochem. 2021. [Google Scholar] [CrossRef]
- Lopez, T.; Mustafa, Z.; Chen, C.; Lee, K.B.; Ramirez, A.; Benitez, C.; Luo, X.; Ji, R.-R.; Ge, X. Functional selection of protease inhibitory antibodies. Proc. Natl. Acad. Sci. USA 2019, 116, 16314–16319. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sojka, D.; Šnebergerová, P.; Robbertse, L. Protease Inhibition—An Established Strategy to Combat Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 5762. https://doi.org/10.3390/ijms22115762
Sojka D, Šnebergerová P, Robbertse L. Protease Inhibition—An Established Strategy to Combat Infectious Diseases. International Journal of Molecular Sciences. 2021; 22(11):5762. https://doi.org/10.3390/ijms22115762
Chicago/Turabian StyleSojka, Daniel, Pavla Šnebergerová, and Luïse Robbertse. 2021. "Protease Inhibition—An Established Strategy to Combat Infectious Diseases" International Journal of Molecular Sciences 22, no. 11: 5762. https://doi.org/10.3390/ijms22115762
APA StyleSojka, D., Šnebergerová, P., & Robbertse, L. (2021). Protease Inhibition—An Established Strategy to Combat Infectious Diseases. International Journal of Molecular Sciences, 22(11), 5762. https://doi.org/10.3390/ijms22115762