
Pathophysiological Aspects of Alcohol Metabolism in the Liver


Abstract
1. Introduction
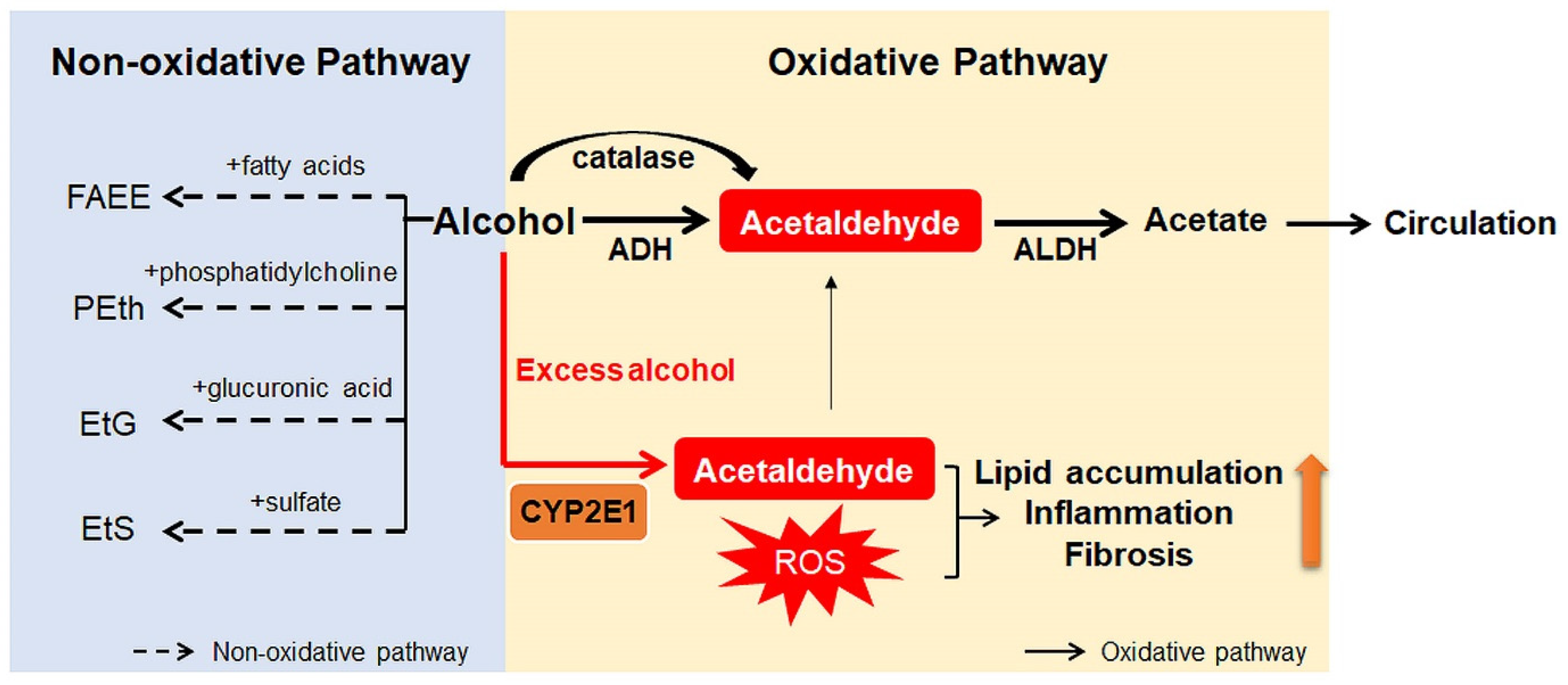
2. Alcohol Metabolism
3. Oxidative Ethanol Metabolites Are Involved in ALD Pathogenesis
3.1. Excessive Lipid Accumulation in Hepatocytes
3.2. Dysregulated Immune System
3.3. Induction of Fibrosis
3.4. Development of Cancer
4. Models Used in the Study of Pathophysiology Related to Alcohol Metabolism
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sudhinaraset, M.; Wigglesworth, C.; Takeuchi, D.T. Social and Cultural Contexts of Alcohol Use: Influences in a Social-Ecological Framework. Alcohol Res. 2016, 38, 35–45. [Google Scholar]
- Khaderi, S.A. Introduction: Alcohol and Alcoholism. Clin. Liver Dis. 2019, 23, 1–10. [Google Scholar] [CrossRef]
- Stockwell, T.; Zhao, J.; Panwar, S.; Roemer, A.; Naimi, T.; Chikritzhs, T. Do “Moderate” Drinkers Have Reduced Mortality Risk? A Systematic Review and Meta-Analysis of Alcohol Consumption and All-Cause Mortality. J. Stud. Alcohol Drugs 2016, 77, 185–198. [Google Scholar] [CrossRef]
- Zakhari, S.; Li, T.K. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology 2007, 46, 2032–2039. [Google Scholar] [CrossRef]
- World Health Organization. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-156563-9. [Google Scholar]
- Shield, K.D.; Parry, C.; Rehm, J. Chronic diseases and conditions related to alcohol use. Alcohol Res. 2013, 35, 155–173. [Google Scholar] [PubMed]
- Rehm, J.; Baliunas, D.; Borges, G.L.; Graham, K.; Irving, H.; Kehoe, T.; Parry, C.D.; Patra, J.; Popova, S.; Poznyak, V.; et al. The relation between different dimensions of alcohol consumption and burden of disease: An overview. Addiction 2010, 105, 817–843. [Google Scholar] [CrossRef] [PubMed]
- Corrao, G.; Bagnardi, V.; Zambon, A.; La Vecchia, C. A meta-analysis of alcohol consumption and the risk of 15 diseases. Prev. Med. 2004, 38, 613–619. [Google Scholar] [CrossRef]
- Lieber, C.S. Alcoholic liver disease: New insights in pathogenesis lead to new treatments. J. Hepatol. 2000, 32, 113–128. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- Altamirano, J.; Bataller, R. Alcoholic liver disease: Pathogenesis and new targets for therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.M.; Reinus, J.F. Prevalence and natural history of alcoholic liver disease. Clin. Liver Dis. 2012, 16, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017, 38, 147–161. [Google Scholar]
- Askgaard, G.; Grønbæk, M.; Kjær, M.S.; Tjønneland, A.; Tolstrup, J.S. Alcohol drinking pattern and risk of alcoholic liver cirrhosis: A prospective cohort study. J. Hepatol. 2015, 62, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Hadland, S.E.; Xuan, Z.; Blanchette, J.G.; Heeren, T.C.; Swahn, M.H.; Naimi, T.S. Alcohol Policies and Alcoholic Cirrhosis Mortality in the United States. Prev. Chronic Dis. 2015, 12, E177. [Google Scholar] [CrossRef]
- Sims, O.T.; Pollio, D.E.; Hong, B.A.; Jain, M.K.; Brown, G.R.; North, C.S. An assessment of concurrent drug and alcohol use among patients seeking treatment for hepatitis C. Ann. Clin. Psychiatry 2016, 28, 31–36. [Google Scholar]
- Dolganiuc, A. Alcohol and Viral Hepatitis: Role of Lipid Rafts. Alcohol Res. 2015, 37, 299–309. [Google Scholar]
- Addolorato, G.; Mirijello, A.; Leggio, L.; Ferrulli, A.; Landolfi, R. Management of alcohol dependence in patients with liver disease. CNS Drugs 2013, 27, 287–299. [Google Scholar] [CrossRef]
- Anantharaju, A.; Van Thiel, D.H. Liver transplantation for alcoholic liver disease. Alcohol Res. Health 2003, 27, 257–268. [Google Scholar]
- Singal, A.K.; Bataller, R.; Ahn, J.; Kamath, P.S.; Shah, V.H. ACG Clinical Guideline: Alcoholic Liver Disease. Am. J. Gastroenterol. 2018, 113, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; DiMartini, A.; Feng, S.; Brown, R., Jr.; Fallon, M. Evaluation for liver transplantation in adults: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Hepatology 2014, 59, 1144–1165. [Google Scholar] [CrossRef] [PubMed]
- Vuittonet, C.L.; Halse, M.; Leggio, L.; Fricchione, S.B.; Brickley, M.; Haass-Koffler, C.L.; Tavares, T.; Swift, R.M.; Kenna, G.A. Pharmacotherapy for alcoholic patients with alcoholic liver disease. Am. J. Health Syst. Pharm. 2014, 71, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Holford, N.H. Clinical pharmacokinetics of ethanol. Clin. Pharm. 1987, 13, 273–292. [Google Scholar] [CrossRef]
- Lieber, C.S. Metabolism of alcohol. Clin. Liver Dis. 2005, 9, 1–35. [Google Scholar] [CrossRef]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar]
- Lieber, C.S. Ethanol metabolism, cirrhosis and alcoholism. Clin. Chim. Acta. 1997, 257, 59–84. [Google Scholar] [CrossRef]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef]
- Leung, T.M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef]
- Heier, C.; Xie, H.; Zimmermann, R. Nonoxidative ethanol metabolism in humans-from biomarkers to bioactive lipids. IUBMB Life 2016, 68, 916–923. [Google Scholar] [CrossRef]
- Maenhout, T.M.; De Buyzere, M.L.; Delanghe, J.R. Non-oxidative ethanol metabolites as a measure of alcohol intake. Clin. Chim. Acta. 2013, 415, 322–329. [Google Scholar] [CrossRef]
- Laposata, E.A.; Lange, L.G. Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 1986, 231, 497–499. [Google Scholar] [CrossRef]
- Lieber, C.S. Hepatic and metabolic effects of ethanol: Pathogenesis and prevention. Ann. Med. 1994, 26, 325–330. [Google Scholar] [CrossRef]
- Lieber, C.S. Mechanism of ethanol induced hepatic injury. Pharmacol. Ther. 1990, 46, 1–41. [Google Scholar] [CrossRef]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef]
- Tuma, D.J.; Casey, C.A. Dangerous byproducts of alcohol breakdown--focus on adducts. Alcohol Res. Health 2003, 27, 285–290. [Google Scholar]
- Israel, Y.; Hurwitz, E.; Niemelä, O.; Arnon, R. Monoclonal and polyclonal antibodies against acetaldehyde-containing epitopes in acetaldehyde-protein adducts. Proc. Natl. Acad. Sci. USA 1986, 83, 7923–7927. [Google Scholar] [CrossRef] [PubMed]
- Niemelä, O.; Parkkila, S.; Pasanen, M.; Iimuro, Y.; Bradford, B.; Thurman, R.G. Early alcoholic liver injury: Formation of protein adducts with acetaldehyde and lipid peroxidation products, and expression of CYP2E1 and CYP3A. Alcohol Clin. Exp. Res. 1998, 22, 2118–2124. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol Res. Health 2003, 27, 277–284. [Google Scholar]
- Holstege, A.; Bedossa, P.; Poynard, T.; Kollinger, M.; Chaput, J.C.; Houglum, K.; Chojkier, M. Acetaldehyde-modified epitopes in liver biopsy specimens of alcoholic and nonalcoholic patients: Localization and association with progression of liver fibrosis. Hepatology 1994, 19, 367–374. [Google Scholar] [CrossRef]
- Israel, Y.; Orrego, H.; Carmichael, F.J. Acetate-mediated effects of ethanol. Alcohol Clin. Exp. Res. 1994, 18, 144–148. [Google Scholar] [CrossRef]
- Seth, D.; Haber, P.S.; Syn, W.K.; Diehl, A.M.; Day, C.P. Pathogenesis of alcohol-induced liver disease: Classical concepts and recent advances. J. Gastroenterol. Hepatol. 2011, 26, 1089–1105. [Google Scholar] [CrossRef]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: A clear and present danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J. Oxidative and Non-Oxidative Metabolomics of Ethanol. Curr. Drug Metab. 2016, 17, 327–335. [Google Scholar] [CrossRef]
- Jeon, S.; Carr, R. Alcohol effects on hepatic lipid metabolism. J. Lipid Res. 2020, 61, 470–479. [Google Scholar] [CrossRef]
- Wei, X.; Shi, X.; Zhong, W.; Zhao, Y.; Tang, Y.; Sun, W.; Yin, X.; Bogdanov, B.; Kim, S.; McClain, C.; et al. Chronic alcohol exposure disturbs lipid homeostasis at the adipose tissue-liver axis in mice: Analysis of triacylglycerols using high-resolution mass spectrometry in combination with in vivo metabolite deuterium labeling. PLoS ONE 2013, 8, e55382. [Google Scholar] [CrossRef]
- Doege, H.; Baillie, R.A.; Ortegon, A.M.; Tsang, B.; Wu, Q.; Punreddy, S.; Hirsch, D.; Watson, N.; Gimeno, R.E.; Stahl, A. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: Alterations in hepatic lipid homeostasis. Gastroenterology 2006, 130, 1245–1258. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar] [CrossRef]
- Marmier, S.; Dentin, R.; Daujat-Chavanieu, M.; Guillou, H.; Bertrand-Michel, J.; Gerbal-Chaloin, S.; Girard, J.; Lotersztajn, S.; Postic, C. Novel role for carbohydrate responsive element binding protein in the control of ethanol metabolism and susceptibility to binge drinking. Hepatology 2015, 62, 1086–1100. [Google Scholar] [CrossRef]
- Bi, L.; Jiang, Z.; Zhou, J. The role of lipin-1 in the pathogenesis of alcoholic fatty liver. Alcohol Alcohol. 2015, 50, 146–151. [Google Scholar] [CrossRef]
- You, M.; Arteel, G.E. Effect of ethanol on lipid metabolism. J. Hepatol. 2019, 70, 237–248. [Google Scholar] [CrossRef]
- Yin, H.; Hu, M.; Liang, X.; Ajmo, J.M.; Li, X.; Bataller, R.; Odena, G.; Stevens, S.M., Jr.; You, M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014, 146, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Zhang, R.; Shen, Z.; Flatow, L.; You, M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J. Biol. Chem. 2012, 287, 9817–9826. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J. Biol. Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef] [PubMed]
- Souza-Mello, V. Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease. World J. Hepatol. 2015, 7, 1012–1019. [Google Scholar] [CrossRef]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef]
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B.; et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Basuroy, S.; Sheth, P.; Mansbach, C.M.; Rao, R.K. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: Protection by EGF and L-glutamine. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G367–G375. [Google Scholar] [CrossRef]
- Rao, R.K. Acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Alcohol Clin. Exp. Res. 1998, 22, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Banan, A.; Forsyth, C.B.; Fields, J.Z.; Lau, C.K.; Zhang, L.J.; Keshavarzian, A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol Clin. Exp. Res. 2008, 32, 355–364. [Google Scholar] [CrossRef]
- Duddempudi, A.T. Immunology in alcoholic liver disease. Clin. Liver Dis. 2012, 16, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef]
- Lee, J.Y.; Ye, J.; Gao, Z.; Youn, H.S.; Lee, W.H.; Zhao, L.; Sizemore, N.; Hwang, D.H. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J. Biol. Chem. 2003, 278, 37041–37051. [Google Scholar] [CrossRef]
- Jia, L.; Chang, X.; Qian, S.; Liu, C.; Lord, C.C.; Ahmed, N.; Lee, C.E.; Lee, S.; Gautron, L.; Mitchell, M.C.; et al. Hepatocyte toll-like receptor 4 deficiency protects against alcohol-induced fatty liver disease. Mol. Metab. 2018, 14, 121–129. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Liu, X.Q.; Zhang, C.; He, W.; Wang, H.; Chen, Y.H.; Liu, X.J.; Chen, X.; Xu, D.X. Tlr4-mutant mice are resistant to acute alcohol-induced sterol-regulatory element binding protein activation and hepatic lipid accumulation. Sci. Rep. 2016, 6, 33513. [Google Scholar] [CrossRef]
- Gao, B.; Ahmad, M.F.; Nagy, L.E.; Tsukamoto, H. Inflammatory pathways in alcoholic steatohepatitis. J. Hepatol. 2019, 70, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Gao, B.; Zakhari, S.; Nagy, L.E. Inflammation in alcoholic liver disease. Annu. Rev. Nutr. 2012, 32, 343–368. [Google Scholar] [CrossRef]
- Shen, Z.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Le, L.; Murr, M.M.; Peng, Y.; You, M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1047–G1053. [Google Scholar] [CrossRef]
- Ramaiah, S.K.; Jaeschke, H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol. Pathol. 2007, 35, 757–766. [Google Scholar] [CrossRef]
- Albano, E.; Vidali, M. Immune mechanisms in alcoholic liver disease. Genes Nutr. 2010, 5, 141–147. [Google Scholar] [CrossRef]
- D’Souza, A.J.; Desai, S.D.; Rudner, X.L.; Kelly, M.N.; Ruan, S.; Shellito, J.E. Suppression of the macrophage proteasome by ethanol impairs MHC class I antigen processing and presentation. PLoS ONE 2013, 8, e56890. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eken, A.; Ortiz, V.; Wands, J.R. Ethanol inhibits antigen presentation by dendritic cells. Clin. Vaccine Immunol. 2011, 18, 1157–1166. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pascual, M.; Fernández-Lizarbe, S.; Guerri, C. Role of TLR4 in ethanol effects on innate and adaptive immune responses in peritoneal macrophages. Immunol. Cell Biol. 2011, 89, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Barreyro, F.J.; Isomoto, H.; Bronk, S.F.; Gores, G.J. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 2007, 56, 1124–1131. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef]
- Sergent, O.; Pereira, M.; Belhomme, C.; Chevanne, M.; Huc, L.; Lagadic-Gossmann, D. Role for membrane fluidity in ethanol-induced oxidative stress of primary rat hepatocytes. J. Pharmacol. Exp. Ther. 2005, 313, 104–111. [Google Scholar] [CrossRef]
- Brenner, D.A. Molecular pathogenesis of liver fibrosis. Trans. Am. Clin. Climatol. Assoc. 2009, 120, 361–368. [Google Scholar]
- Kwon, H.J.; Won, Y.S.; Park, O.; Chang, B.; Duryee, M.J.; Thiele, G.E.; Matsumoto, A.; Singh, S.; Abdelmegeed, M.A.; Song, B.J.; et al. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology 2014, 60, 146–157. [Google Scholar] [CrossRef]
- Purohit, V.; Brenner, D.A. Mechanisms of alcohol-induced hepatic fibrosis: A summary of the Ron Thurman Symposium. Hepatology 2006, 43, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Ridolfi, F.; Di Sario, A.; Saccomanno, S.; Bendia, E.; Benedetti, A.; Greenwel, P. Intracellular signaling pathways involved in acetaldehyde-induced collagen and fibronectin gene expression in human hepatic stellate cells. Hepatology 2001, 33, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. Acetaldehyde stimulates the activation of latent transforming growth factor-beta1 and induces expression of the type II receptor of the cytokine in rat cultured hepatic stellate cells. Biochem. J. 2002, 368, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Inagaki, Y.; Rincon-Sanchez, A.R.; Else, C.; Saccomanno, S.; Benedetti, A.; Ramirez, F.; Rojkind, M. Early response of alpha2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology 2005, 42, 343–352. [Google Scholar] [CrossRef]
- Casini, A.; Cunningham, M.; Rojkind, M.; Lieber, C.S. Acetaldehyde increases procollagen type I and fibronectin gene transcription in cultured rat fat-storing cells through a protein synthesis-dependent mechanism. Hepatology 1991, 13, 758–765. [Google Scholar] [CrossRef]
- Quiroz, S.C.; Bucio, L.; Souza, V.; Hernández, E.; González, E.; Gómez-Quiroz, L.; Kershenobich, D.; Vargas-Vorackova, F.; Gutiérrez-Ruiz, M.C. Effect of endotoxin pretreatment on hepatic stellate cell response to ethanol and acetaldehyde. J. Gastroenterol. Hepatol. 2001, 16, 1267–1273. [Google Scholar] [CrossRef]
- Chen, M.; Liu, J.; Yang, W.; Ling, W. Lipopolysaccharide mediates hepatic stellate cell activation by regulating autophagy and retinoic acid signaling. Autophagy 2017, 13, 1813–1827. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Arellanes-Robledo, J.; Reyes-Gordillo, K.; Shah, R.; Domínguez-Rosales, J.A.; Hernández-Nazara, Z.H.; Ramirez, F.; Rojkind, M.; Lakshman, M.R. Fibrogenic actions of acetaldehyde are β-catenin dependent but Wingless independent: A critical role of nucleoredoxin and reactive oxygen species in human hepatic stellate cells. Free Radic. Biol. Med. 2013, 65, 1487–1496. [Google Scholar] [CrossRef]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology 2002, 35, 62–73. [Google Scholar] [CrossRef]
- Greenwel, P.; Domínguez-Rosales, J.A.; Mavi, G.; Rivas-Estilla, A.M.; Rojkind, M. Hydrogen peroxide: A link between acetaldehyde-elicited alpha1(I) collagen gene up-regulation and oxidative stress in mouse hepatic stellate cells. Hepatology 2000, 31, 109–116. [Google Scholar] [CrossRef]
- Ceni, E.; Crabb, D.W.; Foschi, M.; Mello, T.; Tarocchi, M.; Patussi, V.; Moraldi, L.; Moretti, R.; Milani, S.; Surrenti, C.; et al. Acetaldehyde inhibits PPARgamma via H2O2-mediated c-Abl activation in human hepatic stellate cells. Gastroenterology 2006, 131, 1235–1252. [Google Scholar] [CrossRef] [PubMed]
- Viña, J.; Estrela, J.M.; Guerri, C.; Romero, F.J. Effect of ethanol on glutathione concentration in isolated hepatocytes. Biochem. J. 1980, 188, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1428. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.M.; Yang, J.H.; Ki, S.H. Role of the Nrf2-ARE pathway in liver diseases. Oxid. Med. Cell. Longev. 2013, 2013, 763257. [Google Scholar] [CrossRef]
- Jeong, W.I.; Gao, B. Innate immunity and alcoholic liver fibrosis. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. 1), S112–S118. [Google Scholar] [CrossRef]
- Ni, Y.H.; Huo, L.J.; Li, T.T. Antioxidant axis Nrf2-keap1-ARE in inhibition of alcoholic liver fibrosis by IL-22. World J. Gastroenterol. 2017, 23, 2002–2011. [Google Scholar] [CrossRef]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef]
- Xiang, X.; Hwang, S.; Feng, D.; Shah, V.H.; Gao, B. Interleukin-22 in alcoholic hepatitis and beyond. Hepatol. Int. 2020, 14, 667–676. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, S.; Huang, R.; Wei, L.; Tan, S.; Liang, S.; Tian, Y.; Wu, X.; Lu, Z.; Huang, Q. Helenalin attenuates alcohol-induced hepatic fibrosis by enhancing ethanol metabolism, inhibiting oxidative stress and suppressing HSC activation. Fitoterapia 2014, 95, 203–213. [Google Scholar] [CrossRef] [PubMed]
- El-Sisi, A.E.E.; Sokar, S.S.; Shebl, A.M.; Mohamed, D.Z. Antifibrotic effect of diethylcarbamazine combined with hesperidin against ethanol induced liver fibrosis in rats. Biomed. Pharmacother. 2017, 89, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ying, X.; Sun, W.; Zhu, H.; Jiang, X.; Chen, B. The therapeutic effect of fraxetin on ethanol-induced hepatic fibrosis by enhancing ethanol metabolism, inhibiting oxidative stress and modulating inflammatory mediators in rats. Int. Immunopharmacol. 2018, 56, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects. Biomedicines 2018, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Takaki, A. Alcohol and hepatocellular carcinoma. BMJ Open Gastroenterol. 2019, 6, e000260. [Google Scholar] [CrossRef] [PubMed]
- Ganne-Carrié, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef]
- Wang, M.; McIntee, E.J.; Cheng, G.; Shi, Y.; Villalta, P.W.; Hecht, S.S. Identification of DNA adducts of acetaldehyde. Chem. Res. Toxicol. 2000, 13, 1149–1157. [Google Scholar] [CrossRef]
- Heymann, H.M.; Gardner, A.M.; Gross, E.R. Aldehyde-Induced DNA and Protein Adducts as Biomarker Tools for Alcohol Use Disorder. Trends Mol. Med. 2018, 24, 144–155. [Google Scholar] [CrossRef]
- Brooks, P.J. DNA damage, DNA repair, and alcohol toxicity—A review. Alcohol Clin. Exp. Res. 1997, 21, 1073–1082. [Google Scholar] [PubMed]
- Matsuda, T.; Terashima, I.; Matsumoto, Y.; Yabushita, H.; Matsui, S.; Shibutani, S. Effective utilization of N2-ethyl-2′-deoxyguanosine triphosphate during DNA synthesis catalyzed by mammalian replicative DNA polymerases. Biochemistry 1999, 38, 929–935. [Google Scholar] [CrossRef]
- Collier, J.D.; Bassendine, M.F.; Burt, A.D.; Major, G.N. Characterisation of the DNA repair enzyme for O(6)-methylguanine in cirrhosis. J. Hepatol. 1996, 25, 158–165. [Google Scholar] [CrossRef]
- He, S.M.; Lambert, B. Acetaldehyde-induced mutation at the hprt locus in human lymphocytes in vitro. Environ. Mol. Mutagen. 1990, 16, 57–63. [Google Scholar] [CrossRef]
- Druesne-Pecollo, N.; Tehard, B.; Mallet, Y.; Gerber, M.; Norat, T.; Hercberg, S.; Latino-Martel, P. Alcohol and genetic polymorphisms: Effect on risk of alcohol-related cancer. Lancet Oncol. 2009, 10, 173–180. [Google Scholar] [CrossRef]
- Munaka, M.; Kohshi, K.; Kawamoto, T.; Takasawa, S.; Nagata, N.; Itoh, H.; Oda, S.; Katoh, T. Genetic polymorphisms of tobacco- and alcohol-related metabolizing enzymes and the risk of hepatocellular carcinoma. J. Cancer. Res. Clin. Oncol. 2003, 129, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Hara, M.; Higaki, Y.; Ichiba, M.; Horita, M.; Mizuta, T.; Eguchi, Y.; Yasutake, T.; Ozaki, I.; Yamamoto, K.; et al. Influence of alcohol consumption and gene polymorphisms of ADH2 and ALDH2 on hepatocellular carcinoma in a Japanese population. Int. J. Cancer 2006, 118, 1501–1507. [Google Scholar] [CrossRef]
- Huang, P.H.; Hu, C.C.; Chien, C.H.; Chen, L.W.; Chien, R.N.; Lin, Y.S.; Chao, M.; Lin, C.L.; Yeh, C.T. The Defective Allele of Aldehyde Dehydrogenase 2 Gene is Associated with Favorable Postoperative Prognosis in Hepatocellular Carcinoma. J. Cancer 2019, 10, 5735–5743. [Google Scholar] [CrossRef] [PubMed]
- Seo, W.; Gao, Y.; He, Y.; Sun, J.; Xu, H.; Feng, D.; Park, S.H.; Cho, Y.E.; Guillot, A.; Ren, T.; et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J. Hepatol. 2019, 71, 1000–1011. [Google Scholar] [CrossRef]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, J.L.; Yu, K.K.; Xu, M.; Xu, Y.Z.; Chen, L.; Lu, Y.M.; Fang, H.S.; Wang, X.Y.; Hu, Z.Q.; et al. Activation of the NF-κB pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol. Cancer 2015, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E. Animal models of alcoholic liver disease. Dig. Dis. 2010, 28, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Brandon-Warner, E.; Schrum, L.W.; Schmidt, C.M.; McKillop, I.H. Rodent models of alcoholic liver disease: Of mice and men. Alcohol 2012, 46, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.S.; Duley, J.A.; Algar, E.M.; Mather, P.B.; Rout, U.K. Biochemical and genetic studies on enzymes of alcohol metabolism: The mouse as a model organism for human studies. Alcohol Alcohol. 1986, 21, 41–56. [Google Scholar]
- Gao, B.; Xu, M.J.; Bertola, A.; Wang, H.; Zhou, Z.; Liangpunsakul, S. Animal Models of Alcoholic Liver Disease: Pathogenesis and Clinical Relevance. Gene Expr. 2017, 17, 173–186. [Google Scholar] [CrossRef]
- Dilley, J.E.; Nicholson, E.R.; Fischer, S.M.; Zimmer, R.; Froehlich, J.C. Alcohol Drinking and Blood Alcohol Concentration Revisited. Alcohol Clin. Exp. Res. 2018, 42, 260–269. [Google Scholar] [CrossRef]
- Lamas-Paz, A.; Hao, F.; Nelson, L.J.; Vázquez, M.T.; Canals, S.; Gómez Del Moral, M.; Martínez-Naves, E.; Nevzorova, Y.A.; Cubero, F.J. Alcoholic liver disease: Utility of animal models. World J. Gastroenterol. 2018, 24, 5063–5075. [Google Scholar] [CrossRef]
- DeCarli, L.M.; Lieber, C.S. Fatty liver in the rat after prolonged intake of ethanol with a nutritionally adequate new liquid diet. J. Nutr. 1967, 91, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. The feeding of ethanol in liquid diets. Alcohol Clin. Exp. Res. 1986, 10, 550–553. [Google Scholar] [CrossRef]
- Wilkin, R.J.; Lalor, P.F.; Parker, R.; Newsome, P.N. Murine Models of Acute Alcoholic Hepatitis and Their Relevance to Human Disease. Am. J. Pathol. 2016, 186, 748–760. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Reidelberger, R.D.; French, S.W.; Largman, C. Long-term cannulation model for blood sampling and intragastric infusion in the rat. Am. J. Physiol. 1984, 247, R595–R599. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Lazaro, R.; Wang, P.Y.; Higashiyama, R.; Machida, K.; Tsukamoto, H. Mouse intragastric infusion (iG) model. Nat. Protoc. 2012, 7, 771–781. [Google Scholar] [CrossRef]
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, B.P.; Stice, C.P.; Liu, C.; Greenberg, A.S.; Ausman, L.M.; Wang, X.D. Inhibition of diethylnitrosamine-initiated alcohol-promoted hepatic inflammation and precancerous lesions by flavonoid luteolin is associated with increased sirtuin 1 activity in mice. Hepatobiliary Surg. Nutr. 2015, 4, 124–134. [Google Scholar] [CrossRef]
- Muñoz, N.M.; Katz, L.H.; Shina, J.H.; Gi, Y.J.; Menon, V.K.; Gagea, M.; Rashid, A.; Chen, J.; Mishra, L. Generation of a mouse model of T-cell lymphoma based on chronic LPS challenge and TGF-β signaling disruption. Genes Cancer 2014, 5, 348–352. [Google Scholar] [CrossRef]
- Karaca, G.; Xie, G.; Moylan, C.; Swiderska-Syn, M.; Guy, C.D.; Krüger, L.; Machado, M.V.; Choi, S.S.; Michelotti, G.A.; Burkly, L.C.; et al. Role of Fn14 in acute alcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G325–G334. [Google Scholar] [CrossRef] [PubMed]
- Takase, S.; Yasuhara, M.; Takada, A.; Ueshima, Y. Changes in blood acetaldehyde levels after ethanol administration in alcoholics. Alcohol 1990, 7, 37–41. [Google Scholar] [CrossRef]
- Korsten, M.A.; Matsuzaki, S.; Feinman, L.; Lieber, C.S. High blood acetaldehyde levels after ethanol administration. Difference between alcoholic and nonalcoholic subjects. N. Engl. J. Med. 1975, 292, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Lindros, K.O.; Stowell, A.; Pikkarainen, P.; Salaspuro, M. Elevated blood acetaldehyde in alcoholics with accelerated ethanol elimination. Pharmacol. Biochem. Behav. 1980, 13, 119–124. [Google Scholar] [CrossRef]
- Nuutinen, H.; Lindros, K.O.; Salaspuro, M. Determinants of blood acetaldehyde level during ethanol oxidation in chronic alcoholics. Alcohol Clin. Exp. Res. 1983, 7, 163–168. [Google Scholar] [CrossRef]
- Palmer, K.R.; Jenkins, W.J. Impaired acetaldehyde oxidation in alcoholics. Gut 1982, 23, 729–733. [Google Scholar] [CrossRef]
- Isse, T.; Matsuno, K.; Oyama, T.; Kitagawa, K.; Kawamoto, T. Aldehyde dehydrogenase 2 gene targeting mouse lacking enzyme activity shows high acetaldehyde level in blood, brain, and liver after ethanol gavages. Alcohol Clin. Exp. Res. 2005, 29, 1959–1964. [Google Scholar] [CrossRef]
- Chaudhry, K.K.; Samak, G.; Shukla, P.K.; Mir, H.; Gangwar, R.; Manda, B.; Isse, T.; Kawamoto, T.; Salaspuro, M.; Kaihovaara, P.; et al. ALDH2 Deficiency Promotes Ethanol-Induced Gut Barrier Dysfunction and Fatty Liver in Mice. Alcohol Clin. Exp. Res. 2015, 39, 1465–1475. [Google Scholar] [CrossRef]
- Deltour, L.; Foglio, M.H.; Duester, G. Metabolic deficiencies in alcohol dehydrogenase Adh1, Adh3, and Adh4 null mutant mice. Overlapping roles of Adh1 and Adh4 in ethanol clearance and metabolism of retinol to retinoic acid. J. Biol. Chem. 1999, 274, 16796–16801. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wu, D.; Wang, X.; Ward, S.C.; Cederbaum, A.I. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radic. Biol. Med. 2010, 49, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, B.; No da, Y.; Lee, G.; Lee, S.R.; Oh, H.; Lee, S.H. A 3D alcoholic liver disease model on a chip. Integr. Biol. 2016, 8, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, X.; Tan, Z.; Su, Y.; Liu, J.; Chang, M.; Yan, F.; Chen, J.; Chen, T.; Li, C.; et al. Human ESC-derived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury. Cell Res. 2019, 29, 1009–1026. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Models | Administration | Characteristics | Feasibility |
|---|---|---|---|
| Ad libitum alcohol-drinking water | Oral alcohol consumption by drinking water | Low BAL; minimal elevation of ALT; mild steatosis | Easy to perform |
| Ad libitum liquid diet (Lieber–DeCarli diet) | Oral alcohol consumption with alcohol-containing liquid diet formula but with no other food or drink | Variable elevation range of ALT; marked steatosis; mild inflammation | Easy to perform |
| Intragastric infusion (The Tsukamoto-French model) | Direct enteral feeding through a surgically implanted intragastric cannula | High BAL; marked elevation of ALT; severe steatosis; mild inflammation; fibrosis | Difficult to perform |
| Chronic and binge alcohol feeding (Gao-binge model) | A single or repeated intragastric gavage of alcohol following chronic feeding with the Lieber–DeCarli liquid diet | High BAL; marked elevation of ALT; steatosis; neutrophil infiltration; necrosis; no fibrosis | Easy to perform |
| Lieber–DeCarli diet + other hepatotoxins (Second hit model) | Addition of hepatotoxins such as DEN, LPS or CCl4 during the chronic feeding phase of the Lieber–DeCarli liquid diet | Marked elevation of ALT; high mortality rate; significant liver fibrosis | Easy to perform |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyun, J.; Han, J.; Lee, C.; Yoon, M.; Jung, Y. Pathophysiological Aspects of Alcohol Metabolism in the Liver. Int. J. Mol. Sci. 2021, 22, 5717. https://doi.org/10.3390/ijms22115717
Hyun J, Han J, Lee C, Yoon M, Jung Y. Pathophysiological Aspects of Alcohol Metabolism in the Liver. International Journal of Molecular Sciences. 2021; 22(11):5717. https://doi.org/10.3390/ijms22115717
Chicago/Turabian StyleHyun, Jeongeun, Jinsol Han, Chanbin Lee, Myunghee Yoon, and Youngmi Jung. 2021. "Pathophysiological Aspects of Alcohol Metabolism in the Liver" International Journal of Molecular Sciences 22, no. 11: 5717. https://doi.org/10.3390/ijms22115717
APA StyleHyun, J., Han, J., Lee, C., Yoon, M., & Jung, Y. (2021). Pathophysiological Aspects of Alcohol Metabolism in the Liver. International Journal of Molecular Sciences, 22(11), 5717. https://doi.org/10.3390/ijms22115717