
Understanding Lung Carcinogenesis from a Morphostatic Perspective: Prevention and Therapeutic Potential of Phytochemicals for Targeting Cancer Stem Cells



Abstract
1. Introduction
2. A Close Connection between Normal Lung Tissue Repair Mechanism and Carcinogenesis
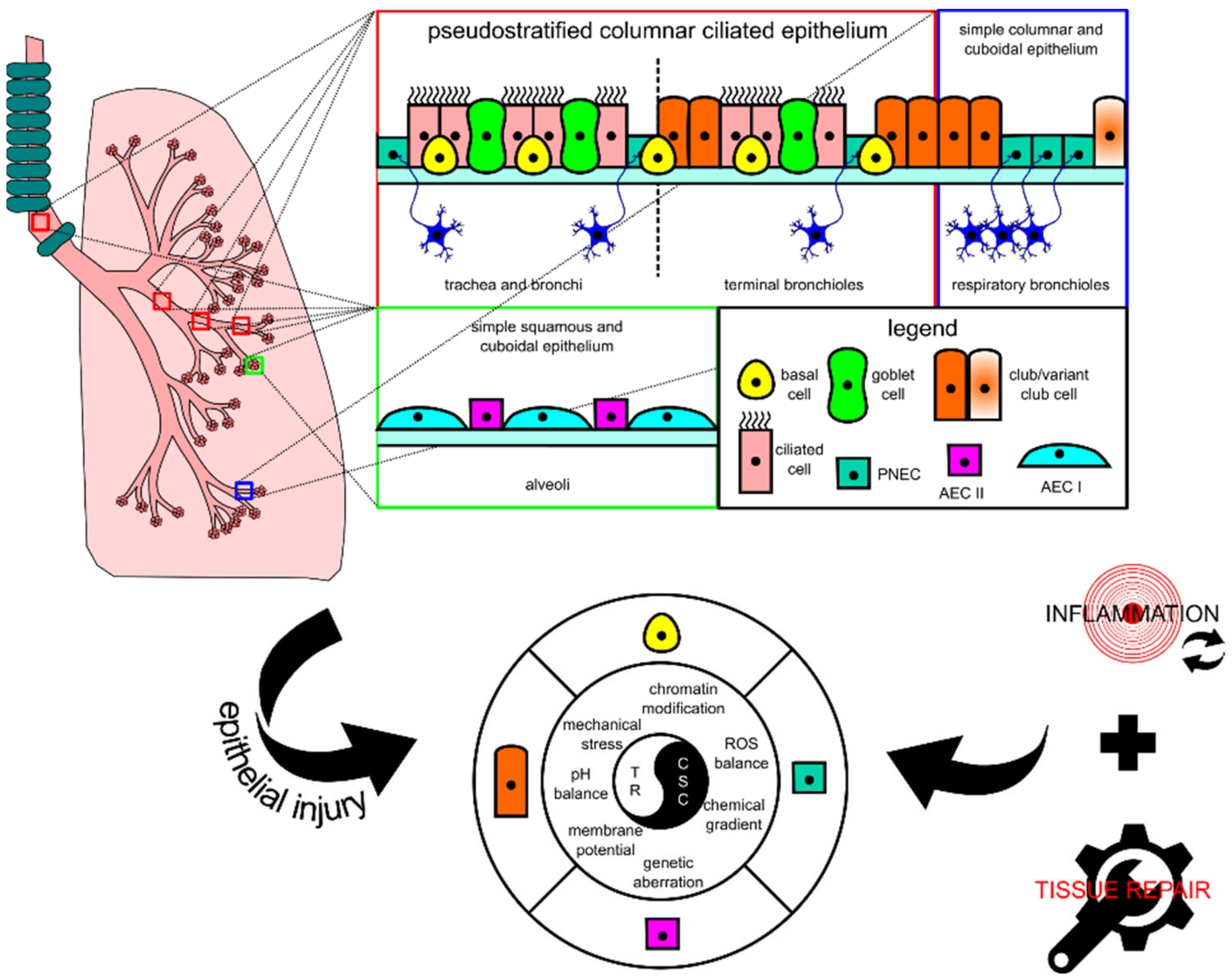
2.1. Lung Tissue, Progenitor Cell, Cancer Stem Cell, and Injury-Triggered Carcinogenesis
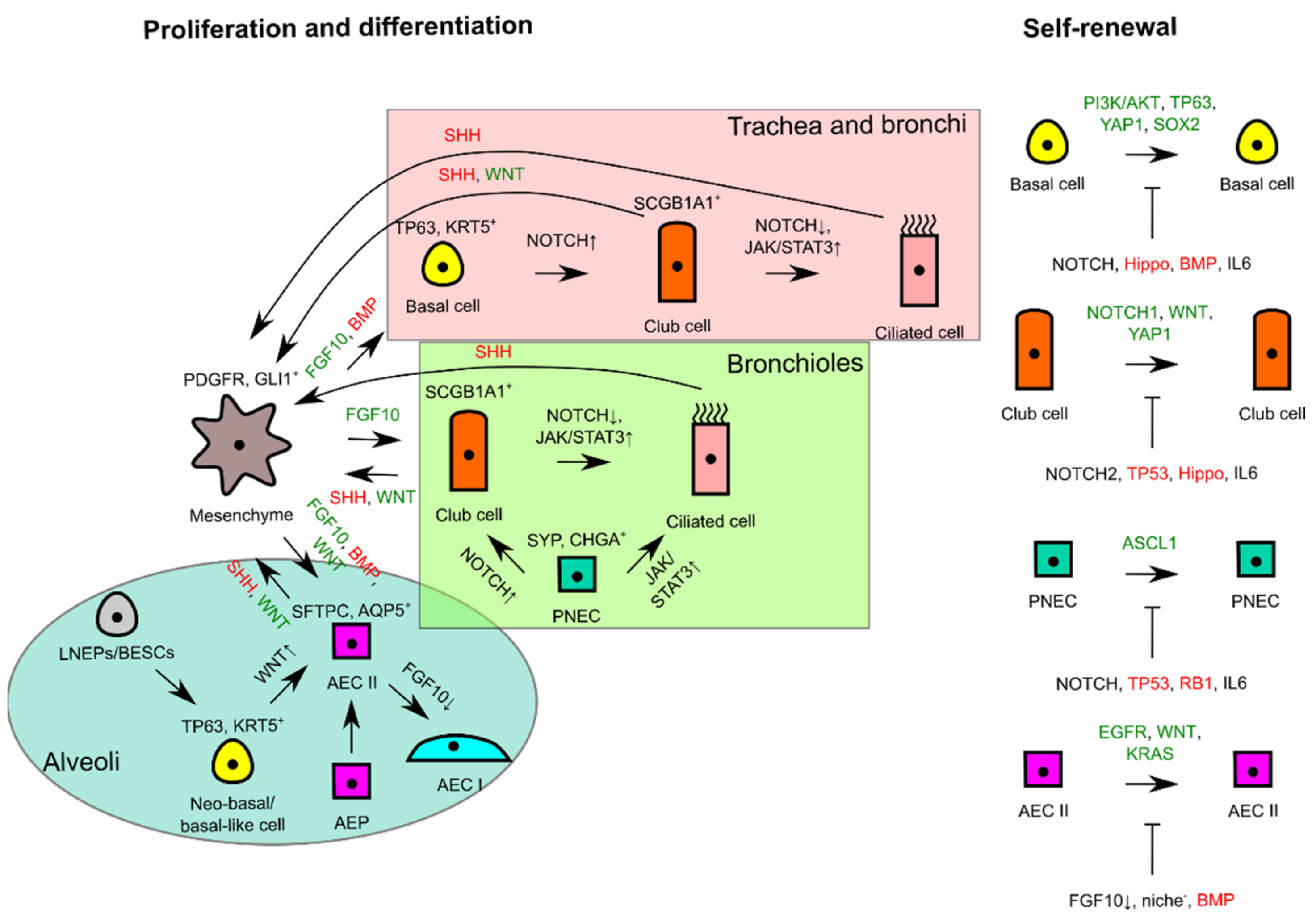
2.2. Normal Physiological Responses for Homeostasis and Injury Repair in Lung Tissue
2.2.1. Basal Cell
2.2.2. Club Cell
2.2.3. PNEC
2.2.4. AEC II
3. Dysregulations and Aberrations Supporting Lung Carcinogenesis
3.1. Molecular Aberrations Driving Tumor Initiation and Lung CSC Maintenance
3.2. Dysregulations of Signals from Microenvironment during Lung Tissue Repair
3.3. Cellular Reprogramming Involving Transitioning to Mesenchymal Phenotype
4. Current Use of Phytochemical Compounds in Treating Lung Cancer
5. The Potential Use of Phytochemical Compounds for Prevention and Targeting CSCs in Lung Cancer
5.1. Curcumin
5.2. Resveratrol
5.3. Quercetin
5.4. Epigallocatechin-3-Gallate
5.5. Luteolin
5.6. Sulforaphane
5.7. Berberine
5.8. Genistein
5.9. Capsaicin
6. Discussion and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kassa, M.; Grace, J. The Global Burden and Perspectives on Non-Communicable Diseases (NCDs) and the Prevention, Data Availability and Systems Approach of NCDs in Low-resource Countries. In Public Health in Developing Countries—Challenges and Opportunities; IntechOpen: London, UK, 2020. [Google Scholar]
- Mikkelsen, B.; Williams, J.; Rakovac, I.; Wickramasinghe, K.; Hennis, A.; Shin, H.-R.; Farmer, M.; Weber, M.; Berdzuli, N.; Borges, C.; et al. Life course approach to prevention and control of non-communicable diseases. BMJ 2019, 364, l257. [Google Scholar] [CrossRef] [PubMed]
- Powell, H.S.; Greenberg, D.L. Screening for unhealthy diet and exercise habits: The electronic health record and a healthier population. Prev. Med. Rep. 2019, 14, 100816. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Hunter, D.J.; Spiegelman, D.; Albanes, D.; Beeson, W.L.; Brandt, P.A.V.D.; Colditz, G.A.; Feskanich, D.; Folsom, A.R.; Fraser, G.E.; et al. Intakes of vitamins A, C and E and folate and multivitamins and lung cancer: A pooled analysis of 8 prospective studies. Int. J. Cancer 2005, 118, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Shareck, M.; Rousseau, M.-C.; Koushik, A.; Siemiatycki, J.; Parent, M.-E. Inverse Association between Dietary Intake of Selected Carotenoids and Vitamin C and Risk of Lung Cancer. Front. Oncol. 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.W.; Baird, P.; Davis, R.H., Jr.; Ferreri, S.; Knudtson, M.; Koraym, A.; Waters, V.; Williams, C.L. Health benefits of dietary fiber. Nutr. Rev. 2009, 67, 188–205. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Mukhtar, H. Dietary agents for prevention and treatment of lung cancer. Cancer Lett. 2015, 359, 155–164. [Google Scholar] [CrossRef]
- Heng, W.S.; Gosens, R.; Kruyt, F.A. Lung cancer stem cells: Origin, features, maintenance mechanisms and therapeutic targeting. Biochem. Pharmacol. 2019, 160, 121–133. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016, 372, 147–156. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef]
- Kotton, D.N.; Morrisey, E.E. Lung regeneration: Mechanisms, applications and emerging stem cell populations. Nat. Med. 2014, 20, 822–832. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Song, H.; Yao, E.; Lin, C.; Gacayan, R.; Chen, M.-H.; Chuang, P.-T. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 17531–17536. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nat. Cell Biol. 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Yuan, T.; Volckaert, T.; Redente, E.F.; Hopkins, S.; Klinkhammer, K.; Wasnick, R.; Chao, C.-M.; Yuan, J.; Zhang, J.-S.; Yao, C.; et al. FGF10-FGFR2B Signaling Generates Basal Cells and Drives Alveolar Epithelial Regeneration by Bronchial Epithelial Stem Cells after Lung Injury. Stem Cell Rep. 2019, 12, 1041–1055. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nat. Cell Biol. 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Levin, M. Morphogenetic fields in embryogenesis, regeneration, and cancer: Non-local control of complex patterning. Biosystems 2012, 109, 243–261. [Google Scholar] [CrossRef]
- Afify, S.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nat. Cell Biol. 2015, 526, 578–582. [Google Scholar] [CrossRef]
- Yuan, T.; Volckaert, T.; Chanda, D.; Thannickal, V.J.; De Langhe, S.P. Fgf10 Signaling in Lung Development, Homeostasis, Disease, and Repair After Injury. Front. Genet. 2018, 9, 9. [Google Scholar] [CrossRef]
- Watson, J.K.; Rulands, S.; Wilkinson, A.C.; Wuidart, A.; Ousset, M.; Van Keymeulen, A.; Göttgens, B.; Blanpain, C.; Simons, B.D.; Rawlins, E.L. Clonal Dynamics Reveal Two Distinct Populations of Basal Cells in Slow-Turnover Airway Epithelium. Cell Rep. 2015, 12, 90–101. [Google Scholar] [CrossRef]
- Ochieng, J.K.; Schilders, K.; Kool, H.; Munck, A.B.-D.; Kempen, M.B.-V.; Gontan, C.; Smits, R.; Grosveld, F.G.; Wijnen, R.M.; Tibboel, D.; et al. Sox2 Regulates the Emergence of Lung Basal Cells by Directly Activating the Transcription of Trp63. Am. J. Respir. Cell Mol. Biol. 2014, 51, 311–322. [Google Scholar] [CrossRef]
- Zhao, R.; Fallon, T.R.; Saladi, S.V.; Pardo-Saganta, A.; Villoria, J.; Mou, H.; Vinarsky, V.; Gonzalez-Celeiro, M.; Nunna, N.; Hariri, L.P.; et al. Yap Tunes Airway Epithelial Size and Architecture by Regulating the Identity, Maintenance, and Self-Renewal of Stem Cells. Dev. Cell 2014, 30, 151–165. [Google Scholar] [CrossRef]
- Balasooriya, G.I.; Goschorska, M.; Piddini, E.; Rawlins, E.L. FGFR2 is required for airway basal cell self-renewal and terminal differentiation. Development 2017, 144, 1600–1606. [Google Scholar] [CrossRef]
- Teixeira, V.H.; Nadarajan, P.A.; Graham, T.; Pipinikas, C.P.; Brown, J.M.; Falzon, M.; Nye, E.; Poulsom, R.; Lawrence, D.A.; Wright, N.; et al. Stochastic homeostasis in human airway epithelium is achieved by neutral competition of basal cell progenitors. eLife 2013, 2, e00966. [Google Scholar] [CrossRef]
- Mori, M.; Mahoney, J.E.; Stupnikov, M.R.; Paez-Cortez, J.R.; Szymaniak, A.D.; Varelas, X.; Herrick, D.B.; Schwob, J.; Zhang, H.; Cardoso, W.V. Notch3-Jagged signaling controls the pool of undifferentiated airway progenitors. Development 2015, 142, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Wang, Y.; Barak, L.S.; Bai, Y.; Randell, S.H.; Hogan, B.L.M. IL-6/STAT3 promotes regeneration of airway ciliated cells from basal stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E3641–E3649. [Google Scholar] [CrossRef]
- Tadokoro, T.; Gao, X.; Hong, C.C.; Hotten, D.; Hogan, B.L.M. BMP signaling and cellular dynamics during regeneration of airway epithelium from basal progenitors. Development 2016, 143, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Spella, M.; Lilis, I.; Pepe, M.A.; Chen, Y.; Armaka, M.; Lamort, A.-S.E.; Zazara, D.; Roumelioti, F.; Vreka, M.I.; Kanellakis, N.; et al. Club cells form lung adenocarcinomas and maintain the alveoli of adult mice. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, S.D.; Giangreco, A.; Power, J.H.; Stripp, B.R. Neuroepithelial Bodies of Pulmonary Airways Serve as a Reservoir of Progenitor Cells Capable of Epithelial Regeneration. Am. J. Pathol. 2000, 156, 269–278. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef]
- Tompkins, D.H.; Besnard, V.; Lange, A.W.; Wert, S.E.; Keiser, A.R.; Smith, A.N.; Lang, R.; Whitsett, J.A. Sox2 Is Required for Maintenance and Differentiation of Bronchiolar Clara, Ciliated, and Goblet Cells. PLoS ONE 2009, 4, e8248. [Google Scholar] [CrossRef]
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; De Langhe, S.P. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Investig. 2011, 121, 4409–4419. [Google Scholar] [CrossRef]
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The Role of Scgb1a1+ Clara Cells in the Long-Term Maintenance and Repair of Lung Airway, but Not Alveolar, Epithelium. Cell Stem Cell 2009, 4, 525–534. [Google Scholar] [CrossRef]
- Kiyokawa, H.; Morimoto, M. Notch signaling in the mammalian respiratory system, specifically the trachea and lungs, in development, homeostasis, regeneration, and disease. Dev. Growth Differ. 2020, 62, 67–79. [Google Scholar] [CrossRef]
- Zheng, D.; Soh, B.-S.; Yin, L.; Hu, G.; Chen, Q.; Choi, H.; Han, J.; Chow, V.T.K.; Chen, J. Differentiation of Club Cells to Alveolar Epithelial Cells In Vitro. Sci. Rep. 2017, 7, srep41661. [Google Scholar] [CrossRef]
- McConnell, A.M.; Yao, C.; Yeckes, A.R.; Wang, Y.; Selvaggio, A.S.; Tang, J.; Kirsch, D.G.; Stripp, B.R. p53 Regulates Progenitor Cell Quiescence and Differentiation in the Airway. Cell Rep. 2016, 17, 2173–2182. [Google Scholar] [CrossRef]
- Tata, P.R.; Mou, H.; Pardo-Saganta, A.; Zhao, R.; Prabhu, M.; Law, B.M.; Vinarsky, V.; Cho, J.L.; Breton, S.; Sahay, A.; et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nat. Cell Biol. 2013, 503, 218–223. [Google Scholar] [CrossRef]
- Lange, A.W.; Sridharan, A.; Xu, Y.; Stripp, B.R.; Perl, A.-K.; Whitsett, J.A. Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J. Mol. Cell Biol. 2014, 7, 35–47. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, S.; Dai, H.; Hu, X.; Li, C.; Xing, Y. The role of pulmonary mesenchymal cells in airway epithelium regeneration during injury repair. Stem Cell Res. Ther. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Xing, Y.; Li, A.; Borok, Z.; Li, C.; Minoo, P. NOTCH1 Is Required for Regeneration of Clara Cells During Repair of Airway Injury. Stem Cells 2012, 30, 946–955. [Google Scholar] [CrossRef]
- Yao, E.; Lin, C.; Wu, Q.; Zhang, K.; Song, H.; Chuang, P.-T. Notch Signaling Controls Transdifferentiation of Pulmonary Neuroendocrine Cells in Response to Lung Injury. Stem Cells 2018, 36, 377–391. [Google Scholar] [CrossRef]
- Ouadah, Y.; Rojas, E.R.; Riordan, D.P.; Capostagno, S.; Kuo, C.S.; Krasnow, M.A. Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch. Cell 2019, 179, 403–416.e23. [Google Scholar] [CrossRef]
- Ray, S.; Chiba, N.; Yao, C.; Guan, X.; McConnell, A.M.; Brockway, B.; Que, L.; McQualter, J.L.; Stripp, B.R. Rare SOX2 + Airway Progenitor Cells Generate KRT5 + Cells that Repopulate Damaged Alveolar Parenchyma following Influenza Virus Infection. Stem Cell Rep. 2016, 7, 817–825. [Google Scholar] [CrossRef]
- Yang, Y.; Riccio, P.; Schotsaert, M.; Mori, M.; Lu, J.; Lee, D.-K.; García-Sastre, A.; Xu, J.; Cardoso, W.V. Spatial-Temporal Lineage Restrictions of Embryonic p63+ Progenitors Establish Distinct Stem Cell Pools in Adult Airways. Dev. Cell 2018, 44, 752–761.e4. [Google Scholar] [CrossRef]
- Xi, Y.; Kim, T.; Brumwell, A.N.; Driver, I.H.; Wei, Y.; Tan, V.; Jackson, J.R.; Xu, J.; Lee, D.-K.; Gotts, J.E.; et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol. 2017, 19, 904–914. [Google Scholar] [CrossRef]
- Volckaert, T.; Yuan, T.; Chao, C.-M.; Bell, H.; Sitaula, A.; Szimmtenings, L.; El Agha, E.; Chanda, D.; Majka, S.; Bellusci, S.; et al. Fgf10-Hippo Epithelial-Mesenchymal Crosstalk Maintains and Recruits Lung Basal Stem Cells. Dev. Cell 2017, 43, 48–59.e5. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nat. Cell Biol. 2014, 507, 190–194. [Google Scholar] [CrossRef]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef]
- Chung, M.-I.; Bujnis, M.; Barkauskas, C.E.; Kobayashi, Y.; Hogan, B.L.M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development 2018, 145, 145. [Google Scholar] [CrossRef]
- Ferone, G.; Song, J.-Y.; Sutherland, K.D.; Bhaskaran, R.; Monkhorst, K.; Lambooij, J.-P.; Proost, N.; Gargiulo, G.; Berns, A. SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell 2016, 30, 519–532. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef]
- Xu, X.; Huang, L.; Futtner, C.; Schwab, B.; Rampersad, R.R.; Lu, Y.; Sporn, T.A.; Hogan, B.L.; Onaitis, M.W. The cell of origin and subtype of K-Ras-induced lung tumors are modified by Notch and Sox2. Genes Dev. 2014, 28, 1929–1939. [Google Scholar] [CrossRef]
- Kashima, J.; Kitadai, R.; Okuma, Y. Molecular and Morphological Profiling of Lung Cancer: A Foundation for “Next-Generation” Pathologists and Oncologists. Cancers 2019, 11, 599. [Google Scholar] [CrossRef]
- Gong, M.; Li, Y.; Ye, X.; Zhang, L.; Wang, Z.; Xu, X.; Shen, Y.; Zheng, C. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell Commun. Signal. 2020, 18, 1–11. [Google Scholar] [CrossRef]
- Ryan, S.-L.; Beard, S.; Barr, M.P.; Umezawa, K.; Heavey, S.; Godwin, P.; Gray, S.G.; Cormican, D.; Finn, S.P.; Gately, K.A.; et al. Targeting NF-κB-mediated inflammatory pathways in cisplatin-resistant NSCLC. Lung Cancer 2019, 135, 217–227. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, M.; Li, Y.; Wang, Y.; Wang, K.; Chen, Q.; Zhang, R.; Song, W.; Huang, Q.; Zhao, W.; et al. YAP manipulates proliferation via PTEN/AKT/mTOR-mediated autophagy in lung adenocarcinomas. Cancer Cell Int. 2021, 21, 1–13. [Google Scholar] [CrossRef]
- Jia, D.; Augert, A.; Kim, N.-W.; Eastwood, E.; Wu, N.; Ibrahim, A.H.; Kim, K.-B.; Dunn, C.T.; Pillai, S.P.; Gazdar, A.F.; et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov. 2018, 8, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Shire, N.J.; Klein, A.B.; Golozar, A.; Collins, J.M.; Fraeman, K.H.; Nordstrom, B.L.; McEwen, R.; Hembrough, T.; Rizvi, N.A. STK11 (LKB1) mutations in metastatic NSCLC: Prognostic value in the real world. PLoS ONE 2020, 15, e0238358. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Yao, C.; Qi, X.; Stripp, B.R.; Tang, N. STK11 is required for the normal program of ciliated cell differentiation in airways. Cell Discov. 2019, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Willemse, B.W.M.; Hacken, N.H.T.T.; Rutgers, B.; Lesman-Leegte, I.G.A.T.; Postma, D.S.; Timens, W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur. Respir. J. 2005, 26, 835–845. [Google Scholar] [CrossRef]
- Birru, R.; Kahkonon, B.; Di, Y.P. Chronic Inflammation in the Pathogenesis of COPD and Lung Cancer. Proc. Am. Thorac. Soc. 2012, 9, 81. [Google Scholar] [CrossRef]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette Smoking and Inflammation. J. Dent. Res. 2011, 91, 142–149. [Google Scholar] [CrossRef]
- Xu, Y.; Li, H.; Bajrami, B.; Kwak, H.; Cao, S.; Liu, P.; Zhou, J.; Zhou, Y.; Zhu, H.; Ye, K.; et al. Cigarette smoke (CS) and nicotine delay neutrophil spontaneous death via suppressing production of diphosphoinositol pentakisphosphate. Proc. Natl. Acad. Sci. USA 2013, 110, 7726–7731. [Google Scholar] [CrossRef]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Heal. 2018, 15, 1033. [Google Scholar] [CrossRef]
- Miglino, N.; Roth, M.; Lardinois, D.; Sadowski, C.; Tamm, M.; Borger, P. Cigarette smoke inhibits lung fibroblast proliferation by translational mechanisms. Eur. Respir. J. 2011, 39, 705–711. [Google Scholar] [CrossRef]
- Lacanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Investig. 2019, 129, 2107–2122. [Google Scholar] [CrossRef]
- Xiong, W.; Wang, L.; Yu, F. Expression of bone morphogenetic protein 6 in non-small cell lung cancer and its significance. Oncol. Lett. 2018, 17, 1946–1952. [Google Scholar] [CrossRef]
- Shen, W.; Pang, H.; Xin, B.; Duan, L.; Liu, L.; Zhang, H. Biological effects of BMP7 on small-cell lung cancer cells and its bone metastasis. Int. J. Oncol. 2018, 53, 1354–1362. [Google Scholar] [CrossRef]
- Li, R.; Ong, S.L.; Tran, L.M.; Jing, Z.; Liu, B.; Park, S.J.; Huang, Z.L.; Walser, T.C.; Heinrich, E.L.; Lee, G.; et al. Chronic IL-1β-induced inflammation regulates epithelial-to-mesenchymal transition memory phenotypes via epigenetic modifications in non-small cell lung cancer. Sci. Rep. 2020, 10, 377. [Google Scholar] [CrossRef]
- Adams, D.S.; Levin, M. Endogenous voltage gradients as mediators of cell-cell communication: Strategies for investigating bioelectrical signals during pattern formation. Cell Tissue Res 2012, 352, 95–122. [Google Scholar] [CrossRef]
- Levin, M. Molecular bioelectricity: How endogenous voltage potentials control cell behavior and instruct pattern regulation in vivo. Mol. Biol. Cell 2014, 25, 3835–3850. [Google Scholar] [CrossRef]
- McLaughlin, K.A.; Levin, M. Bioelectric signaling in regeneration: Mechanisms of ionic controls of growth and form. Dev. Biol. 2018, 433, 177–189. [Google Scholar] [CrossRef]
- Ferreira, F.; Luxardi, G.; Reid, B.; Zhao, M. Early bioelectric activities mediate redox-modulated regeneration. Development 2016, 143, 4582–4594. [Google Scholar] [CrossRef]
- Fogarty, C.E.; Bergmann, A. Killers creating new life: Caspases drive apoptosis-induced proliferation in tissue repair and disease. Cell Death Differ. 2017, 24, 1390–1400. [Google Scholar] [CrossRef]
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- Blackiston, D.; Adams, D.S.; Lemire, J.M.; Lobikin, M.; Levin, M. Transmembrane potential of GlyCl-expressing instructor cells induces a neoplastic-like conversion of melanocytes via a serotonergic pathway. Dis. Model. Mech. 2011, 4, 67–85. [Google Scholar] [CrossRef]
- Zhu, Y.; Lu, S.; Gold, M.S. Persistent inflammation increases GABA-induced depolarization of rat cutaneous dorsal root ganglion neurons in vitro. Neuroscience 2012, 220, 330–340. [Google Scholar] [CrossRef][Green Version]
- Chernet, B.T.; Levin, M. Transmembrane voltage potential is an essential cellular parameter for the detection and control of tumor development in a Xenopus model. Dis. Model. Mech. 2013, 6, 595–607. [Google Scholar] [CrossRef]
- Scharrer, B. Insect tumors induced by nerve severance: Incidence and mortality. Cancer Res. 1953, 13, 73–76. [Google Scholar]
- Minutti, C.M.; Knipper, J.A.; Allen, J.E.; Zaiss, D.M. Tissue-specific contribution of macrophages to wound healing. Semin. Cell Dev. Biol. 2017, 61, 3–11. [Google Scholar] [CrossRef]
- Aiello, N.M.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef]
- Ruiz-Ceja, K.A.; Chirino, Y.I. Current FDA-approved treatments for non-small cell lung cancer and potential biomarkers for its detection. Biomed. Pharmacother. 2017, 90, 24–37. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.Y.S.; Nazre, M. Vinca Alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235. [Google Scholar]
- Faller, B.A.; Pandit, T.N. Safety and Efficacy of Vinorelbine in the Treatment of Non-Small Cell Lung Cancer. Clin. Med. Insights Oncol. 2011, 5, CMO-S5074. [Google Scholar] [CrossRef]
- Ojima, I.; Lichtenthal, B.; Lee, S.; Wang, C.; Wang, X. Taxane anticancer agents: A patent perspective. Expert Opin. Ther. Patents 2016, 26, 1–20. [Google Scholar] [CrossRef]
- Verweij, J.; Clavel, M.; Chevalier, B. Paclitaxel (TaxolTM) and docetaxel (TaxotereTM): Not simply two of a kind. Ann. Oncol. 1994, 5, 495–505. [Google Scholar] [CrossRef]
- Ardalani, H.; Avan, A.; Ghayour-Mobarhan, M. Podophyllotoxin: A novel potential natural anticancer agent. Avicenna J. Phytomed. 2017, 7, 285–294. [Google Scholar]
- Sabari, J.K.; Lok, B.H.; Laird, J.H.; Poirier, J.T.; Rudin, J.K.S.J.T.P.C.M. Unravelling the biology of SCLC: Implications for therapy. Nat. Rev. Clin. Oncol. 2017, 14, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-Damaging Agents in Cancer Chemotherapy: Serendipity and Chemical Biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Shanker, M.; Willcutts, D.A.; Roth, J.; Ramesh, R. Drug resistance in lung cancer. Lung Cancer Targets Ther. 2010, 1, 23–36. [Google Scholar]
- Tajuddin, W.N.B.W.M.; Lajis, N.H.; Abas, F.; Othman, I.; Naidu, R. Mechanistic Understanding of Curcumin’s Therapeutic Effects in Lung Cancer. Nutrients 2019, 11, 2989. [Google Scholar] [CrossRef]
- Lee, J.C.; Kinniry, P.A.; Arguiri, E.; Serota, M.; Kanterakis, S.; Chatterjee, S.; Solomides, C.C.; Javvadi, P.; Koumenis, C.; Cengel, K.A.; et al. Dietary Curcumin Increases Antioxidant Defenses in Lung, Ameliorates Radiation-Induced Pulmonary Fibrosis, and Improves Survival in Mice. Radiat. Res. 2010, 173, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Rennolds, J.; Malireddy, S.; Hassan, F.; Tridandapani, S.; Parinandi, N.; Boyaka, P.N.; Cormet-Boyaka, E. Curcumin regulates airway epithelial cell cytokine responses to the pollutant cadmium. Biochem. Biophys. Res. Commun. 2012, 417, 256–261. [Google Scholar] [CrossRef]
- Liu, F.; Gao, S.; Yang, Y.; Zhao, X.; Fan, Y.; Ma, W.; Yang, D.; Yang, A.; Yu, Y. Antitumor activity of curcumin by modulation of apoptosis and autophagy in human lung cancer A549 cells through inhibiting PI3K/Akt/mTOR pathway. Oncol. Rep. 2018, 39, 1523–1531. [Google Scholar] [CrossRef]
- Jiao, D.; Wang, J.; Lu, W.; Tang, X.; Chen, J.; Mou, H.; Chen, Q.-Y. Curcumin inhibited HGF-induced EMT and angiogenesis through regulating c-Met dependent PI3K/Akt/mTOR signaling pathways in lung cancer. Mol. Ther. Oncolytics 2016, 3, 16018. [Google Scholar] [CrossRef]
- Wu, G.-Q.; Chai, K.-Q.; Zhu, X.-M.; Jiang, H.; Wang, X.; Xue, Q.; Zheng, A.-H.; Zhou, H.-Y.; Chen, Y.; Chen, X.-C.; et al. Anti-cancer effects of curcumin on lung cancer through the inhibition of EZH2 and NOTCH1. Oncotarget 2016, 7, 26535–26550. [Google Scholar] [CrossRef]
- Wu, L.; Guo, L.; Liang, Y.; Liu, X.; Jiang, L.; Wang, L. Curcumin suppresses stem-like traits of lung cancer cells via inhibiting the JAK2/STAT3 signaling pathway. Oncol. Rep. 2015, 34, 3311–3317. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Yang, X.; Chen, Y.; Jiang, Y.; Wang, S.-J.; Li, Y.; Wang, X.-Q.; Meng, Y.; Zhu, M.-M.; Ma, X.; et al. Curcumin Suppresses Lung Cancer Stem Cells via Inhibiting Wnt/β-catenin and Sonic Hedgehog Pathways. Phytother. Res. 2017, 31, 680–688. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Calderón-Montaño, J.M.; Salvador, J.; Robles, A.; López-Lázaro, M. The dark side of curcumin. Int. J. Cancer 2010, 126, 1771–1775. [Google Scholar] [CrossRef]
- Smagurauskaite, G.; Mahale, J.; Brown, K.; Thomas, A.L.; Howells, L.M. New Paradigms to Assess Consequences of Long-Term, Low-Dose Curcumin Exposure in Lung Cancer Cells. Molecules 2020, 25, 366. [Google Scholar] [CrossRef]
- Chen, P.; Huang, H.-P.; Wang, Y.; Jin, J.; Long, W.-G.; Chen, K.; Zhao, X.-H.; Chen, C.-G.; Li, J. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J. Exp. Clin. Cancer Res. 2019, 38, 1–17. [Google Scholar] [CrossRef]
- Su, W.; Wei, T.; Lu, M.; Meng, Z.; Chen, X.; Jing, J.; Li, J.; Yao, W.; Zhu, H.; Fu, T. Treatment of metastatic lung cancer via inhalation administration of curcumin composite particles based on mesoporous silica. Eur. J. Pharm. Sci. 2019, 134, 246–255. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Hamza, R.Z.; El-Shenawy, N.S. Anti-inflammatory and antioxidant role of resveratrol on nicotine-induced lung changes in male rats. Toxicol. Rep. 2017, 4, 399–407. [Google Scholar] [CrossRef]
- Rasheduzzaman, M.; Jeong, J.-K.; Park, S.-Y. Resveratrol sensitizes lung cancer cell to TRAIL by p53 independent and suppression of Akt/NF-κB signaling. Life Sci. 2018, 208, 208–220. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Tang, L.; Chen, H.; Wu, C.; Zhao, M.; Yang, Y.; Chen, X.; Liu, G. Resveratrol inhibits TGF-β1-induced epithelial-to-mesenchymal transition and suppresses lung cancer invasion and metastasis. Toxicology 2013, 303, 139–146. [Google Scholar] [CrossRef]
- Li, X.; Wang, D.; Zhao, Q.C.; Shi, T.; Chen, J. Resveratrol Inhibited Non–small Cell Lung Cancer Through Inhibiting STAT-3 Signaling. Am. J. Med Sci. 2016, 352, 524–530. [Google Scholar] [CrossRef]
- Liu, D.; He, B.; Lin, L.; Malhotra, A.; Yuan, N. Potential of curcumin and resveratrol as biochemical and biophysical modulators during lung cancer in rats. Drug Chem. Toxicol. 2018, 42, 328–334. [Google Scholar] [CrossRef]
- Ko, J.-C.; Syu, J.-J.; Chen, J.-C.; Wang, T.-J.; Chang, P.-Y.; Chen, C.-Y.; Jian, Y.-T.; Jian, Y.-J.; Lin, Y.-W. Resveratrol Enhances Etoposide-Induced Cytotoxicity through Down-Regulating ERK1/2 and AKT-Mediated X-ray Repair Cross-Complement Group 1 (XRCC1) Protein Expression in Human Non-Small-Cell Lung Cancer Cells. Basic Clin. Pharmacol. Toxicol. 2015, 117, 383–391. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Hoti, S.L.; Prasad, N.R. Resveratrol loaded gelatin nanoparticles synergistically inhibits cell cycle progression and constitutive NF-kappaB activation, and induces apoptosis in non-small cell lung cancer cells. Biomed. Pharmacother. 2015, 70, 274–282. [Google Scholar] [CrossRef]
- Parasuraman, S.; David, A.V.A.; Arulmoli, R. Overviews of biological importance of quercetin: A bioactive flavonoid. Pharmacogn. Rev. 2016, 10, 84–89. [Google Scholar] [CrossRef]
- Chen, W.; Padilla, M.T.; Xu, X.; Desai, D.; Krzeminski, J.; Amin, S.; Lin, Y. Quercetin inhibits multiple pathways involved in interleukin 6 secretion from human lung fibroblasts and activity in bronchial epithelial cell transformation induced by benzo[a]pyrene diol epoxide. Mol. Carcinog. 2015, 55, 1858–1866. [Google Scholar] [CrossRef]
- Xu, D.; Hu, M.-J.; Wang, Y.-Q.; Cui, Y.-L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef]
- Yeh, S.-L.; Yeh, C.-L.; Chan, S.-T.; Chuang, C.-H. Plasma Rich in Quercetin Metabolites Induces G2/M Arrest by Upregulating PPAR-γExpression in Human A549 Lung Cancer Cells. Planta Medica 2011, 77, 992–998. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, X. Stimulatory effects of curcumin and quercetin on posttranslational modifications of p53 during lung carcinogenesis. Hum. Exp. Toxicol. 2017, 37, 618–625. [Google Scholar] [CrossRef]
- Arbain, N.H.; Salim, N.; Masoumi, H.R.F.; Wong, T.W.; Basri, M.; Rahman, M.B.A. In vitro evaluation of the inhalable quercetin loaded nanoemulsion for pulmonary delivery. Drug Deliv. Transl. Res. 2018, 9, 497–507. [Google Scholar] [CrossRef]
- Chu, C.; Deng, J.; Man, Y.; Qu, Y. Green Tea Extracts Epigallocatechin-3-gallate for Different Treatments. BioMed Res. Int. 2017, 2017, 5615647. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Wu, N.; Liu, Z.; Zhao, H.; Zhao, M. Epigallocatechin-3-gallate alleviates paraquat-induced acute lung injury and inhibits upregulation of toll-like receptors. Life Sci. 2017, 170, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Liu, F.; Zhang, W.; Liu, X.; Lin, B.; Tang, X. Epigallocatechin-3-gallate inhibits nicotine-induced migration and invasion by the suppression of angiogenesis and epithelial-mesenchymal transition in non-small cell lung cancer cells. Oncol. Rep. 2015, 33, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.; Mandal, A.K.A. Next-Generation Sequencing Reveals the Role of Epigallocatechin-3-Gallate in Regulating Putative Novel and Known microRNAs Which Target the MAPK Pathway in Non-Small-Cell Lung Cancer A549 Cells. Molecules 2019, 24, 368. [Google Scholar] [CrossRef]
- Zhu, J.; Jiang, Y.; Yang, X.; Wang, S.; Xie, C.; Li, X.; Li, Y.; Chen, Y.; Wang, X.; Meng, Y.; et al. Wnt/β-catenin pathway mediates (−)-Epigallocatechin-3-gallate (EGCG) inhibition of lung cancer stem cells. Biochem. Biophys. Res. Commun. 2017, 482, 15–21. [Google Scholar] [CrossRef]
- Relat, J.; Blancafort, A.; Oliveras, G.; Cufí, S.; Haro, D.; Marrero, P.F.; Puig, T. Different fatty acid metabolism effects of (−)-Epigallocatechin-3-Gallate and C75 in Adenocarcinoma lung cancer. BMC Cancer 2012, 12, 280. [Google Scholar] [CrossRef]
- Deng, Y.-T.; Lin, J.-K. EGCG Inhibits the Invasion of Highly Invasive CL1-5 Lung Cancer Cells through Suppressing MMP-2 Expression via JNK Signaling and Induces G2/M Arrest. J. Agric. Food Chem. 2011, 59, 13318–13327. [Google Scholar] [CrossRef]
- Chen, A.; Jiang, P.; Zeb, F.; Wu, X.; Xu, C.; Chen, L.; Feng, Q. EGCG regulates CTR1 expression through its pro-oxidative property in non-small-cell lung cancer cells. J. Cell. Physiol. 2020, 235. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Han, L.; Zhou, Y.; Sun, S. Green tea polyphenol EGCG reverse cisplatin resistance of A549/DDP cell line through candidate genes demethylation. Biomed. Pharmacother. 2015, 69, 285–290. [Google Scholar] [CrossRef]
- Wang, P.; Heber, D.; Henning, S.M. Quercetin increased bioavailability and decreased methylation of green tea polyphenols in vitro and in vivo. Food Funct. 2012, 3, 635–642. [Google Scholar] [CrossRef]
- Chen, B.-H.; Hsieh, C.-H.; Tsai, S.-Y.; Wang, C.-Y.; Wang, C.-C. Anticancer effects of epigallocatechin-3-gallate nanoemulsion on lung cancer cells through the activation of AMP-activated protein kinase signaling pathway. Sci. Rep. 2020, 10, 5163. [Google Scholar] [CrossRef]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a Flavonoid with Potential for Cancer Prevention and Therapy. Curr. Cancer Drug Targets 2008, 8, 634–646. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Son, Y.-O.; Divya, S.P.; Roy, R.V.; Hitron, J.A.; Wang, L.; Kim, D.; Dai, J.; Asha, P.; Zhang, Z.; et al. Luteolin inhibits Cr(VI)-induced malignant cell transformation of human lung epithelial cells by targeting ROS mediated multiple cell signaling pathways. Toxicol. Appl. Pharmacol. 2014, 281, 230–241. [Google Scholar] [CrossRef]
- Kasala, E.R.; Bodduluru, L.N.; Barua, C.C.; Gogoi, R. Antioxidant and antitumor efficacy of Luteolin, a dietary flavone on benzo(a)pyrene-induced experimental lung carcinogenesis. Biomed. Pharmacother. 2016, 82, 568–577. [Google Scholar] [CrossRef]
- Meng, G.; Chai, K.; Li, X.; Zhu, Y.; Huang, W. Luteolin exerts pro-apoptotic effect and anti-migration effects on A549 lung adenocarcinoma cells through the activation of MEK/ERK signaling pathway. Chem. Interact. 2016, 257, 26–34. [Google Scholar] [CrossRef]
- Chen, K.-C.; Chen, C.-Y.; Lin, C.-J.; Yang, T.-Y.; Chen, T.-H.; Wu, L.-C.; Wu, C.-C. Luteolin attenuates TGF-β1-induced epithelial–mesenchymal transition of lung cancer cells by interfering in the PI3K/Akt–NF-κB–Snail pathway. Life Sci. 2013, 93, 924–933. [Google Scholar] [CrossRef]
- Bai, L.; Xu, X.; Wang, Q.; Xu, S.; Ju, W.; Wang, X.; Chen, W.; He, W.; Tang, H.; Lin, Y. A Superoxide-Mediated Mitogen-Activated Protein Kinase Phosphatase-1 Degradation and c-Jun NH2-Terminal Kinase Activation Pathway for Luteolin-Induced Lung Cancer Cytotoxicity. Mol. Pharmacol. 2012, 81, 549–555. [Google Scholar] [CrossRef]
- Attoub, S.; Hassan, A.H.; Vanhoecke, B.; Iratni, R.; Takahashi, T.; Gaben, A.-M.; Bracke, M.; Awad, S.; John, A.; Kamalboor, H.A.; et al. Inhibition of cell survival, invasion, tumor growth and histone deacetylase activity by the dietary flavonoid luteolin in human epithelioid cancer cells. Eur. J. Pharmacol. 2011, 651, 18–25. [Google Scholar] [CrossRef]
- Amin, A.R.M.R.; Wang, D.; Zhang, H.; Peng, S.; Shin, H.J.C.; Brandes, J.C.; Tighiouart, M.; Khuri, F.R.; Chen, Z.G.; Shin, D.M. Enhanced Anti-tumor Activity by the Combination of the Natural Compounds (−)-Epigallocatechin-3-gallate and Luteolin: POTENTIAL ROLE OF p53. J. Biol. Chem. 2010, 285, 34557–34565. [Google Scholar] [CrossRef]
- Li, J.; Cheng, X.; Chen, Y.; He, W.; Ni, L.; Xiong, P.; Wei, M. Vitamin E TPGS modified liposomes enhance cellular uptake and targeted delivery of luteolin: An in vivo/in vitro evaluation. Int. J. Pharm. 2016, 512, 262–272. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, Y.; Ma, L.; Ji, R.; Qu, Y.; Xin, Y.; Lv, G. Chemopreventive activity of sulforaphane. Drug Des. Dev. Ther. 2018, 12, 2905–2913. [Google Scholar] [CrossRef]
- Xie, C.; Zhu, J.; Jiang, Y.; Chen, J.; Wang, X.; Geng, S.; Wu, J.; Zhong, C.; Li, X.; Meng, Z. Sulforaphane Inhibits the Acquisition of Tobacco Smoke-Induced Lung Cancer Stem Cell-Like Properties via the IL-6/ΔNp63α/Notch Axis. Theranostics 2019, 9, 4827–4840. [Google Scholar] [CrossRef]
- Geng, Y.; Zhou, Y.; Wu, S.; Hu, Y.; Lin, K.; Wang, Y.; Zheng, Z.; Wu, W. Sulforaphane Induced Apoptosis via Promotion of Mitochondrial Fusion and ERK1/2-Mediated 26S Proteasome Degradation of Novel Pro-survival Bim and Upregulation of Bax in Human Non-Small Cell Lung Cancer Cells. J. Cancer 2017, 8, 2456–2470. [Google Scholar] [CrossRef]
- Tsai, J.-Y.; Tsai, S.-H.; Wu, C.-C. The chemopreventive isothiocyanate sulforaphane reduces anoikis resistance and anchorage-independent growth in non-small cell human lung cancer cells. Toxicol. Appl. Pharmacol. 2019, 362, 116–124. [Google Scholar] [CrossRef]
- Wang, T.-H.; Chen, C.-C.; Huang, K.-Y.; Shih, Y.-M.; Chen, C.-Y. High levels of EGFR prevent sulforaphane-induced reactive oxygen species-mediated apoptosis in non-small-cell lung cancer cells. Phytomedicine 2019, 64, 152926. [Google Scholar] [CrossRef]
- Wang, Y.; Mandal, A.K.; Son, Y.-O.; Pratheeshkumar, P.; Wise, J.T.; Wang, L.; Zhang, Z.; Shi, X.; Chen, Z. Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol. Appl. Pharmacol. 2018, 353, 23–30. [Google Scholar] [CrossRef]
- Gao, L.; Cheng, D.; Yang, J.; Wu, R.; Li, W.; Kong, A.-N. Sulforaphane epigenetically demethylates the CpG sites of the miR-9-3 promoter and reactivates miR-9-3 expression in human lung cancer A549 cells. J. Nutr. Biochem. 2018, 56, 109–115. [Google Scholar] [CrossRef]
- Wang, F.; Wang, W.; Li, J.; Zhang, J.; Wang, X.; Wang, M. Sulforaphane reverses gefitinib tolerance in human lung cancer cells via modulation of sonic hedgehog signaling. Oncol. Lett. 2017, 15, 109–114. [Google Scholar] [CrossRef]
- Rakariyatham, K.; Yang, X.; Gao, Z.; Song, M.; Han, Y.; Chen, X.; Xiao, H. Synergistic chemopreventive effect of allyl isothiocyanate and sulforaphane on non-small cell lung carcinoma cells. Food Funct. 2019, 10, 893–902. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Du, X.; Ma, H.; Yao, J. The Anti-Cancer Mechanisms of Berberine: A Review. Cancer Manag. Res. 2020, 12, 695–702. [Google Scholar] [CrossRef]
- Fu, L.; Chen, W.; Guo, W.; Wang, J.; Tian, Y.; Shi, D.; Zhang, X.; Qiu, H.; Xiao, X.; Kang, T.; et al. Berberine Targets AP-2/hTERT, NF-κB/COX-2, HIF-1α/VEGF and Cytochrome-c/Caspase Signaling to Suppress Human Cancer Cell Growth. PLoS ONE 2013, 8, e69240. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Q.; Shi, J.-M.; Ding, Z.; Xia, Q.; Zheng, T.-S.; Ren, Y.-B.; Li, M.; Fan, L.-H. Berberine induces apoptosis in non-small-cell lung cancer cells by upregulating miR-19a targeting tissue factor. Cancer Manag. Res. 2019, 11, 9005–9015. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, F.; Jiang, S.; Chen, X.; Zhang, S.; Zhao, H. Berberine hydrochloride inhibits cell proliferation and promotes apoptosis of non-small cell lung cancer via the suppression of the MMP2 and Bcl-2/Bax signaling pathways. Oncol. Lett. 2018, 15, 7409–7414. [Google Scholar] [CrossRef] [PubMed]
- Kalaiarasi, A.; Anusha, C.; Sankar, R.; Rajasekaran, S.; Marshal, J.J.; Muthusamy, K.; Ravikumar, V. Plant Isoquinoline Alkaloid Berberine Exhibits Chromatin Remodeling by Modulation of Histone Deacetylase to Induce Growth Arrest and Apoptosis in the A549 Cell Line. J. Agric. Food Chem. 2016, 64, 9542–9550. [Google Scholar] [CrossRef]
- Zheng, F.; Tang, Q.; Wu, J.; Zhao, S.; Liang, Z.; Li, L.; Wu, W.; Hann, S. p38α MAPK-mediated induction and interaction of FOXO3a and p53 contribute to the inhibited-growth and induced-apoptosis of human lung adenocarcinoma cells by berberine. J. Exp. Clin. Cancer Res. 2014, 33, 36. [Google Scholar] [CrossRef]
- Qi, H.-W.; Xin, L.-Y.; Xu, X.; Ji, X.-X.; Fan, L.-H. Epithelial-to-mesenchymal transition markers to predict response of Berberine in suppressing lung cancer invasion and metastasis. J. Transl. Med. 2014, 12, 22. [Google Scholar] [CrossRef]
- Kumar, R.; Awasthi, M.; Sharma, A.; Padwad, Y.; Sharma, R. Berberine induces dose-dependent quiescence and apoptosis in A549 cancer cells by modulating cell cyclins and inflammation independent of mTOR pathway. Life Sci. 2020, 244, 117346. [Google Scholar] [CrossRef]
- Zhu, T.; Li, L.-L.; Xiao, G.-F.; Luo, Q.-Z.; Liu, Q.-Z.; Yao, K.-T.; Xiao, G.-H. Berberine Increases Doxorubicin Sensitivity by Suppressing STAT3 in Lung Cancer. Am. J. Chin. Med. 2015, 43, 1487–1502. [Google Scholar] [CrossRef]
- Kabary, D.M.; Helmy, M.W.; Elkhodairy, K.A.; Fang, J.-Y.; Elzoghby, A.O. Hyaluronate/lactoferrin layer-by-layer-coated lipid nanocarriers for targeted co-delivery of rapamycin and berberine to lung carcinoma. Colloids Surfaces B Biointerfaces 2018, 169, 183–194. [Google Scholar] [CrossRef]
- Tuli, H.S.; Tuorkey, M.J.; Thakral, F.; Sak, K.; Kumar, M.; Sharma, A.K.; Sharma, U.; Jain, A.; Aggarwal, V.; Bishayee, A. Molecular Mechanisms of Action of Genistein in Cancer: Recent Advances. Front. Pharmacol. 2019, 10, 1336. [Google Scholar] [CrossRef]
- Zhang, J.; Su, H.; Li, Q.; Li, J.; Zhao, Q. Genistein decreases A549 cell viability via inhibition of the PI3K/AKT/HIF-1α/VEGF and NF-κB/COX-2 signaling pathways. Mol. Med. Rep. 2017, 15, 2296–2302. [Google Scholar] [CrossRef]
- Yang, Y.; Zang, A.; Jia, Y.; Shang, Y.; Zhang, Z.; Ge, K.; Zhang, J.; Fan, W.; Wang, B. Genistein inhibits A549 human lung cancer cell proliferation via miR-27a and MET signaling. Oncol. Lett. 2016, 12, 2189–2193. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, C.; Qing, Y.; Cheng, Y.; Jiang, X.; Li, M.; Yang, Z.; Wang, N. Genistein induces apoptosis by stabilizing intracellular p53 protein through an APE1-mediated pathway. Free. Radic. Biol. Med. 2015, 86, 209–218. [Google Scholar] [CrossRef]
- Wu, T.-C.; Lin, Y.-C.; Chen, H.-L.; Huang, P.-R.; Liu, S.-Y.; Yeh, S.-L. The enhancing effect of genistein on apoptosis induced by trichostatin A in lung cancer cells with wild type p53 genes is associated with upregulation of histone acetyltransferase. Toxicol. Appl. Pharmacol. 2016, 292, 94–102. [Google Scholar] [CrossRef]
- Zhou, R.-J.; Yang, X.-Q.; Wang, N.; Zhou, Q.; Xia, L.; Li, M.-X.; Zeng, L.-L.; Wang, G.; Yang, Z.-Z. Anti-tumor Effects of All-trans Retinoic Acid are Enhanced by Genistein. Cell Biophys. 2012, 62, 177–184. [Google Scholar] [CrossRef]
- Sacko, K.; Thangavel, K.; Shoyele, S.A. Codelivery of Genistein and miRNA-29b to A549 Cells Using Aptamer-Hybrid Nanoparticle Bioconjugates. Nanomaterials 2019, 9, 1052. [Google Scholar] [CrossRef]
- Clark, R.; Lee, S.-H. Anticancer Properties of Capsaicin Against Human Cancer. Anticancer Res. 2016, 36, 837–843. [Google Scholar] [PubMed]
- Anandakumar, P.; Kamaraj, S.; Jagan, S.; Ramakrishnan, G.; Asokkumar, S.; Naveenkumar, C.; Raghunandhakumar, S.; Devaki, T. Capsaicin inhibits benzo(a)pyrene-induced lung carcinogenesis in an in vivo mouse model. Inflamm. Res. 2012, 61, 1169–1175. [Google Scholar] [CrossRef]
- Chakraborty, S.; Adhikary, A.; Mazumdar, M.; Mukherjee, S.; Bhattacharjee, P.; Guha, D.; Choudhuri, T.; Chattopadhyay, S.; Sa, G.; Sen, A.; et al. Capsaicin-Induced Activation of p53-SMAR1 Auto-Regulatory Loop Down-Regulates VEGF in Non-Small Cell Lung Cancer to Restrain Angiogenesis. PLoS ONE 2014, 9, e99743. [Google Scholar] [CrossRef]
- Lau, J.K.; Brown, K.C.; Dom, A.M.; Witte, T.R.; Thornhill, B.A.; Crabtree, C.M.; Perry, H.E.; Brown, J.M.; Ball, J.G.; Creel, R.G.; et al. Capsaicin induces apoptosis in human small cell lung cancer via the TRPV6 receptor and the calpain pathway. Apoptosis 2014, 19, 1190–1201. [Google Scholar] [CrossRef]
- Chen, J.-C.; Ko, J.-C.; Yen, T.-C.; Chen, T.-Y.; Lin, Y.-C.; Ma, P.-F.; Lin, Y.-W. Capsaicin enhances erlotinib-induced cytotoxicity via AKT inactivation and excision repair cross-complementary 1 (ERCC1) down-regulation in human lung cancer cells. Toxicol. Res. 2019, 8, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Parashar, P.; Tripathi, C.B.; Arya, M.; Kanoujia, J.; Singh, M.; Yadav, A.; Kaithwas, G.A.; Saraf, S. A synergistic approach for management of lung carcinoma through folic acid functionalized co-therapy of capsaicin and gefitinib nanoparticles: Enhanced apoptosis and metalloproteinase-9 down-regulation. Phytomedicine 2019, 53, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Chlapek, P.; Slavikova, V.; Mazanek, P.; Sterba, J.; Veselska, R. Why Differentiation Therapy Sometimes Fails: Molecular Mechanisms of Resistance to Retinoids. Int. J. Mol. Sci. 2018, 19, 132. [Google Scholar] [CrossRef] [PubMed]
- Qin, A.; Reddy, H.G.; Weinberg, F.D.; Kalemkerian, G.P. Cyclin-dependent kinase inhibitors for the treatment of lung cancer. Expert Opin. Pharmacother. 2020, 21, 941–952. [Google Scholar] [CrossRef]
- Zhang, J.; Si, J.; Gan, L.; Di, C.; Xie, Y.; Sun, C.; Li, H.; Guo, M.; Zhang, H. Research progress on therapeutic targeting of quiescent cancer cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2810–2819. [Google Scholar] [CrossRef]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-Inflammatory Drugs as Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef]
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef]
- Rawangkan, A.; Wongsirisin, P.; Namiki, K.; Iida, K.; Kobayashi, Y.; Shimizu, Y.; Fujiki, H.; Suganuma, M. Green Tea Catechin Is an Alternative Immune Checkpoint Inhibitor that Inhibits PD-L1 Expression and Lung Tumor Growth. Molecules 2018, 23, 2071. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Zhang, N.; Yin, M.; Dong, J.; Zeng, Q.; Mao, G.; Song, D.; Liu, L.; Deng, H. Berberine diminishes cancer cell PD-L1 expression and facilitates antitumor immunity via inhibiting the deubiquitination activity of CSN5. Acta Pharm. Sin. B 2020, 10, 2299–2312. [Google Scholar] [CrossRef]
- Xia, Y.; Zhan, C.; Feng, M.; Leblanc, M.; Ke, E.; Yeddula, N.; Verma, I.M. Targeting CREB Pathway Suppresses Small Cell Lung Cancer. Mol. Cancer Res. 2018, 16, 825–832. [Google Scholar] [CrossRef]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef]
- Loira-Pastoriza, C.; Todoroff, J.; Vanbever, R. Delivery strategies for sustained drug release in the lungs. Adv. Drug Deliv. Rev. 2014, 75, 81–91. [Google Scholar] [CrossRef]
- Muller, A.G.; Sarker, S.D.; Saleem, I.Y.; Hutcheon, G.A. Delivery of natural phenolic compounds for the potential treatment of lung cancer. DARU J. Pharm. Sci. 2019, 27, 433–449. [Google Scholar] [CrossRef]
- Heng, W.S.; Cheah, S.-C. Chelerythrine Chloride Downregulates β-Catenin and Inhibits Stem Cell Properties of Non-Small Cell Lung Carcinoma. Molecules 2020, 25, 224. [Google Scholar] [CrossRef]
- Pan, S.-Y.; Zhou, S.-F.; Gao, S.-H.; Yu, Z.-L.; Zhang, S.-F.; Tang, M.-K.; Sun, J.-N.; Ma, D.-L.; Han, Y.-F.; Fong, W.-F.; et al. New Perspectives on How to Discover Drugs from Herbal Medicines: CAM’s Outstanding Contribution to Modern Therapeutics. Evid-Based Compl. Alt. 2013, 2013, 627375. Available online: https://www.hindawi.com/journals/ecam/2013/627375/ (accessed on 14 July 2020). [CrossRef]
- Katiyar, C.; Kanjilal, S.; Gupta, A.; Katiyar, S. Drug discovery from plant sources: An integrated approach. AYU An Int. Q. J. Res. Ayurveda 2012, 33, 10–19. [Google Scholar] [CrossRef]
- Wurtzel, E.T.; Kutchan, T.M. Plant metabolism, the diverse chemistry set of the future. Science 2016, 353, 1232–1236. [Google Scholar] [CrossRef]
- Pott, D.M.; Osorio, S.; Vallarino, J.G. From Central to Specialized Metabolism: An Overview of Some Secondary Compounds Derived from the Primary Metabolism for Their Role in Conferring Nutritional and Organoleptic Characteristics to Fruit. Front. Plant Sci. 2019, 10, 835. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Phytochemical | Antioxidant | Anti-Inflammatory | Signaling Pathway Target | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Wnt | Notch | Hh | JNK | JAK/STAT3 | PI3K/AKT | NF-κB | RTK | TGFβ/BMP | |||
| Curcumin | Yes | Yes | ↓ | ↓ | ↓ | - | ↓ | ↓ | ↓ | ↓ | - |
| Resveratrol | Yes | Yes | - | - | - | - | ↓ | ↓ | ↓ | ↓ | ↓ |
| Quercetin | Yes | Yes | - | - | - | - | ↓ | ↓ | ↓ | ↓ | - |
| EGCG | Yes | Yes | ↓ | - | ↓ | ↓ | - | ↓ | ↓ | ↓ | ↓ |
| Luteolin | Yes | Yes | - | - | - | ↑ | ↓ | ↑↓ | ↓ | ↑↓ | ↓ |
| Sulforaphane | Yes | Yes | ↓ | ↓ | ↓ | - | - | ↓ | ↓ | ↑↓ | - |
| Berberine | Yes | Yes | - | - | - | - | ↓ | ↓ | ↓ | ↑↓ | ↓ |
| Genistein | Yes | Yes | - | - | - | - | - | ↓ | ↓ | ↓ | - |
| Capsaicin | Yes | Yes | - | - | - | - | - | ↓ | ↓ | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heng, W.S.; Kruyt, F.A.E.; Cheah, S.-C. Understanding Lung Carcinogenesis from a Morphostatic Perspective: Prevention and Therapeutic Potential of Phytochemicals for Targeting Cancer Stem Cells. Int. J. Mol. Sci. 2021, 22, 5697. https://doi.org/10.3390/ijms22115697
Heng WS, Kruyt FAE, Cheah S-C. Understanding Lung Carcinogenesis from a Morphostatic Perspective: Prevention and Therapeutic Potential of Phytochemicals for Targeting Cancer Stem Cells. International Journal of Molecular Sciences. 2021; 22(11):5697. https://doi.org/10.3390/ijms22115697
Chicago/Turabian StyleHeng, Win Sen, Frank A. E. Kruyt, and Shiau-Chuen Cheah. 2021. "Understanding Lung Carcinogenesis from a Morphostatic Perspective: Prevention and Therapeutic Potential of Phytochemicals for Targeting Cancer Stem Cells" International Journal of Molecular Sciences 22, no. 11: 5697. https://doi.org/10.3390/ijms22115697
APA StyleHeng, W. S., Kruyt, F. A. E., & Cheah, S.-C. (2021). Understanding Lung Carcinogenesis from a Morphostatic Perspective: Prevention and Therapeutic Potential of Phytochemicals for Targeting Cancer Stem Cells. International Journal of Molecular Sciences, 22(11), 5697. https://doi.org/10.3390/ijms22115697