
Breast Cancer Predisposition Genes and Synthetic Lethality


Abstract
1. Introduction
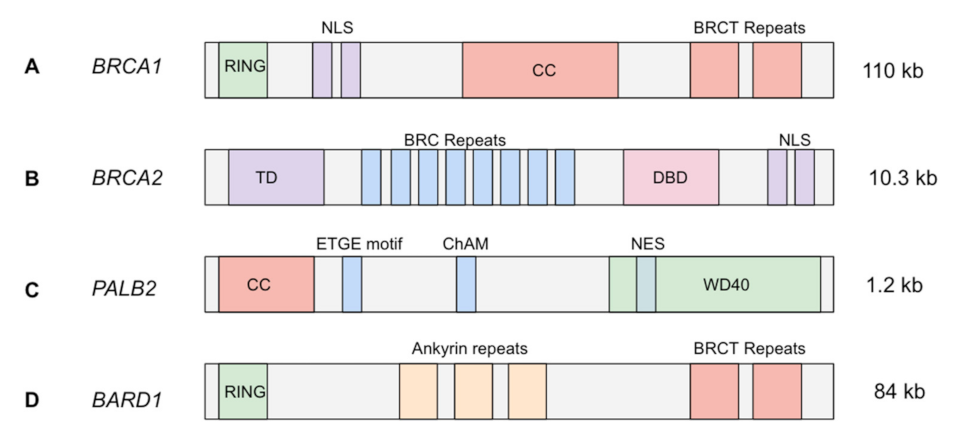
2. Breast CPGs Affecting HR
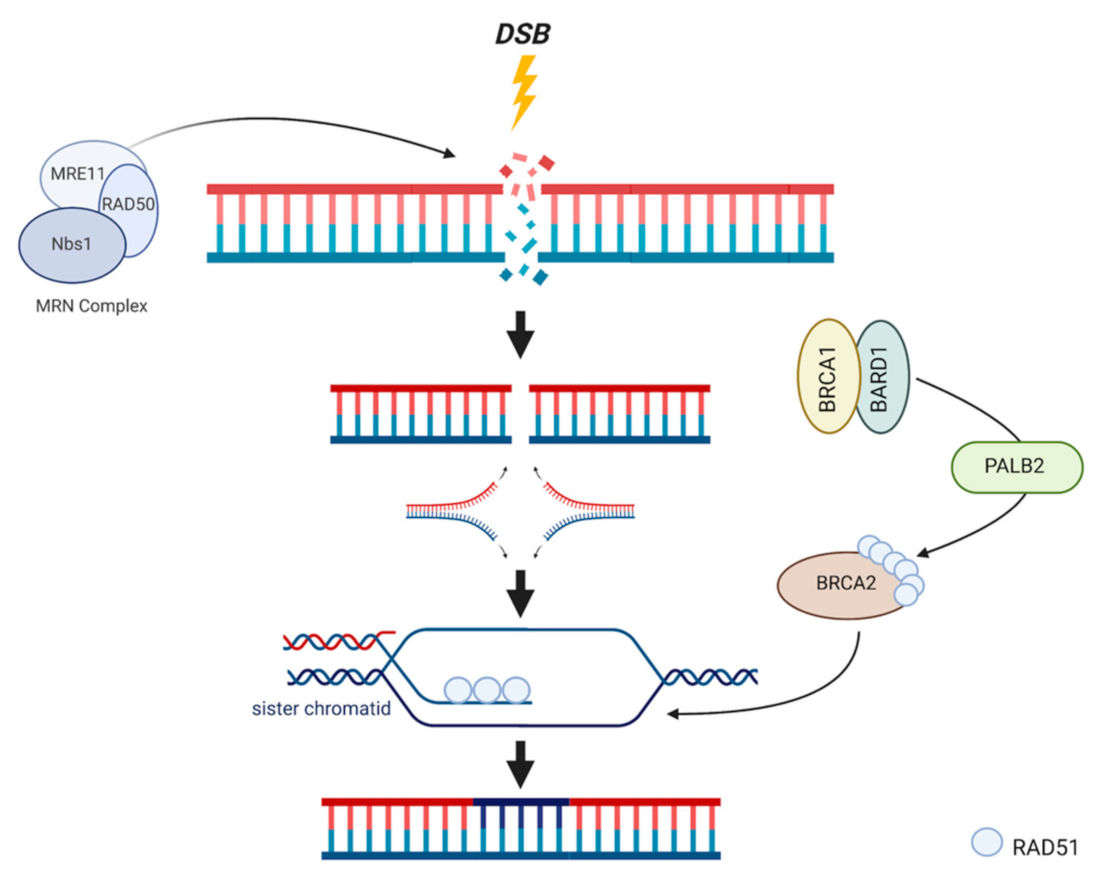
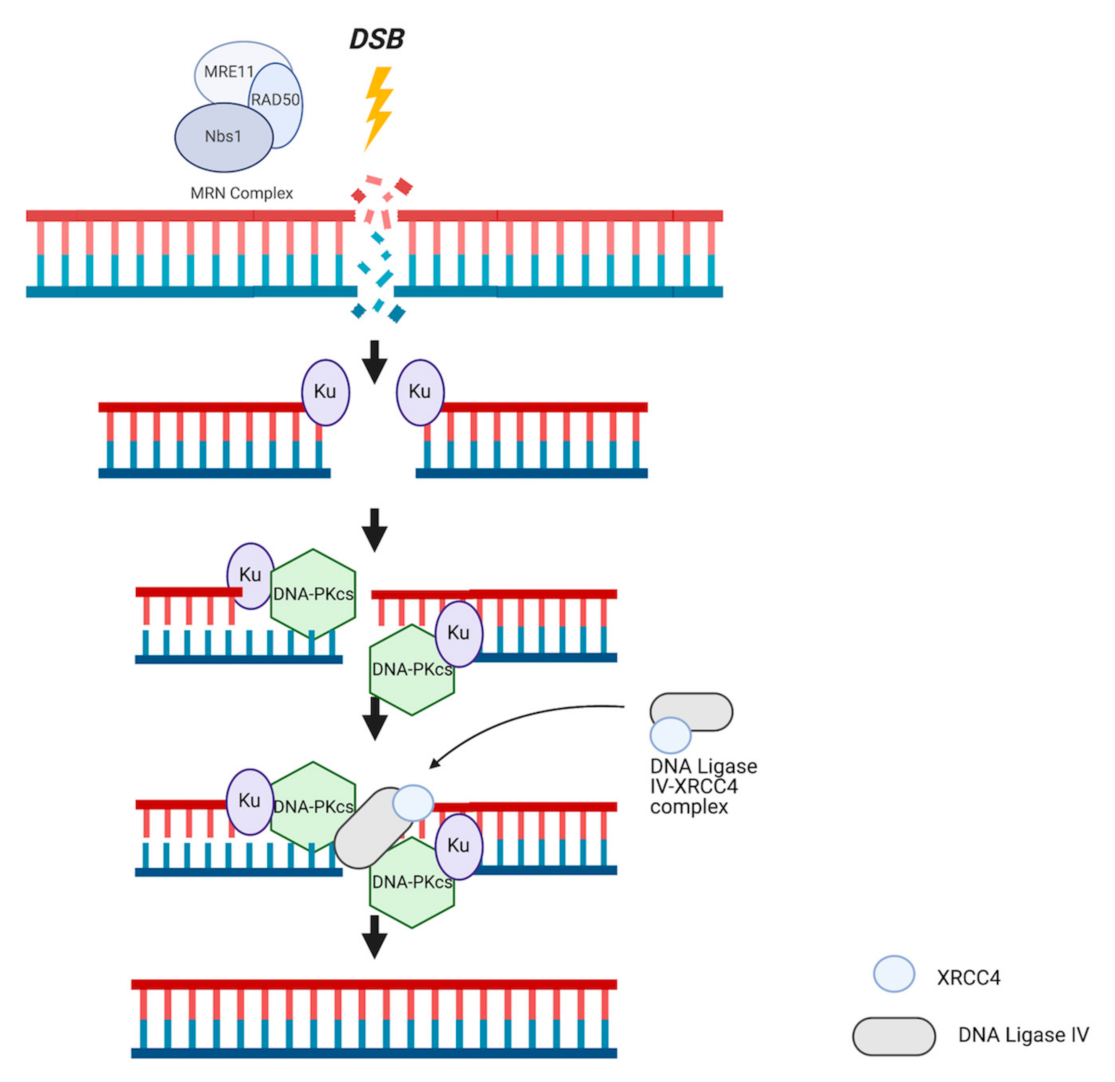
3. DNA Repair Systems
4. Repair-Independent BRCA1/2 Involvement
5. Traditional Therapeutic Options
6. Immune Checkpoint Blockade Therapy
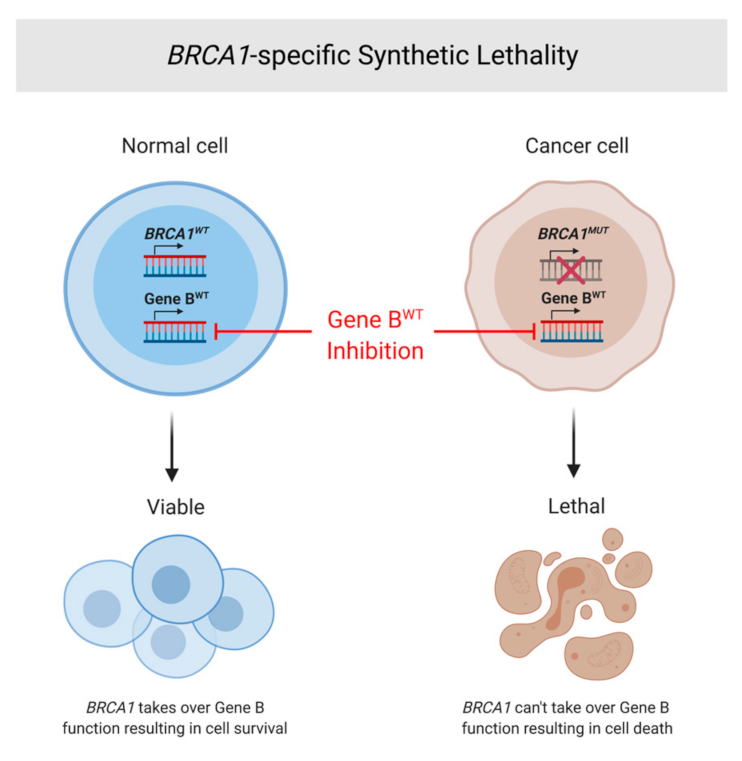
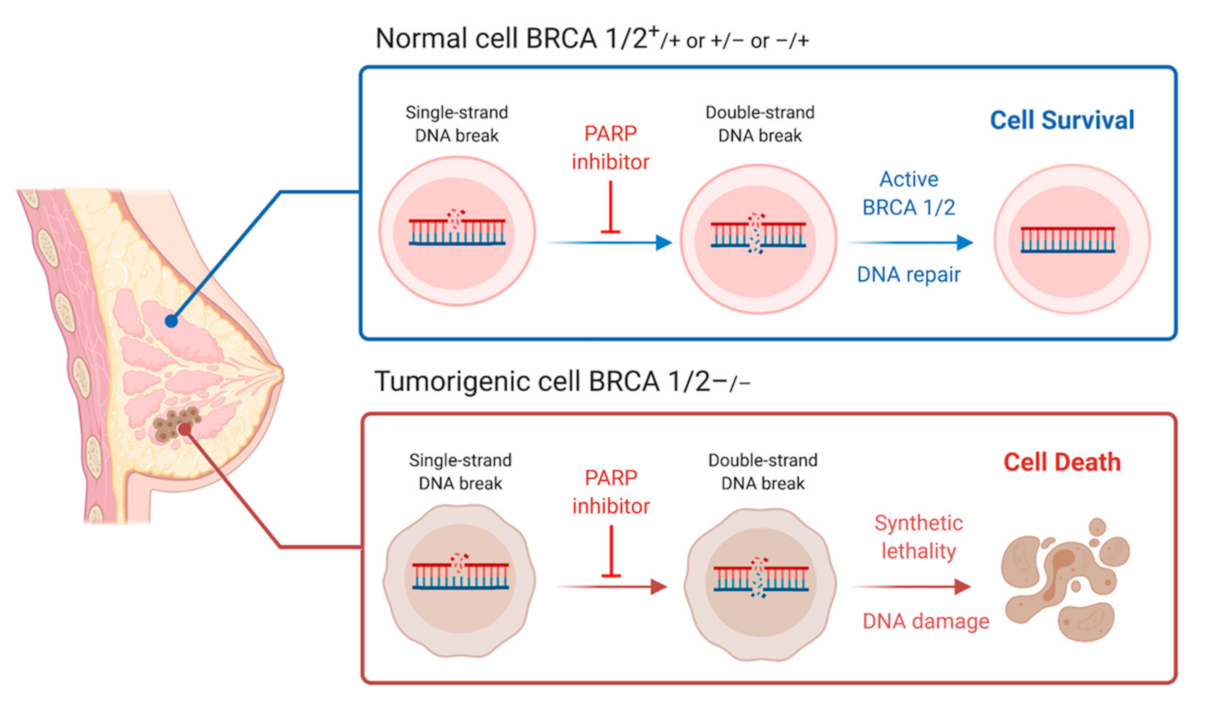
7. Synthetic Lethality and Therapeutic Options
8. BRCA1/2 Ovarian Cancer
9. PARP Inhibitor Clinical Trails and Use in Practice
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dossus, L.; Benusiglio, P.R. Lobular breast cancer: Incidence and genetic and non-genetic risk factors. Breast Cancer Res. 2015, 17, 1–8. [Google Scholar] [CrossRef]
- Bayraktar, S.; Glück, S. Systemic therapy options in BRCA mutation-associated breast cancer. Breast Cancer Res. Treat. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA J. Am. Med. Assoc. 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-Cancer Risk in Families with Mutations in PALB2. N. Engl. J. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999. [Google Scholar] [CrossRef]
- Sato, K.; Koyasu, M.; Nomura, S. Mutation status of RAD51C, PALB2 and BRIP1 in 100 Japanese familial breast cancer cases without BRCA1 and BRCA2 mutations. Cancer Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shimelis, H.; LaDuca, H.; Hu, C. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J. Natl. Cancer Inst. 2018. [Google Scholar] [CrossRef]
- Filippini, S.E.; Vega, A. Breast cancer genes: Beyond BRCA1 and BRCA2. Front. Biosci. 2013, 18, 1358–1372. [Google Scholar] [CrossRef]
- Pharoah, P.D.P.; Antoniou, A.C.; Easton, D.F.; Ponder, B.A.J. Polygenes, Risk Prediction, and Targeted Prevention of Breast Cancer. N. Engl. J. Med. 2008, 358, 2796–2803. [Google Scholar] [CrossRef]
- Śniadecki, M.; Brzeziński, M.; Darecka, K. Bard1 and breast cancer: The possibility of creating screening tests and new preventive and therapeutic pathways for predisposed women. Genes 2020, 11, 1251. [Google Scholar] [CrossRef] [PubMed]
- Silwal-Pandit, L.; Langerød, A.; Børresen-Dale, A.L. TP53 mutations in breast and ovarian cancer. Cold Spring Harb. Perspect Med. 2017, 7. [Google Scholar] [CrossRef]
- Børresen-Dale, A.L. TP53 and breast cancer. Hum. Mutat. 2003, 21, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Jackson, S.A.; Susswein, L.R. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Penson, A.V.; Taylor, B.S.; Petrini, J.H.J. Nbn−Mre11 interaction is required for tumor suppression and genomic integrity. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Murthy, P.; Muggia, F. Women’s cancers: How the discovery of BRCA genes is driving current concepts of cancer biology and therapeutics. Ecancermedicalscience 2019. [Google Scholar] [CrossRef]
- Liu, E.Y.; Xu, N.; O’Prey, J. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004. [Google Scholar] [CrossRef] [PubMed]
- Epstein, R.J. The unpluggable in pursuit of the undruggable: Tackling the dark matter of the cancer therapeutics universe. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, H.; Lord, C.J. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010. [Google Scholar] [CrossRef]
- Deng, C.X.; Brodie, S.G. Roles of BRCA1 and its interacting proteins. BioEssays 2000. [Google Scholar] [CrossRef]
- Bielinska, B.; Bielinska, B.A. The BRCA1 tumor suppressor: Potential long-range interactions of the BRCA1 promoter and the risk of breast cancer. Rev. Artic J. Transl. Sci. J. Transl. Sci. 2017, 3, 1–10. [Google Scholar] [CrossRef][Green Version]
- Semmler, L.; Reiter-Brennan, C.; Klein, A. BRCA1 and breast cancer: A review of the underlying mechanisms resulting in the tissue-specific tumorigenesis in mutation carriers. J. Breast Cancer 2019. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Christou, C.M.; Kyriacou, K. BRCA1 and its network of interacting partners. Biology 2013, 2, 40–63. [Google Scholar] [CrossRef]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA genes: The role in genome stability, cancer stemness and therapy resistance. J. Cancer 2019. [Google Scholar] [CrossRef]
- Clark, S.L.; Rodriguez, A.M.; Snyder, R.R.; Hankins, G.D.V.; Boehning, D. Structure-function of the tumor suppressor BRCA1. Comput. Struct. Biotechnol. J. 2012. [Google Scholar] [CrossRef]
- Godet, I.; MGilkes, D. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr. Cancer Sci. Ther. 2017. [Google Scholar] [CrossRef]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ma, J.; Wu, J. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr. Biol. 2009. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, J.; Zhang, K. Molecular Mechanisms of PALB2 Function and Its Role in Breast Cancer Management. Front. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, A.I.; Banerjee, T.; Wingo, M. Functional analysis of BARD1 missense variants in homology-directed repair and damage sensitivity. PLoS Genet. 2019. [Google Scholar] [CrossRef]
- Ayi, T.C.; Tsan, J.T.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Conservation of function and primary structure in the BRCA1-associated RING domain (BARD1) protein. Oncogene 1998. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; de Antoni, A.; Técher, H.; Baldi, G.; Costanzo, V. Moonlighting at replication forks – a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.N.; Kachnic, L.A. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Xia, B. PALB2/FANCN: Recombining cancer and fanconi anemia. Cancer Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Nepomuceno, T.C.; De Gregoriis, G.; de Oliveira, F.M.B.; Suarez-Kurtz, G.; Monteiro, A.N.; Carvalho, M.A. The role of PALB2 in the DNA damage response and cancer predisposition. Int. J. Mol. Sci. 2017, 18, 1886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell. 2006. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Van Der Sluis, P.C.; Boulware, D.; Hazlehurst, L.A.; Dalton, W.S. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood 2005. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F.; Formicola, D.; Capasso, M. Dualistic role of BARD1 in cancer. Genes 2017, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Jefford, C.E. Is there more to BARD1 than BRCA1? Nat. Rev. Cancer 2006. [Google Scholar] [CrossRef]
- Daza-Martin, M.; Densham, R.M.; Morris, J.R. BRCA1-BARD1: The importance of being in shape. Mol. Cell Oncol. 2019. [Google Scholar] [CrossRef]
- Densham, R.M.; Garvin, A.J.; Stone, H.R. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct Mol. Biol. 2016. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Wong, N. 53BP1 inhibits homologous recombination in BRCA1-deficient cells by blocking resection of DNA breaks. Cell 2010. [Google Scholar] [CrossRef]
- Levran, O.; Attwooll, C.; Henry, R.T. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat. Genet. 2005. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C.S.; He, M.; Mer, G.; Chen, J. The BRCT Domain Is a Phospho-Protein Binding Domain. Science 2003. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.B.; Bell, D.W.; Ganesan, S. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001. [Google Scholar] [CrossRef]
- Cantor, S.; Drapkin, R.; Zhang, F. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA damage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lundin, C.; Erixon, K.; Arnaudeau, C. Different Roles for Nonhomologous End Joining and Homologous Recombination following Replication Arrest in Mammalian Cells. Mol. Cell Biol. 2002. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018. [Google Scholar] [CrossRef]
- Ingram, S.P.; Warmenhoven, J.W.; Henthorn, N.T. Mechanistic modelling supports entwined rather than exclusively competitive DNA double-strand break repair pathway. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Onyango, D.O.; Lee, G.; Stark, J.M. PRPF8 is important for BRCA1-mediated homologous recombination. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the Choice between Recombination and End Joining at DNA Double-Strand Breaks. Mol. Cell Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, L.Y. BRCA1 and homologous recombination: Implications from mouse embryonic development. Cell Biosci. 2020. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.C.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006. [Google Scholar] [CrossRef]
- Smith, G.C.M.; Jackson, S.P. The DNA-dependent protein kinase. Genes Dev. 1999. [Google Scholar] [CrossRef]
- Doherty, A.J.; Jackson, S.P. DNA repair: How Ku makes ends meet. Curr. Biol. 2001. [Google Scholar] [CrossRef]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to dna and its implications for double-strand break repair. Nature 2001. [Google Scholar] [CrossRef]
- Martin, I.V.; MacNeill, S.A. ATP-dependent DNA ligases. Genome Biol. 2002. [Google Scholar] [CrossRef]
- Calsou, P.; Delteil, C.; Frit, P.; Drouet, J.; Salles, B. Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J. Mol. Biol. 2003. [Google Scholar] [CrossRef]
- Zhuang, J.; Zhang, J.; Willers, H. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res. 2006. [Google Scholar] [CrossRef] [PubMed]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G. HHS Public Access. Restoration 2018, 68, 414–430. [Google Scholar] [CrossRef]
- Daboussi, F.; Courbet, S.; Benhamou, S. A homologous recombination defect affects replication-fork progression in mammalian cells. J. Cell Sci. 2008, 121, 162–166. [Google Scholar] [CrossRef]
- Costanzo, V. Brca2, Rad51 and Mre11: Performing balancing acts on replication forks. DNA Repair 2011, 10, 1060–1065. [Google Scholar] [CrossRef]
- Lehmann, A.R.; Fuchs, R.P. Gaps and forks in DNA replication: Rediscovering old models. DNA Repair 2006, 5, 1495–1498. [Google Scholar] [CrossRef]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lemaçon, D.; Jackson, J.; Quinet, A. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1–BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef]
- Zimmer, J.; Tacconi, E.M.C.; Folio, C. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell. 2016, 61, 449–460. [Google Scholar] [CrossRef]
- Lai, X.; Broderick, R.; Bergoglio, V. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Demaria, S.; Formenti, S.C.; Galluzzi, L. Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 2018, 34, 361–378. [Google Scholar] [CrossRef]
- Zhu, Y.; Wu, J.; Zhang, C. BRCA mutations and survival in breast cancer: An updated systematic review and meta-analysis. Oncotarget 2016, 7, 70113–70127. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.K.; Van Den Broek, A.J.; Tollenaar, R.A.E.M. Breast Cancer Survival of BRCA1/BRCA2 Mutation Carriers in a Hospital-Based Cohort of Young Women. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Lee, A.; Moon, B.I.; Kim, T.H. BRCA1/BRCA2 pathogenic variant breast cancer: Treatment and prevention strategies. Ann. Lab. Med. 2020. [Google Scholar] [CrossRef]
- Pierce, L.J.; Phillips, K.A.; Griffith, K.A. Local therapy in BRCA1 and BRCA2 mutation carriers with operable breast cancer: Comparison of breast conservation and mastectomy. Breast Cancer Res. Treat. 2010. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.; Giannakakou, P. BRCA1 regulates microtubule dynamics and taxane-induced apoptotic cell signaling. Oncogene 2014. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Fumet, J.D.; Limagne, E.; Thibaudin, M.; Ghiringhelli, F. Immunogenic cell death and elimination of immunosuppressive cells: A double-edged sword of chemotherapy. Cancers 2020, 12, 2637. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef]
- Tan, T.J.; Chan, J.J.; Kamis, S.; Dent, R.A. What is the role of immunotherapy in breast cancer? Chin. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Saenger, Y. The Mechanism of Anti-CTLA-4 Activity and the Negative Regulation of T-Cell Activation. Oncologist 2008. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018. [Google Scholar] [CrossRef]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.; Alimzhanov, M.B.; Rajewsky, N.; Rajewsky, K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igα/β heterodimer. Cell 2004. [Google Scholar] [CrossRef]
- Lee, J.M.; Ledermann, J.A.; Kohn, E.C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 2014, 25, 32–40. [Google Scholar] [CrossRef]
- Morales, J.C.; Li, L.; Fattah, F.J. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014. [Google Scholar] [CrossRef]
- Bochum, S.; Berger, S.; Martens, U.M. Olaparib. Recent Results Cancer Res. 2018. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Thomas, A.; Murai, J.; Pommier, Y. The evolving landscape of predictive biomarkers of response to PARP inhibitors. J. Clin. Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Kersbergen, A.; Boon, U. Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 2013. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008. [Google Scholar] [CrossRef]
- Booth, L.; Cruickshanks, N.; Ridder, T.; Dai, Y.; Grant, S.; Dent, P. PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells. Cancer Biol. Ther. 2013, 14, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.; Poklepovic, A.; Dent, P. The CHK1 inhibitor SRA737 synergizes with PARP1 inhibitors to kill carcinoma cells. Cancer Biol. Ther. 2018, 19, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Kennedy, R.D.; Sidi, S.; Look, A.T.; D’Andrea, A. CHK1 inhibition as a strategy for targeting fanconi anemia (FA) DNA repair pathway deficient tumors. Mol. Cancer 2009, 8. [Google Scholar] [CrossRef] [PubMed]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Ghosh, M.; Kim, T.H. Synergism of AZD6738, an ATR inhibitor, in combination with belotecan, a camptothecin analogue, in chemotherapy-resistant ovarian cancer. Int. J. Mol. Sci. 2021, 22, 1223. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.L. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Krajewska, M.; Fehrmann, R.S.N.; Schoonen, P.M. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene 2015, 34, 3474–3481. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell. 2019, 73, 885–899.e6. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.; Park, M.; Eulitt, P.; Yang, C.; Yacoub, A.; Dent, P. Poly(ADP-Ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in carcinoma cells. Mol. Pharmacol. 2010, 78, 909–917. [Google Scholar] [CrossRef] [PubMed]
- O’Carrigan, B.; de Miguel Luken, M.J.; Papadatos-Pastos, D. Phase I trial of a first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combination (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies. J. Clin. Oncol. 2016, 34 (Suppl. 15), 2504. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Xie, X. BRCA mutations in the manifestation and treatment of ovarian cancer. Oncotarget 2017, 8, 97657–97670. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Ford, D.; Bishop, D.T. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Am. J. Hum. Genet. 1995, 56, 265–271. [Google Scholar]
- Ford, D.; Easton, D.F.; Stratton, M. Genetic Heterogeneity and Penetrance Analysis of the BRCA1 and BRCA2 Genes in Breast Cancer Families. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef]
- Toss, A.; Tomasello, C.; Razzaboni, E. Hereditary ovarian cancer: Not only BRCA1 and 2 Genes. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Coats, K.H. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. J. Am. Med. Assoc. 2006, 295, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Nevanlinna, H.; Bartek, J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 2006, 25, 5912–5919. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, E.S.; Lallemand, F.; Petitalot, A.; Caputo, S.M.; Rouleau, E. Hrness in breast and ovarian cancers. Int. J. Mol. Sci. 2020, 21, 3850. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; McCluggage, W.G.; Gilks, C.B. High-grade serous carcinoma of tubo-ovarian origin: Recent developments. Histopathology 2017, 71, 339–356. [Google Scholar] [CrossRef]
- George, S.H.L.; Shaw, P. BRCA and early events in the development of serous ovarian cancer. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Scully, R.; Chen, J.; Plug, A. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997, 88, 265–275. [Google Scholar] [CrossRef]
- Kim, J.; Park, E.Y.; Kim, O. Cell origins of high-grade serous ovarian cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef]
- Piek, J.M.J.; Van Diest, P.J.; Zweemer, R.P. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol. 2001, 195, 451–456. [Google Scholar] [CrossRef]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-grade serous ovarian cancer: Basic sciences, clinical and therapeutic standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef]
- Bast, R.C.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Wei, W.; Li, Y.; Lv, S.; Zhang, C.; Tian, Y. PARP-1 may be involved in angiogenesis in epithelial ovarian cancer. Oncol. Lett. 2016, 12, 4561–4567. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J. Panel to Discuss How Combination Therapies Could Create Better, Longer-Lasting Outcomes. 2021. Available online: https://www.aacrnews.org/news/panel-to-discuss-how-combination-therapies-could-create-better-longer-lasting-outcomes/ (accessed on 24 May 2021).
- Zhang, X.; Chiang, H.C.; Wang, Y. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Pillay, N.; Tighe, A.; Nelson, L. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019, 35, 519–533.e8. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Arend, R.; Westin, S.N.; Coleman, R. Decision analysis for secondline maintenance treatment of platinum sensitive recurrent ovarian cancer: A review. Int. J. Gynecol. Cancer 2020, 30. [Google Scholar] [CrossRef] [PubMed]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Walker, L.C.; Lattimore, V.L.; Kvist, A. Comprehensive Assessment of BARD1 Messenger Ribonucleic Acid Splicing With Implications for Variant Classification. Front Genet. 2019. [Google Scholar] [CrossRef]
- Taylor, A.M.; Chan, D.L.H.; Tio, M. PARP (Poly ADP-Ribose Polymerase) inhibitors for locally advanced or metastatic breast cancer. Cochrane Database Syst. Rev. 2021, 2021. [Google Scholar] [CrossRef]
- Mirza, M.R.; Coleman, R.L.; González-Martín, A. The forefront of ovarian cancer therapy: Update on PARP inhibitors. Ann. Oncol. 2020, 31, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Liu, W.; Xu, S. PARP inhibitors as maintenance therapy in newly diagnosed advanced ovarian cancer: A meta-analysis. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 485–493. [Google Scholar] [CrossRef]
- Cancer Statistics Review, 1975-2017-SEER Statistics. Available online: https://seer.cancer.gov/archive/csr/1975_2017/ (accessed on 16 May 2021).
- Kenny, L.M.; Orsi, F.; Adam, A. Interventional radiology in breast cancer. Breast 2017, 35, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Clinical Challenges: PARP Inhibitors in Ovarian Cancer | MedPage Today. Available online: https://www.medpagetoday.com/clinical-challenges/asco-ovarian-cancer/87068 (accessed on 16 May 2021).
- Wang, T.; Yu, H.; Hughes, N.W. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903.e15. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Thomas, D.; Chan, S. Systematic discovery of mutation-specific synthetic lethals by mining pan-cancer human primary tumor data. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Xiao, Y.; Thakkar, K.N.; Zhao, H. The m6A RNA demethylase FTO is a HIF-independent synthetic lethal partner with the VHL tumor suppressor. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Predisposition Genes (CPGs) | |||
|---|---|---|---|
| Risk Level | Gene | Estimated Incidence of Mutations in Hereditary Breast Cancer | Associated Process |
| High Risk | BRCA1 | ~50% between BRCA1 and BRCA2 [4] | HR (several functions) [6,8] |
| BRCA2 | ~50% between BRCA1 and BRCA2 [4] | HR (loading and leading RAD51) [6,8] | |
| PALB2 | <3% [9,10] | HR (recruitment of BRCA2 and DNA replication via ICL) [6] | |
| BARD1 | <1% [8,11] | HR (ubiquitin ligase) [6] | |
| Moderate Risk | TP53 | 25–30% [12,13] | Cell cycle checkpoint protein (triggered by DNA damage) [7] |
| BRIP1 | <3% [9,10] | HR (complexes with BRCA1) [7] | |
| RAD51C | <3% [9,10] | HR (promotes invading strand exchange) [8] | |
| MSH6 | <1% [8] | Mismatch repair (MMR)-associated protein [14] | |
| Low Risk | ATM | <3% [9,10] | Cell cycle checkpoint protein [7] |
| CHEK2 | <3% [9,10] | Cell cycle checkpoint protein (checkpoint kinase-2) [7] | |
| Nbn | <1% [8] | DDR (complexes with MRE11 and RAD50 in DSB) [15] | |
| RAD50 | <1% [8] | DDR (complexes with MRE11 and Nbn in DSB) [15] | |
| Trial Name | Status | Conditions | Phase | Results Summary |
|---|---|---|---|---|
| Efficacy and Safety of PARPi to Treat Pancreatic Cancer | Unknown | Pancreatic Cancer | Phase II | None available yet |
| Survival Data and Characteristics of Finisterian Patients Treated with PARP Inhibitors for Ovarian Cancer Between 2014 and 2019 | Completed | Ovarian Neoplasm | Does not specify | None available |
| Pamiparib in Fusion Positive, Reversion Negative HGSOC or Carcinosarcoma With BRCA1/2 Gene Mutations If Progression on PARPI or Chemotherapy | Recruiting | Ovarian Cancer Carcinosarcoma | Phase II | None available yet |
| A Study to Examine Olaparib Maintenance Retreatment in Patients with Epithelial Ovarian Cancer | Active, not recruiting | Epithelial Ovarian Cancer | Phase III | None available yet |
| Study of M4344 in Combination with Niraparib | Not yet recruiting | Advanced Solid Tumor Breast Cancer | Phase I Phase II | None available yet |
| Window of Opportunity Trial, PARP Inhibitor Rucaparib Affect on PD-L1 Expression in Triple Negative Breast Tumors | Recruiting | Breast Cancer | Early Phase I | None available yet |
| Olaparib Arsenic Trioxide Platinum Resistance Relapsed Ovarian Cancer | Active, not recruiting | Ovarian Cancer | Phase I Phase II | None available yet |
| Recurrent Ovarian Carcinosarcoma Anti-pd-1 Niraparib | Recruiting | Ovarian Carcinosarcoma Endometrial Carcinosarcoma | Phase II Phase III | None available yet |
| ATR Inhibitor BAY 1895344 Plus Niraparib Phase 1b Study in Advanced Solid Tumors and Ovarian Cancer | Recruiting | Advanced Solid Tumors (excluding prostate cancer) Ovarian Cancer | Phase I | None available yet |
| The Clinical Markers for PARP Inhibitors-related Efficacy in Ovarian Cancer | Recruiting | Ovarian Cancer | Does not specify | None available yet |
| Platinum and PARPI for Neoadjuvant Treatment of Triple Negative Breast Cancer (TNBC) and/or Germline BRCA (gBRCA) Positive Breast Cancer | Recruiting | Breast Cancer | Phase II Phase III | None available yet |
| Induction and Maintenance Treatment with PARP Inhibitor and Immunotherapy in HPV-negative HNSCC | Recruiting | Head and Neck Squamous Cell Carcinoma | Phase II | None available yet |
| Stratified Evaluation of PDS and NACT-IDS in Ovarian Cancer (FOCUS) | Not yet recruiting | Epithelial Ovarian Cancer Fallopian Tube Cancer Primary Peritoneal Carcinoma | Phase III | None available yet |
| Niraparib Maintenance in Patients with Advanced Ovarian Cancer at Neoadjuvant Setting | Recruiting | Ovarian Cancer | Phase II | None available yet |
| Combination of HX008 and Niraparib in Germ-line-mutated Metastatic Breast Cancer | Not yet recruiting | Breast Cancer | Phase II | None available yet |
| Pembrolizumab and Olaparib in Recurrent/Metastatic, Platinum Resistant Nasopharyngeal Cancer | Not yet recruiting | Nasopharyngeal Carcinoma | Phase II | None available yet |
| Anlotinib and Niraparib Dual Therapy Evaluation in Platinum-resistant Recurrent Ovarian Cancer | Recruiting | Platinum-Resistant Ovarian Cancer | Phase II | None available yet |
| Multi-maintenance Olaparib After Disease Recurrence in Participants with Platinum Sensitive BRCA HGSOC | Active, not recruiting | Ovarian Cancer | Early Phase I | None available yet |
| Olaparib After Response to Trabectedin-pegylated Liposomal Doxorubicin in Recurrent Ovarian Carcinoma | Active, not recruiting | Ovarian Cancer | Phase II | None available yet |
| Investigation of 2X-121 in Patients with Advanced Ovarian Cancer Selected by the 2X-121 DRP | Recruiting | Advanced Ovarian Cancer | Phase II | None available yet |
| DVAC/OvCa and Standard of Care (SoC) in Relapsed Ovarian, Fallopian Tube, and Primary Peritoneal Carcinoma | Not yet recruiting | Ovarian Cancer Fallopian Tube Cancer Peritoneal Cancer | Phase III | None available yet |
| A Study of Niraparib in Patients with Ovarian Cancer Who Have Received Three or Four Previous Chemotherapy Regimens | Active, not recruiting | Ovarian Neoplasms Ovarian Cancer | Phase II | Of 463 participants who received Niraparib, 122 completed study. Of 47 patients in primary efficacy population, 13 (28%) achieved overall response [159] |
| Olaparib and Temozolomide in Treating Patients with Advanced, Metastatic, or Unresectable Uterine Leiomyosarcoma | Active, not recruiting | Stage III–IV Uterine Corpus Leiomyosarcoma | Phase II | None available yet |
| Analysis of the Clinical Experience with Rucaparib in the Rucaparib Access Program (RAP) in Spain—A GEICO Study | Recruiting | Epithelial Ovarian Cancer Fallopian Tube Cancer Primary Peritoneal Cancer | Does not specify | None available yet |
| A Study to Evaluate Rucaparib in Patients with Solid Tumors and With Deleterious Mutations in HRR Genes | Recruiting | Solid Tumor | Phase II | None available yet |
| A Study in Ovarian Cancer Patients Evaluating Rucaparib and Nivolumab as Maintenance Treatment Following Response to Front-Line Platinum-Based Chemotherapy | Active, not recruiting | Epithelial Ovarian Cancer Primary Peritoneal Fallopian Tube Cancer | Phase III | None available yet |
| A Study of Rucaparib Versus Physician’s Choice of Therapy in Patients with Metastatic Castration-resistant Prostate Cancer and Homologous Recombination Gene Deficiency | Recruiting | Metastatic Castration-Resistant Prostate Cancer | Phase III | None available yet |
| A Study of Rucaparib in Patients with Metastatic Castration-resistant Prostate Cancer and Homologous Recombination Gene Deficiency | Active, not recruiting | Metastatic Castration-Resistant Prostate Cancer | Phase II | None available yet |
| A Study of ZEN003694 and Talazoparib in Patients With Triple Negative Breast Cancer | Recruiting | Triple -Negative Breast Cancer | Phase II | None available yet |
| An Efficacy and Safety Study of Niraparib in Men With Metastatic Castration-Resistant Prostate Cancer and DNA-Repair Anomalies | Active, not recruiting | Prostatic neoplasms | Phase II | None available yet |
| Ascending Doses of Ceralasertib in Combination With Chemotherapy and/or Novel Anti Cancer Agents | Recruiting | Advanced Solid Malignancies—Head and Neck Squamous Cell Carcinoma, ATM Pro/Deficient Non-Small-Cell Lung Cancer, Gastric, Breast, and Ovarian Cancer | Phase I Phase II | None available yet |
| Olaparib With or Without Atezolizumab in Treating Patients With Locally Advanced Unresectable or Metastatic Non-HER2-Positive Breast Cancer | Recruiting | Locally Advanced Unresectable Breast Carcinoma Metastatic Breast Carcinoma Stage III–IV Breast Cancer | Phase II | None available yet |
| Niraparib and Dostarlimab for the Treatment of Germline or Somatic BRCA1/2 and PALB2 Mutated Metastatic Pancreatic Cancer | Recruiting | Metastatic Pancreatic Ductal Adenocarcinoma Stage IV Pancreatic Cancer | Phase II | None available yet |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neiger, H.E.; Siegler, E.L.; Shi, Y. Breast Cancer Predisposition Genes and Synthetic Lethality. Int. J. Mol. Sci. 2021, 22, 5614. https://doi.org/10.3390/ijms22115614
Neiger HE, Siegler EL, Shi Y. Breast Cancer Predisposition Genes and Synthetic Lethality. International Journal of Molecular Sciences. 2021; 22(11):5614. https://doi.org/10.3390/ijms22115614
Chicago/Turabian StyleNeiger, Hannah E., Emily L. Siegler, and Yihui Shi. 2021. "Breast Cancer Predisposition Genes and Synthetic Lethality" International Journal of Molecular Sciences 22, no. 11: 5614. https://doi.org/10.3390/ijms22115614
APA StyleNeiger, H. E., Siegler, E. L., & Shi, Y. (2021). Breast Cancer Predisposition Genes and Synthetic Lethality. International Journal of Molecular Sciences, 22(11), 5614. https://doi.org/10.3390/ijms22115614