
Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease


{kind=link}
Abstract
1. Introduction
2. Neuroinflammation in PD
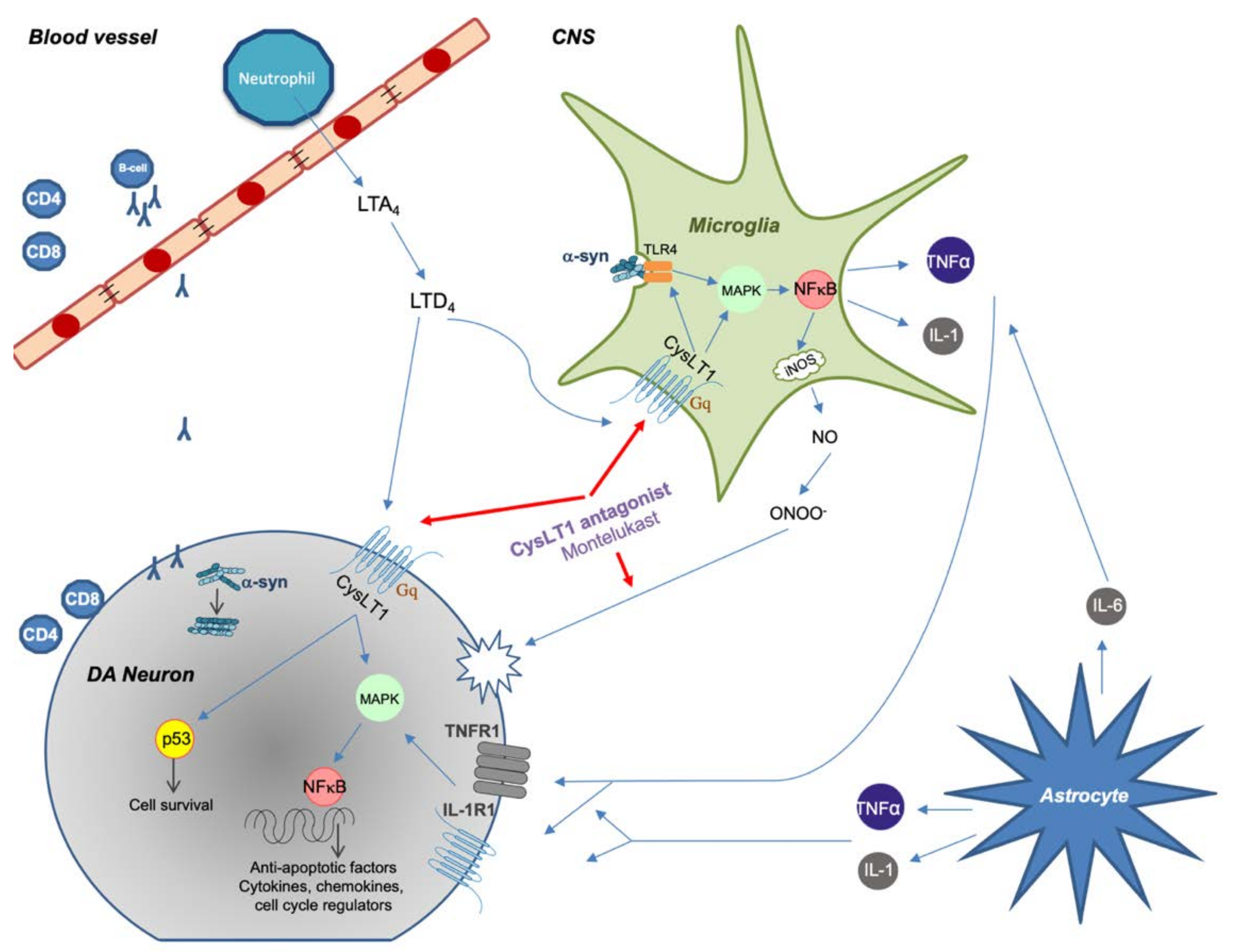
2.1. Astrocytes
2.2. Microglia
2.3. Toll-Like Receptors
2.4. Autophagy
2.5. Monocytes
2.6. Adaptive Immunity
2.7. Cytokines
2.8. Genetic and Environmental Factors
3. Immunomodulatory Treatment Studies
4. Leukotriene Signaling Pathway
4.1. Leukotrienes
4.2. Leukotrienes in CNS Disorders
4.3. 5-Lipoxygenase
5. Montelukast
5.1. Discovery and Current Use
5.2. Montelukast as a Treatment for PD
5.3. Montelukast as a Treatment for other CNS Disorders
6. Conclusions
Funding
Conflicts of Interest
References
- Calabrese, V.P.; Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; et al. Projected Number of People With Parkinson Disease in the Most Populous Nations, 2005 Through 2030. Neurology 2007, 69, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS Clinical Diagnostic Criteria for Parkinson’s Disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Postuma, R.B.; Adler, C.H.; Bloem, B.R.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.; Joseph, L.; et al. MDS Research Criteria for Prodromal Parkinson’s Disease. Mov. Disord. 2015, 30, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, S.; Chang, S.C.; Lee, J. Significant Roles of Neuroinflammation in Parkinson’s Disease: Therapeutic Targets for PD Prevention. Arch. Pharm. Res. 2019, 42, 416–425. [Google Scholar] [CrossRef]
- Fox, S.H.; Katzenschlager, R.; Lim, S.Y.; Barton, B.; de Bie, R.M.A.; Seppi, K.; Coelho, M.; Sampaio, C. International Parkinson and Movement Disorder Society Evidence-Based Medicine Review: Update on Treatments for the Motor Symptoms of Parkinson’s Disease. Mov. Disord. 2018, 33, 1248–1266. [Google Scholar] [CrossRef]
- Kaur, K.; Gill, J.S.; Bansal, P.K.; Deshmukh, R. Neuroinflammation—A Major Cause for Striatal Dopaminergic Degeneration in Parkinson’s Disease. J. Neurol. Sci. 2017, 381, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2020, 36, 16–24. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Vardjan, N.; Zorec, R. Physiology of astroglia. In Advances in Experimental Medicine and Biology; Springer Nature Singapore: Singapore, 2019; Volume 1175, pp. 45–91. [Google Scholar]
- Verkhratsky, A.; Ho, M.S.; Vardjan, N.; Zorec, R.; Parpura, V. General pathophysiology of astroglia. In Advances in Experimental Medicine and Biology; 2019; Volume 1175, pp. 149–179. [Google Scholar]
- Miller, S.J. Astrocyte Heterogeneity in the Adult Central Nervous System. Front. Cell. Neurosci. 2018, 12, 401. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS Neurodegenerative Diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P. Corticobasal Degeneration: Advances in Clinicopathology and Biomarkers. Curr. Opin. Neurol. 2019, 32, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.S. Microglia in Parkinson’s Disease. In Advances in Experimental Medicine and Biology; Springer Nature Singapore: Singapore, 2019; Volume 640, pp. 335–353. ISBN 9780387097886. [Google Scholar]
- Cerami, C.; Iaccarino, L.; Perani, D. Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of in Vivo PET Imaging. Int. J. Mol. Sci. 2017, 18, 993. [Google Scholar] [CrossRef]
- Jucaite, A.; Svenningsson, P.; Rinne, J.O.; Cselényi, Z.; Varnäs, K.; Johnström, P.; Amini, N.; Kirjavainen, A.; Helin, S.; Minkwitz, M.; et al. Effect of the Myeloperoxidase Inhibitor AZD3241 on Microglia: A PET Study in Parkinson’s Disease. Brain 2015, 138, 2687–2700. [Google Scholar] [CrossRef]
- Best, L.; Ghadery, C.; Pavese, N.; Tai, Y.F.; Strafella, A.P. New and Old TSPO PET Radioligands for Imaging Brain Microglial Activation in Neurodegenerative Disease. Curr. Neurol. Neurosci. Rep. 2019, 19, 24. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Iranzo, A.; Østergaard, K.; Serradell, M.; Otto, M.; Svendsen, K.B.; Garrido, A.; Vilas, D.; Borghammer, P.; Santamaria, J.; et al. Assessment of Neuroinflammation in Patients with Idiopathic Rapid-Eye-Movement Sleep Behaviour Disorder: A Case-Control Study. Lancet Neurol. 2017, 16, 789–796. [Google Scholar] [CrossRef]
- Cumming, P.; Burgher, B.; Patkar, O.; Breakspear, M.; Vasdev, N.; Thomas, P.; Liu, G.J.; Banati, R. Sifting through the Surfeit of Neuroinflammation Tracers. J. Cereb. Blood Flow Metab. 2018, 38, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Varnäs, K.; Cselényi, Z.; Jucaite, A.; Halldin, C.; Svenningsson, P.; Farde, L.; Varrone, A. PET Imaging of [11C]PBR28 in Parkinson’s Disease Patients Does Not Indicate Increased Binding to TSPO despite Reduced Dopamine Transporter Binding. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Kim, J.H.; Greenamyre, J.T. Selective Microglial Activation in the Rat Rotenone Model of Parkinson’s Disease. Neurosci. Lett. 2003, 341, 87–90. [Google Scholar] [CrossRef]
- Zhang, D.; Li, S.; Hou, L.; Jing, L.; Ruan, Z.; Peng, B.; Zhang, X.; Hong, J.-S.; Zhao, J.; Wang, Q. Microglial Activation Contributes to Cognitive Impairments in Rotenone-Induced Mouse Parkinson’s Disease Model. J. Neuroinflammation 2021, 18, 4. [Google Scholar] [CrossRef]
- Kouli, A.; Horne, C.B.; Williams-Gray, C.H. Toll-like Receptors and Their Therapeutic Potential in Parkinson’s Disease and α-Synucleinopathies. Brain. Behav. Immun. 2019, 81, 41–51. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like Receptor 4 Is Required for α-Synuclein Dependent Activation of Microglia and Astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Dzamko, N.; Geczy, C.; Halliday, G. Inflammation Is Genetically Implicated in Parkinson’s Disease. Neuroscience 2015, 302, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 Absence Reduces Neuroinflammation and Inflammasome Activation in Parkinson’s Diseases in Vivo Model. Brain. Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef]
- Siracusa, R.; Paterniti, I.; Cordaro, M.; Crupi, R.; Bruschetta, G.; Campolo, M.; Cuzzocrea, S.; Esposito, E. Neuroprotective Effects of Temsirolimus in Animal Models of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 2403–2419. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. Blood Monocytes and Their Subsets: Established Features and Open Questions. Front. Immunol. 2015, 6, 423. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Microglia and Monocytes: ’Tis Plain the Twain Meet in the Brain. Nat. Neurosci. 2011, 14, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Wijeyekoon, R.S.; Kronenberg-Versteeg, D.; Scott, K.M.; Hayat, S.; Kuan, W.L.; Evans, J.R.; Breen, D.P.; Cummins, G.; Jones, J.L.; Clatworthy, M.R.; et al. Peripheral Innate Immune and Bacterial Signals Relate to Clinical Heterogeneity in Parkinson’s Disease. Brain. Behav. Immun. 2020, 87, 473–488. [Google Scholar] [CrossRef]
- Farmen, K.; Nissen, S.K.; Stokholm, M.G.; Iranzo, A.; Østergaard, K.; Serradell, M.; Otto, M.; Svendsen, K.B.; Garrido, A.; Vilas, D.; et al. Monocyte Markers Correlate with Immune and Neuronal Brain Changes in REM Sleep Behavior Disorder. Proc. Natl. Acad. Sci. USA 2021, 118, e2020858118. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, S.; Liu, J. The Role of T Cells in the Pathogenesis of Parkinson’s Disease. Prog. Neurobiol. 2018, 169, 1–23. [Google Scholar] [CrossRef]
- Kannarkat, G.T.; Boss, J.M.; Tansey, M.G. The Role of Innate and Adaptive Immunity in Parkinson’s Disease. J. Parkinsons. Dis. 2013, 3, 493–514. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Invest. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Maxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human IPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2018, 23, 123–131. [Google Scholar] [CrossRef]
- Green, H.F.; Khosousi, S.; Svenningsson, P. Plasma IL-6 and IL-17A Correlate with Severity of Motor and Non-Motor Symptoms in Parkinson’s Disease. J. Parkinsons. Dis. 2019, 9, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-Specific T Cell Reactivity Is Associated with Preclinical and Early Parkinson’s Disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Gadina, M.; Siegel, R. Cytokines and cytokine receptors. In Clinical Immunology; Elsevier: Amsterdam, The Netherlands, 2013; pp. 108–135. [Google Scholar]
- Murray, M.E.; Kouri, N.; Lin, W.-L.; Jack, C.R.; Dickson, D.W.; Vemuri, P. Clinicopathologic Assessment and Imaging of Tauopathies in Neurodegenerative Dementias. Alzheimers. Res. Ther. 2014, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, Y.; Cao, Z.; Liu, Q.; Cheng, Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front. Immunol. 2018, 9, 2122. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s Disease: A Target for Neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Alam, Q.; Zubair Alam, M.; Mushtaq, G.A.; Damanhouri, G.; Rasool, M.; Amjad Kamal, M.; Haque, A. Inflammatory Process in Alzheimer’s and Parkinson’s Diseases: Central Role of Cytokines. Curr. Pharm. Des. 2016, 22, 541–548. [Google Scholar] [CrossRef]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease among Patients with Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef] [PubMed]
- West, P.K.; Viengkhou, B.; Campbell, I.L.; Hofer, M.J. Microglia Responses to Interleukin-6 and Type I Interferons in Neuroinflammatory Disease. Glia 2019, 67, 1821–1841. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.B.; Graeber, M.B. Towards a Pathway Definition of Parkinson’s Disease: A Complex Disorder with Links to Cancer, Diabetes and Inflammation. Neurogenetics 2008, 9, 1–13. [Google Scholar] [CrossRef]
- Saiki, M.; Baker, A.; Williams-Gray, C.H.; Foltynie, T.; Goodman, R.S.; Taylor, C.J.; Compston, D.A.S.; Barker, R.A.; Sawcer, S.J.; Goris, A. Association of the Human Leucocyte Antigen Region with Susceptibility to Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Vivekanantham, S.; Shah, S.; Dewji, R.; Dewji, A.; Khatri, C.; Ologunde, R. Neuroinflammation in Parkinson’s Disease: Role in Neurodegeneration and Tissue Repair. Int. J. Neurosci. 2015, 125, 717–725. [Google Scholar] [CrossRef]
- Gagliano, S.A.; Pouget, J.G.; Hardy, J.; Knight, J.; Barnes, M.R.; Ryten, M.; Weale, M.E. Genomics Implicates Adaptive and Innate Immunity in Alzheimer’s and Parkinson’s Diseases. Ann. Clin. Transl. Neurol. 2016, 3, 924–933. [Google Scholar] [CrossRef]
- Witoelar, A.; Jansen, I.E.; Wang, Y.; Desikan, R.S.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.K.; Hernandez, D.G.; Djurovic, S.; Schork, A.J.; et al. Genome-Wide Pleiotropy between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017, 74, 780–792. [Google Scholar] [CrossRef]
- Martino, R.; Candundo, H.; van Lieshout, P.; Shin, S.; Crispo, J.A.G.; Barakat-Haddad, C. Onset and Progression Factors in Parkinson’s Disease: A Systematic Review. Neurotoxicology 2017, 61, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kline, E.M.; Houser, M.C.; Herrick, M.K.; Seibler, P.; Klein, C.; West, A.; Tansey, M.G. Genetic and Environmental Factors in Parkinson’s Disease Converge on Immune Function and Inflammation. Mov. Disord. 2020, 36, 25–36. [Google Scholar] [CrossRef]
- Alqasrawi, D.; Abdelli, L.S.; Naser, S.A. Mystery Solved: Why Smoke Extract Worsens Disease in Smokers with Crohn’s Disease and Not Ulcerative Colitis? Gut Map! Microorganisms 2020, 8, 666. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Danielsen, E.H.; Hamilton-Dutoit, S.J.; Borghammer, P. Pathological α-Synuclein in Gastrointestinal Tissues from Prodromal Parkinson Disease Patients. Ann. Neurol. 2016, 79, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Rees, K.; Stowe, R.; Patel, S.; Ives, N.; Breen, K.; Clarke, C.E.; Ben-Shlomo, Y. Non-Steroidal Anti-Inflammatory Drugs as Disease-Modifying Agents for Parkinson’s Disease: Evidence from Observational Studies. Cochrane Database Syst. Rev. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A.; Gross, A.; Vouri, S.M.; Camacho-Soto, A.; Willis, A.W.; Searles Nielsen, S. Immunosuppressants and Risk of Parkinson Disease. Ann. Clin. Transl. Neurol. 2018, 5, 870–875. [Google Scholar] [CrossRef]
- Zhang, Y.-S.; Li, J.-D.; Yan, C. An Update on Vinpocetine: New Discoveries and Clinical Implications. Eur. J. Pharmacol. 2018, 819, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Xiaomu, W.; Xufang, X.; Liang, S. Vinpocetine Regulates Levels of Circulating TLRs in Parkinson’s Disease Patients. Neurol. Sci. 2019, 40, 113–120. [Google Scholar] [CrossRef]
- Offermanns, S. The Nicotinic Acid Receptor GPR109A (HM74A or PUMA-G) as a New Therapeutic Target. Trends Pharmacol. Sci. 2006, 27, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Giri, B.; Belanger, K.; Seamon, M.; Bradley, E.; Purohit, S.; Chong, R.; Morgan, J.C.; Baban, B.; Wakade, C. Niacin Ameliorates Neuro-Inflammation in Parkinson’s Disease via GPR109A. Int. J. Mol. Sci. 2019, 20, 4559. [Google Scholar] [CrossRef]
- Wakade, C.; Chong, R.; Bradley, E.; Morgan, J.C. Low-dose Niacin Supplementation Modulates GPR109A, Niacin Index and Ameliorates Parkinson’s Disease Symptoms without Side Effects. Clin. Case Reports 2015, 3, 635–637. [Google Scholar] [CrossRef]
- Greenland, J.C.; Cutting, E.; Kadyan, S.; Bond, S.; Chhabra, A.; Williams-Gray, C.H. Azathioprine Immunosuppression and Disease Modification in Parkinson’s Disease (AZA-PD): A Randomised Double-Blind Placebo-Controlled Phase II Trial Protocol. BMJ Open 2020, 10, e040527. [Google Scholar] [CrossRef]
- Samuelsson, B. Leukotrienes: Mediators of Immediate Hypersensitivity Reactions and Inflammation. Science 1983, 220, 568–575. [Google Scholar] [CrossRef]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and Leukotriene Pathways: Biochemistry, Biology, and Roles in Disease. Chem. Rev. 2011, 111, 5866–5896. [Google Scholar] [CrossRef]
- Bäck, M.; Powell, W.S.; Dahlén, S.E.; Drazen, J.M.; Evans, J.F.; Serhan, C.N.; Shimizu, T.; Yokomizo, T.; Rovati, G.E. Update on Leukotriene, Lipoxin and Oxoeicosanoid Receptors: IUPHAR Review 7. Br. J. Pharmacol. 2014, 171, 3551–3574. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.R.; O’Neill, G.P.; Liu, Q.; Im, D.S.; Sawyer, N.; Metters, K.M.; Coulombe, N.; Abramovitz, M.; Figueroa, D.J.; Zeng, Z.; et al. Characterization of the Human Cysteinyl Leukotriene CysLT1 Receptor. Nature 1999, 399, 789–793. [Google Scholar] [CrossRef]
- Sasaki, F.; Yokomizo, T. The Leukotriene Receptors as Therapeutic Targets of Inflammatory Diseases. Int. Immunol. 2019, 31, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Y.; Zhang, S.; Li, C.; Zhang, L. Modulation of Neuroinflammation by Cysteinyl Leukotriene 1 and 2 Receptors: Implications for Cerebral Ischemia and Neurodegenerative Diseases. Neurobiol. Aging 2020, 87, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Z.; Zhao, B.; Zhang, X.Y.; Huang, X.Q.; Shi, W.Z.; Liu, H.L.; Fang, S.H.; Lu, Y.B.; Zhang, W.P.; Tang, F.D.; et al. Cysteinyl Leukotriene Receptor 2 Is Spatiotemporally Involved in Neuron Injury, Astrocytosis and Microgliosis after Focal Cerebral Ischemia in Rats. Neuroscience 2011, 189, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.H.; Zhou, Y.; Chu, L.S.; Zhang, W.P.; Wang, M.L.; Yu, G.L.; Peng, F.; Wei, E.Q. Spatio-Temporal Expression of Cysteinyl Leukotriene Receptor-2 MRNA in Rat Brain after Focal Cerebral Ischemia. Neurosci. Lett. 2007, 412, 78–83. [Google Scholar] [CrossRef]
- Fang, S.H.; Wei, E.Q.; Zhou, Y.; Wang, M.L.; Zhang, W.P.; Yu, G.L.; Chu, L.S.; Chen, Z. Increased Expression of Cysteinyl Leukotriene Receptor-1 in the Brain Mediates Neuronal Damage and Astrogliosis after Focal Cerebral Ischemia in Rats. Neuroscience 2006, 140, 969–979. [Google Scholar] [CrossRef]
- Michael, J.; Marschallinger, J.; Aigner, L. The Leukotriene Signaling Pathway: A Druggable Target in Alzheimer’s Disease. Drug Discov. Today 2019, 24, 505–516. [Google Scholar] [CrossRef]
- Wang, H.; Shi, Q.; Shi, W.; Zhang, X.; Wang, X.; Zhang, L.; Fang, S.; Lu, Y.; Zhang, W.; Wei, E. Expression and Distribution of Cysteinyl Leukotriene Receptors CysLT1R and CysLT2R, and GPR17 in Brain of Parkinson Disease Model Mice. J. Zhejiang Univ. Med. Sci. 2013, 42, 52–60. [Google Scholar]
- Zhang, X.Y.; Chen, L.; Yang, Y.; Xu, D.M.; Zhang, S.R.; Li, C.T.; Zheng, W.; Yu, S.Y.; Wei, E.Q.; Zhang, L.H. Regulation of Rotenone-Induced Microglial Activation by 5-Lipoxygenase and Cysteinyl Leukotriene Receptor 1. Brain Res. 2014, 1572, 59–71. [Google Scholar] [CrossRef]
- Gelosa, P.; Colazzo, F.; Tremoli, E.; Sironi, L.; Castiglioni, L. Cysteinyl Leukotrienes as Potential Pharmacological Targets for Cerebral Diseases. Mediators Inflamm. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Chen, F.; Thakur, A.; Hong, H. Cysteinyl Leukotrienes and Their Receptors: Emerging Therapeutic Targets in Central Nervous System Disorders. CNS Neurosci. Ther. 2016, 22, 943–951. [Google Scholar] [CrossRef]
- Rahman, S.O.; Singh, R.K.; Hussain, S.; Akhtar, M.; Najmi, A.K. A Novel Therapeutic Potential of Cysteinyl Leukotrienes and Their Receptors Modulation in the Neurological Complications Associated with Alzheimer’s Disease. Eur. J. Pharmacol. 2019, 842, 208–220. [Google Scholar] [CrossRef]
- Chen, F.; Ghosh, A.; Lin, J.; Zhang, C.; Pan, Y.; Thakur, A.; Singh, K.; Hong, H.; Tang, S. 5-Lipoxygenase Pathway and Its Downstream Cysteinyl Leukotrienes as Potential Therapeutic Targets for Alzheimer’s Disease. Brain. Behav. Immun. 2020, 88, 844–855. [Google Scholar] [CrossRef]
- Sinha, S.; Doble, M.; Manju, S.L. 5-Lipoxygenase as a Drug Target: A Review on Trends in Inhibitors Structural Design, SAR and Mechanism Based Approach. Bioorganic Med. Chem. 2019, 27, 3745–3759. [Google Scholar] [CrossRef] [PubMed]
- Farias, S.E.; Zarini, S.; Precht, T.; Murphy, R.C.; Heidenreich, K.A. Transcellular Biosynthesis of Cysteinyl Leukotrienes in Rat Neuronal and Glial Cells. J. Neurochem. 2007, 103, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Chou, V.P.; Holman, T.R.; Manning-Bog, A.B. Differential Contribution of Lipoxygenase Isozymes to Nigrostriatal Vulnerability. Neuroscience 2013, 228, 73–82. [Google Scholar] [CrossRef]
- Kang, K.H.; Liou, H.H.; Hour, M.J.; Liou, H.C.; Fu, W.M. Protection of Dopaminergic Neurons by 5-Lipoxygenase Inhibitor. Neuropharmacology 2013, 73, 380–387. [Google Scholar] [CrossRef]
- Di Meco, A.; Li, J.G.; Praticò, D. Dissecting the Role of 5-Lipoxygenase in the Homocysteine-Induced Alzheimer’s Disease Pathology. J. Alzheimer’s Dis. 2018, 62, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Tan, Y.Z.; Li, D.D.; Tang, S.S.; Wen, X.A.; Long, Y.; Sun, H.-B.; Hong, H.; Hu, M. Zileuton Ameliorates Depressive-like Behaviors, Hippocampal Neuroinflammation, Apoptosis and Synapse Dysfunction in Mice Exposed to Chronic Mild Stress. Int. Immunopharmacol. 2020, 78, 105947. [Google Scholar] [CrossRef] [PubMed]
- Reiss, T.F.; Altman, L.C.; Chervinsky, P.; Bewtra, A.; Stricker, W.E.; Noonan, G.P.; Kundu, S.; Zhang, J. Effects of Montelukast (MK-0476), a New Potent Cysteinyl Leukotriene (LTD4) Receptor Antagonist, in Patients with Chronic Asthma. J. Allergy Clin. Immunol. 1996, 98, 528–534. [Google Scholar] [CrossRef]
- Malmstrom, K.; Schwartz, J.; Reiss, T.F.; Sullivan, T.J.; Reese, J.H.; Jauregui, L.; Miller, K.; Scott, M.; Shingo, S.; Peszek, I.; et al. Effect of Montelukast on Single-Dose Theophylline Pharmacokinetics. Am. J. Ther. 1998, 5, 189–195. [Google Scholar] [CrossRef]
- Trinh, H.K.T.; Lee, S.H.; Cao, T.B.T.; Park, H.S. Asthma Pharmacotherapy: An Update on Leukotriene Treatments. Expert Rev. Respir. Med. 2019, 13, 1169–1178. [Google Scholar] [CrossRef]
- Aigner, L.; Pietrantonio, F.; Bessa de Sousa, D.M.; Michael, J.; Schuster, D.; Reitsamer, H.A.; Zerbe, H.; Studnicka, M. The Leukotriene Receptor Antagonist Montelukast as a Potential COVID-19 Therapeutic. Front. Mol. Biosci. 2020, 7, 610132. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Kim, S.; Lee, J.M.; Oh, Y.-S.; Park, S.M.; Kim, S.R. Montelukast Treatment Protects Nigral Dopaminergic Neurons against Microglial Activation in the 6-Hydroxydopamine Mouse Model of Parkinson’s Disease. Neuroreport 2017, 28, 242–249. [Google Scholar] [CrossRef]
- Nagarajan, V.B.; Marathe, P.A. Effect of Montelukast in Experimental Model of Parkinson’s Disease. Neurosci. Lett. 2018, 682, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Mansour, R.M.; Ahmed, M.A.E.; El-Sahar, A.E.; El Sayed, N.S. Montelukast Attenuates Rotenone-Induced Microglial Activation/P38 MAPK Expression in Rats: Possible Role of Its Antioxidant, Anti-Inflammatory and Antiapoptotic Effects. Toxicol. Appl. Pharmacol. 2018, 358, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Mei, Z.L.; Wang, H.; Hu, M.; Long, Y.; Miao, M.X.; Li, N.; Hong, H. Montelukast Rescues Primary Neurons against Aβ1-42-Induced Toxicity through Inhibiting CysLT1R-Mediated NF-ΚB Signaling. Neurochem. Int. 2014, 75, 26–31. [Google Scholar] [CrossRef]
- Marschallinger, J.; Schäffner, I.; Klein, B.; Gelfert, R.; Rivera, F.J.; Illes, S.; Grassner, L.; Janssen, M.; Rotheneichner, P.; Schmuckermair, C.; et al. Structural and Functional Rejuvenation of the Aged Brain by an Approved Anti-Asthmatic Drug. Nat. Commun. 2015, 6, 8466. [Google Scholar] [CrossRef]
- Marschallinger, J.; Altendorfer, B.; Rockenstein, E.; Holztrattner, M.; Garnweidner-Raith, J.; Pillichshammer, N.; Leister, I.; Hutter-Paier, B.; Strempfl, K.; Unger, M.S.; et al. The Leukotriene Receptor Antagonist Montelukast Reduces Alpha-Synuclein Load and Restores Memory in an Animal Model of Dementia with Lewy Bodies. Neurotherapeutics 2020, 17, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Rozin, S.I. Case Series Using Montelukast in Patients with Memory Loss and Dementia. Open Neurol. J. 2017, 11, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Grinde, B.; Schirmer, H.; Eggen, A.E.; Aigner, L.; Engdahl, B. A Possible Effect of Montelukast on Neurological Aging Examined by the Use of Register Data. Int. J. Clin. Pharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fatima, N.; Shuaib, A. The Role of Montelukast in Traumatic Brain Injury and Brain Ischemia. Neurosurgery 2020, 67. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallin, J.; Svenningsson, P. Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 5606. https://doi.org/10.3390/ijms22115606
Wallin J, Svenningsson P. Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(11):5606. https://doi.org/10.3390/ijms22115606
Chicago/Turabian StyleWallin, Johan, and Per Svenningsson. 2021. "Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 11: 5606. https://doi.org/10.3390/ijms22115606
APA StyleWallin, J., & Svenningsson, P. (2021). Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. International Journal of Molecular Sciences, 22(11), 5606. https://doi.org/10.3390/ijms22115606