
The Acidic Brain—Glycolytic Switch in the Microenvironment of Malignant Glioma



Abstract
1. Introduction
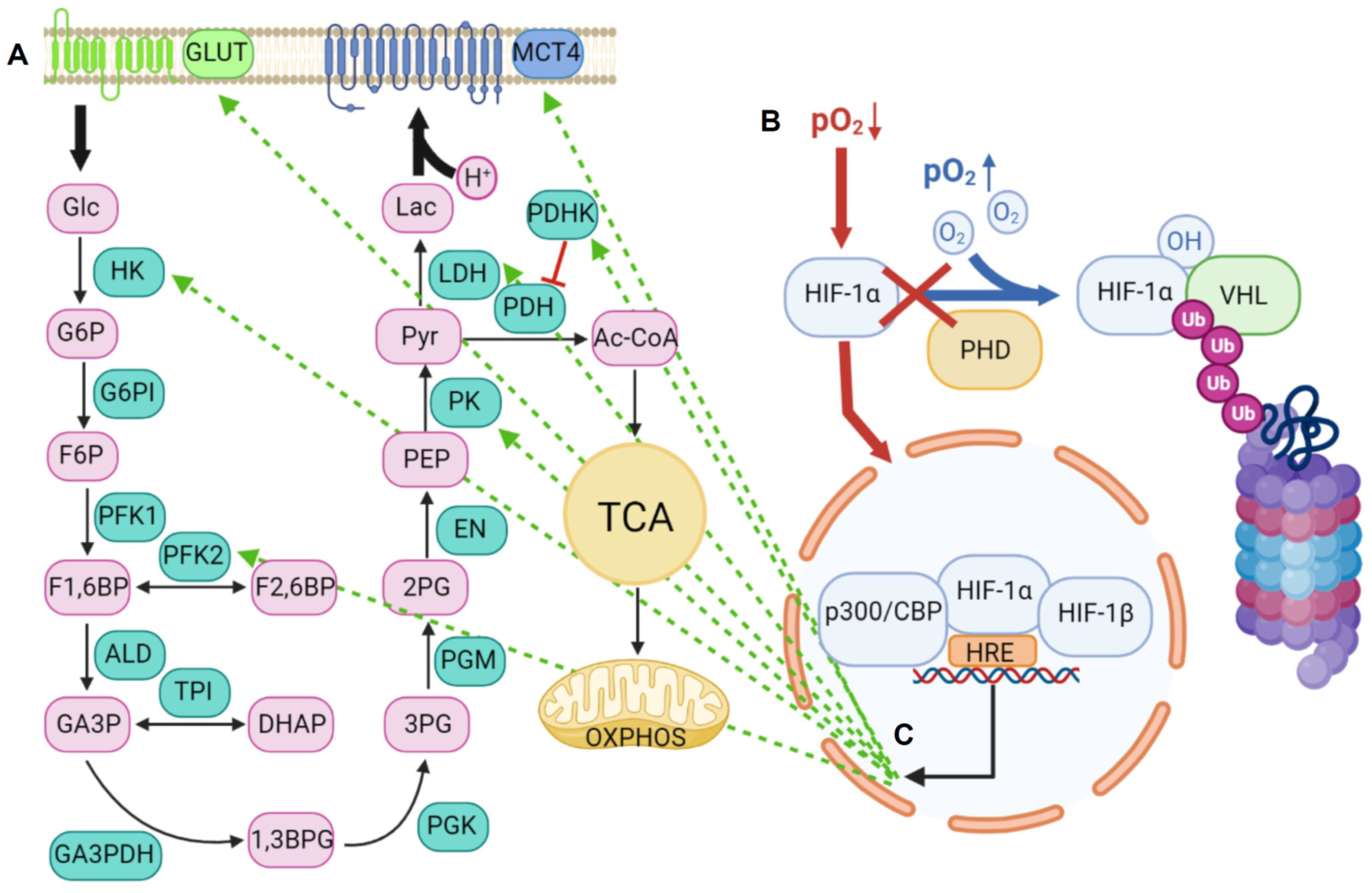
2. Lactic Acid Metabolism within the Tumor Microenvironment
2.1. Lactic Acid Production—A Hallmark of Glycolytic Cancer Cells
2.2. The Role of MCTs in Lactic Acid Metabolism
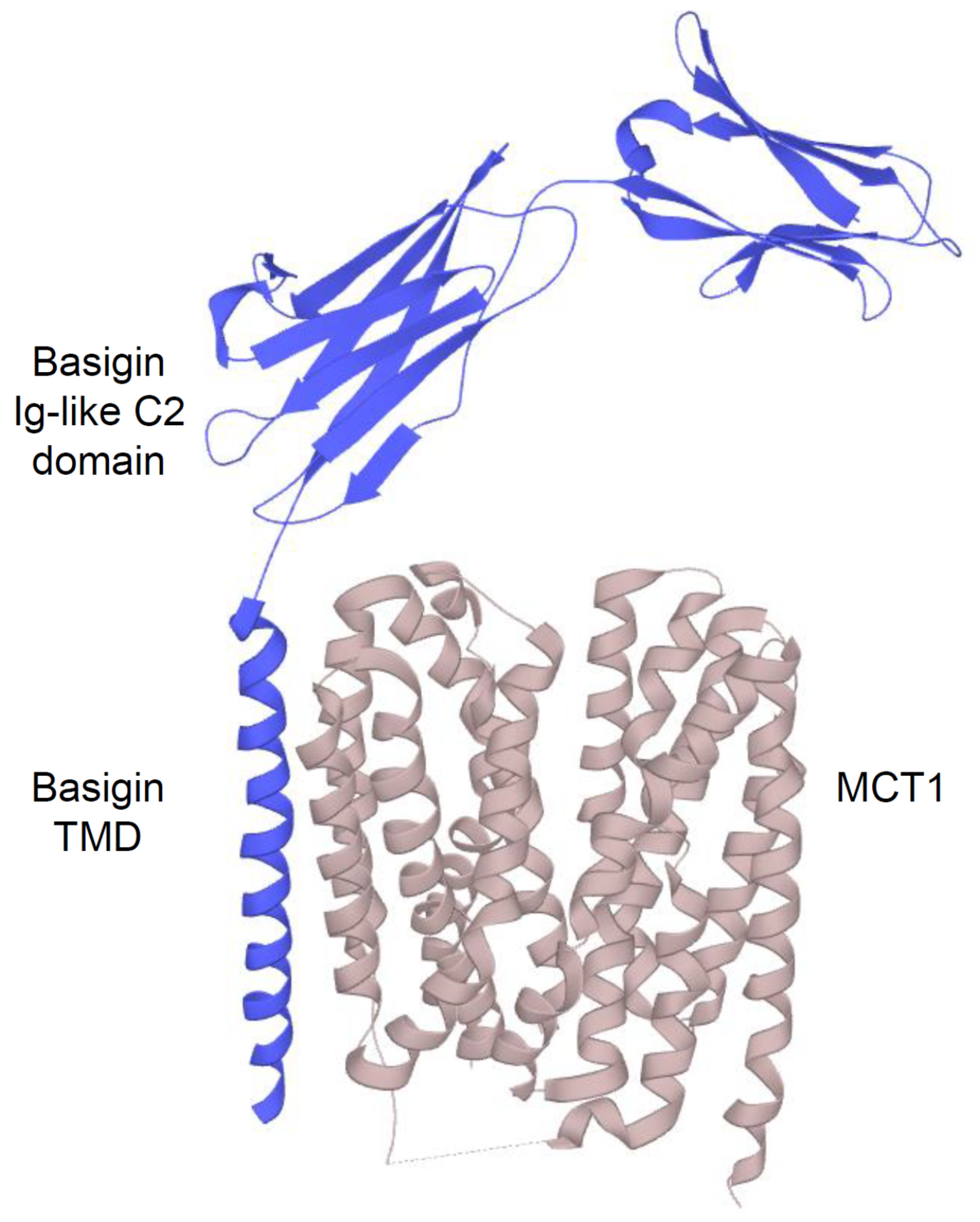
2.2.1. Structure and Function of MCTs
2.2.2. An acidic Tumor Microenvironment and the Metabolic Symbiosis Model
2.3. Lactic Acid Metabolism through MCTs and Functional Consequences on Tumor Malignancy
2.4. Lactic Acid Shuttling and the Induction of Angiogenesis
3. The Role of miRNAs in the Glycolytic Switch
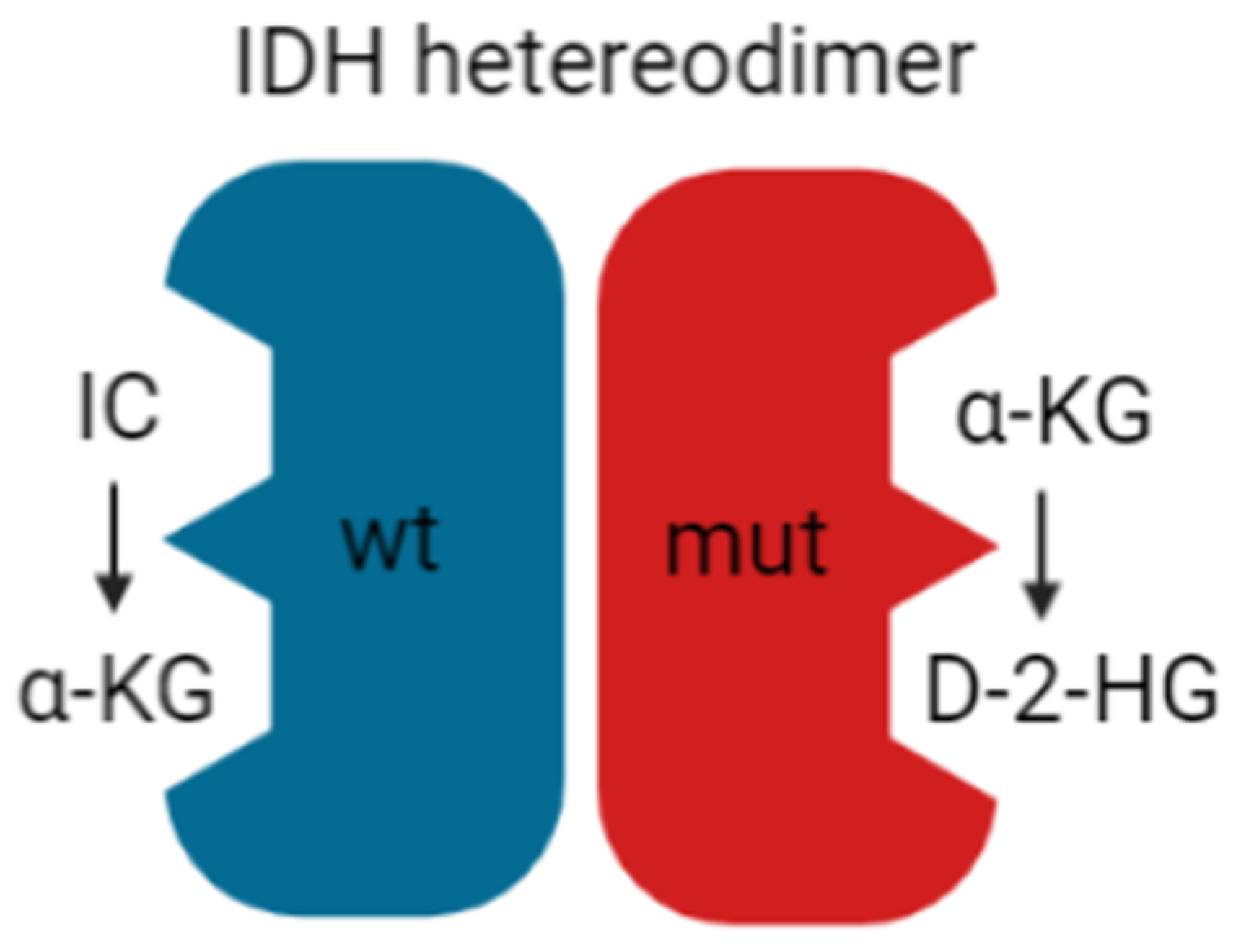
4. The Acidic Tumor Microenvironment in IDH wt Versus IDH Mutant Gliomas
5. Implications for Diagnostics and Therapy
5.1. Diagnostic Approaches for Identifying Glycolytic Tumor Regions
5.2. Glycolytic Players as Targets in Glioma Therapy
5.3. Impact of the Glycolytic Phenotype on Tumor Immunity and Immunotherapy
6. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1,3BPG | 1,3-bis-phospho-gycerate |
| 2PG | 2-phospho-gycerate |
| 3-BP | 3-bromopyruvate |
| 3PG | 3-phospho-gycerate |
| Ac-CoA | Acetyl-coenzyme A |
| AKT | Activates protein kinase B |
| ALD | Aldolase |
| AMPK | Adenosine monophosphate protein kinase |
| ATP | Adenosine triphosphate |
| Bcl-xL | B-cell lymphoma-extra large |
| BIM | Bcl2 like 11 |
| CA | Carbonic anhydrase |
| CAB39 | Calcium-binding protein 39 |
| CAR | Chimeric antigen receptor |
| CBP | CREB-binding protein |
| CD | Cluster of differentiation |
| CELF | CUGBP Elav-like family member |
| CEST | Chemical exchange saturation transfer |
| CHC | Cyano-hydroxycinnamate |
| circ | Circular |
| c-MYC | Cellular myelocytomatosis |
| CO2 | Carbon dioxide |
| CREB | CAMP response element-binding protein |
| CSC | Cancer stem cell |
| CTL | Cytotoxic T lymphocyte |
| CTLA | CTL associated protein |
| CXCL | C-X-C motif chemokine ligand |
| D-2-HG | D-2-hydroxyglutarate |
| DHAP | Di-hydroxy-acetone–phosphate |
| EGFR | Epidermal growth factor receptor |
| EM | Electron microscopy |
| EMMPRIN | Extracellular matrix metalloproteinase inducer |
| EMT | Epithelial-mesenchymal transition |
| EN | Enolase |
| F1,6BP | Fructose-1,6-bis-phosphate |
| F2,6BP | Fructose-2,6-bis-phosphate |
| F6P | Fructose-6-phosphate |
| G6P | Glucose-6-phosphate |
| G6PI | Glucose-6-phosphate-isomerase |
| GA3P | Glycerin-aldehyde-3-phosphate |
| GA3PDH | Glycerin-aldehyde-3-phosphate-dehydrogenase |
| GBM | Glioblastoma multiforme |
| G-CIMP | CpG island methylator phenotype |
| Glc | Glucose |
| GLUT | Glucose transporter |
| GP | Glycoprotein |
| GPR | G-protein receptor |
| GTN | Glyceryl trinitrate |
| H3K4me3 | Histone 3K4 triple methylation |
| HBMEC | Human brain microvascular endothelial cells |
| HDAC | Histone deacetylase |
| HIF | Hypoxia-inducible factor |
| HK | Hexokinase |
| HRE | Hypoxia–response element |
| IC | Isocitrate |
| IDH | Isocitrate dehydrogenase |
| IFN | Interferon |
| Ig | Immunoglobulin |
| IL | Interleukin |
| ING4 | Inhibitor of growth protein 4 |
| IRF | Interferon regulatory factor |
| KDM | Lysine demethylase |
| KG | Ketoglutarate |
| KRAS | Kirsten rat sarcoma virus |
| Lac | Lactate |
| LDH | Lactate dehydrogenase |
| lncRNA | Long non-coding RNA |
| LSINCT5 | Long stress-induced non-coding transcript 5 |
| MAPK | Mitogen-activated protein kinase |
| MAVS | Mitochondrial antiviral signaling protein |
| MCT | Monocarboxylate transporter |
| MIG6 | Mitogen-inducible gene |
| MRI | Magnetic resonance imaging |
| MRSI | Magnetic resonance spectroscopic imaging |
| mTOR | Mammalian target of rapamycin |
| mut | Mutant |
| MV-Edm | Measles virus Edmonston strain |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NAIF1 | Nuclear apoptosis-inducing factor 1 |
| NF | Nuclear factor |
| NHE | Na+/H+ exchanger |
| NO | Nitric oxide |
| NRF | Nuclear respiratory factor |
| OXPHOS | Oxidative phosphorylation |
| pCMBS | p-Chloro-mercuri-benzene sulfonate |
| PDGF | Platelet-derived growth factor |
| PDH | Pyruvate-dehydrogenase |
| PDHK | Pyruvate-dehydrogenase-kinase |
| PDL | Programmed cell death ligand |
| PEP | Phospho-enol-pyruvate |
| PET | Positron emission tomography |
| PFK | Phosphofructokinase |
| PGM | Phospho-glycerate-mutase |
| PGK | Phospho-glycerate-kinase |
| PHD | Prolyl hydroxylase |
| PIK3CD | Phosphatidylinositol-4,5-bisphosphate 3 kinase catalytic subunit delta |
| PK | Pyruvate-kinase |
| PKCδ | Protein kinase Cδ |
| pO2 | Partial pressure oxygen |
| Pyr | Pyruvate |
| RAB10 | Ras-related protein 10 |
| RPN2 | Ribophorin 2 |
| ROS | Reactive oxygen species |
| siRNA | Small interference ribonucleic acid |
| SLC | Solute carrier |
| SMO | Smoothened |
| SNAI | Snail homolog |
| SP1 | Specific protein 1 |
| STAT | Signal transducer and activator of transcription |
| TAM | Tumor-associated macrophage |
| TCA | Tricarboxylic acid |
| TMD | Transmembrane domain |
| TPI | Triose-phosphate-isomerase |
| TSC | Tuberose sclerosis complex |
| Ub | Ubiquitinylated |
| UCP2 | Uncoupling protein 2 |
| VEGF | Vascular endothelial growth factor |
| VHL | Von Hippel–Lindau |
| WHO | World Health Organization |
| wt | Wild-type |
References
- Rasmussen, B.K.; Hansen, S.; Laursen, R.J.; Kosteljanetz, M.; Schultz, H.; Norgard, B.M.; Guldberg, R.; Gradel, K.O. Epidemiology of glioma: Clinical characteristics, symptoms, and predictors of glioma patients grade I-IV in the the Danish Neuro-Oncology Registry. J. Neurooncol. 2017, 135, 571–579. [Google Scholar] [CrossRef]
- Davis, F.G.; Smith, T.R.; Gittleman, H.R.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Glioblastoma incidence rate trends in Canada and the United States compared with England, 1995-2015. Neuro. Oncol. 2019. [Google Scholar] [CrossRef]
- Porter, K.R.; McCarthy, B.J.; Freels, S.; Kim, Y.; Davis, F.G. Prevalence estimates for primary brain tumors in the United States by age, gender, behavior, and histology. Neuro Oncol. 2010, 12, 520–527. [Google Scholar] [CrossRef]
- Castro, M.G.; Cowen, R.; Williamson, I.K.; David, A.; Jimenez-Dalmaroni, M.J.; Yuan, X.; Bigliari, A.; Williams, J.C.; Hu, J.; Lowenstein, P.R. Current and future strategies for the treatment of malignant brain tumors. Pharm. Ther. 2003, 98, 71–108. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef]
- Anton, K.; Baehring, J.M.; Mayer, T. Glioblastoma multiforme: Overview of current treatment and future perspectives. Hematol. Oncol. Clin. N. Am. 2012, 26, 825–853. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef]
- Mashima, T.; Sato, S.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Promotion of glioma cell survival by acyl-CoA synthetase 5 under extracellular acidosis conditions. Oncogene 2009, 28, 9–19. [Google Scholar] [CrossRef]
- De Milito, A.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2010, 127, 207–219. [Google Scholar] [CrossRef]
- Wu, R.; Racker, E. Regulatory mechanisms in carbohydrate metabolism. IV. Pasteur effect and Crabtree effect in ascites tumor cells. J. Biol. Chem. 1959, 234, 1036–1041. [Google Scholar] [CrossRef]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharm. 2011, 2, 49. [Google Scholar] [CrossRef]
- Semenza, G.L.; Artemov, D.; Bedi, A.; Bhujwalla, Z.; Chiles, K.; Feldser, D.; Laughner, E.; Ravi, R.; Simons, J.; Taghavi, P.; et al. ‘The metabolism of tumours’: 70 years later. Novartis Found. Symp. 2001, 240, 251–260; discussion 260–264. [Google Scholar]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef]
- Hirsilä, M.; Koivunen, P.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cox, S.R.; Morita, T.; Kourembanas, S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ. Res. 1995, 77, 638–643. [Google Scholar] [CrossRef]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Curi, R.; Newsholme, P.; Newsholme, E.A. Metabolism of pyruvate by isolated rat mesenteric lymphocytes, lymphocyte mitochondria and isolated mouse macrophages. Biochem. J. 1988, 250, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Racker, E. Bioenergetics and the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar]
- Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell Proteom. 2017, 16, 1906–1921. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.; Linhares, P.; Carvalho, B.; Santos, P.; Costa, B.M.; Vaz, R. Serum lactate levels are associated with glioma malignancy grade. Clin. Neurol. Neurosurg. 2019, 186, 105546. [Google Scholar] [CrossRef]
- Price, N.T.; Jackson, V.N.; Halestrap, A.P. Cloning and sequencing of four new mammalian monocarboxylate transporter (MCT) homologues confirms the existence of a transporter family with an ancient past. Biochem. J. 1998, 329, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H. Computer-aided analyses of transport protein sequences: Gleaning evidence concerning function, structure, biogenesis, and evolution. Microbiol. Rev. 1994, 58, 71–93. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.C.; Halestrap, A.P. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am. J. Physiol. 1993, 264, C761–C782. [Google Scholar] [CrossRef]
- Halestrap, A.P. The monocarboxylate transporter family--Structure and functional characterization. Iubmb Life 2012, 64, 1–9. [Google Scholar] [CrossRef]
- Poole, R.C.; Halestrap, A.P. Interaction of the erythrocyte lactate transporter (monocarboxylate transporter 1) with an integral 70-kDa membrane glycoprotein of the immunoglobulin superfamily. J. Biol. Chem. 1997, 272, 14624–14628. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.; Wilson, M.C.; Heddle, C.; Brown, M.H.; Barclay, A.N.; Halestrap, A.P. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000, 19, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Meredith, D.; Fox, J.E.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: The ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 2005, 280, 27213–27221. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, C.; Wilson, M.C.; Sessions, R.B.; Halestrap, A.P. The role of charged residues in the transmembrane helices of monocarboxylate transporter 1 and its ancillary protein basigin in determining plasma membrane expression and catalytic activity. Mol. Membr. Biol. 2006, 23, 486–498. [Google Scholar] [CrossRef]
- Wilson, M.C.; Meredith, D.; Bunnun, C.; Sessions, R.B.; Halestrap, A.P. Studies on the DIDS-binding site of monocarboxylate transporter 1 suggest a homology model of the open conformation and a plausible translocation cycle. J. Biol. Chem. 2009, 284, 20011–20021. [Google Scholar] [CrossRef] [PubMed]
- Guenette, R.S.; Sridhar, S.; Herley, M.; Mooibroek, M.; Wong, P.; Tenniswood, M. Embigin, a developmentally expressed member of the immunoglobulin super family, is also expressed during regression of prostate and mammary gland. Dev. Genet. 1997, 21, 268–278. [Google Scholar] [CrossRef]
- Froberg, M.K.; Gerhart, D.Z.; Enerson, B.E.; Manivel, C.; Guzman-Paz, M.; Seacotte, N.; Drewes, L.R. Expression of monocarboxylate transporter MCT1 in normal and neoplastic human CNS tissues. Neuroreport 2001, 12, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Reuss, A.M.; Groos, D.; Ghoochani, A.; Buchfelder, M.; Savaskan, N. MCT4 Promotes Tumor Malignancy in F98 Glioma Cells. J. Oncol. 2021, 2021, 6655529. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Liu, Z.J.; Hu, S.Q.; Huo, H.Y.; Xu, Z.R.; Ruan, J.F.; Sun, Y.; Dai, L.P.; Yan, C.B.; Xiong, W.; et al. Lactic acid induces lactate transport and glycolysis/OXPHOS interconversion in glioblastoma. Biochem. Biophys Res. Commun. 2018, 503, 888–894. [Google Scholar] [CrossRef]
- Grillon, E.; Farion, R.; Fablet, K.; De Waard, M.; Tse, C.M.; Donowitz, M.; Remy, C.; Coles, J.A. The spatial organization of proton and lactate transport in a rat brain tumor. PLoS ONE 2011, 6, e17416. [Google Scholar] [CrossRef]
- Cheng, C.; Edin, N.F.; Lauritzen, K.H.; Aspmodal, I.; Christoffersen, S.; Jian, L.; Rasmussen, L.J.; Pettersen, E.O.; Xiaoqun, G.; Bergersen, L.H. Alterations of monocarboxylate transporter densities during hypoxia in brain and breast tumour cells. Cell Oncol. (Dordr.) 2012, 35, 217–227. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- Kaluz, S.; Kaluzová, M.; Liao, S.Y.; Lerman, M.; Stanbridge, E.J. Transcriptional control of the tumor- and hypoxia-marker carbonic anhydrase 9: A one transcription factor (HIF-1) show? Biochim. Biophys. Acta 2009, 1795, 162–172. [Google Scholar] [CrossRef]
- Ames, S.; Andring, J.T.; McKenna, R.; Becker, H.M. CAIX forms a transport metabolon with monocarboxylate transporters in human breast cancer cells. Oncogene 2020, 39, 1710–1723. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Seetharaman, K. Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Res. 1996, 56, 1194–1198. [Google Scholar] [PubMed]
- Ferrauto, G.; Di Gregorio, E.; Auboiroux, V.; Petit, M.; Berger, F.; Aime, S.; Lahrech, H. CEST-MRI for glioma pH quantification in mouse model: Validation by immunohistochemistry. Nmr. Biomed. 2018, 31, e4005. [Google Scholar] [CrossRef]
- Li, X.; Vigneron, D.B.; Cha, S.; Graves, E.E.; Crawford, F.; Chang, S.M.; Nelson, S.J. Relationship of MR-derived lactate, mobile lipids, and relative blood volume for gliomas in vivo. Ajnr Am. J. Neuroradiol. 2005, 26, 760–769. [Google Scholar] [PubMed]
- Kubelt, C.; Peters, S.; Ahmeti, H.; Huhndorf, M.; Huber, L.; Cohrs, G.; Hövener, J.B.; Jansen, O.; Synowitz, M.; Held-Feindt, J. Intratumoral Distribution of Lactate and the Monocarboxylate Transporters 1 and 4 in Human Glioblastoma Multiforme and Their Relationships to Tumor Progression-Associated Markers. Int. J. Mol. Sci. 2020, 21, 6254. [Google Scholar] [CrossRef] [PubMed]
- Reichert, M.; Steinbach, J.P.; Supra, P.; Weller, M. Modulation of growth and radiochemosensitivity of human malignant glioma cells by acidosis. Cancer 2002, 95, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335–46353. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Schneider, H.P.; Bröer, A.; Rahman, B.; Hamprecht, B.; Deitmer, J.W. Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem. J. 1998, 333, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef]
- Nikoobakht, M.; Shamshiripour, P.; Azimi Nekoo, Z.; Fallah Haghmohammadi, S. Elevated Lactate and Total Protein Levels in Stereotactic Brain Biopsy Specimen; Potential Biomarkers of Malignancy and Poor Prognosis. Arch. Iran. Med. 2019, 22, 125–131. [Google Scholar]
- Lim, K.S.; Lim, K.J.; Price, A.C.; Orr, B.A.; Eberhart, C.G.; Bar, E.E. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene 2014, 33, 4433–4441. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Painter, W.B.; Chen, H.; Minamide, L.S.; Abe, H.; Bamburg, J.R. Intracellular pH modulation of ADF/cofilin proteins. Cell Motil. Cytoskelet. 2000, 47, 319–336. [Google Scholar] [CrossRef]
- Magalhaes, M.A.; Larson, D.R.; Mader, C.C.; Bravo-Cordero, J.J.; Gil-Henn, H.; Oser, M.; Chen, X.; Koleske, A.J.; Condeelis, J. Cortactin phosphorylation regulates cell invasion through a pH-dependent pathway. J. Cell Biol. 2011, 195, 903–920. [Google Scholar] [CrossRef]
- Busco, G.; Cardone, R.A.; Greco, M.R.; Bellizzi, A.; Colella, M.; Antelmi, E.; Mancini, M.T.; Dell’Aquila, M.E.; Casavola, V.; Paradiso, A.; et al. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010, 24, 3903–3915. [Google Scholar] [CrossRef]
- Gioia, M.; Fasciglione, G.F.; Monaco, S.; Iundusi, R.; Sbardella, D.; Marini, S.; Tarantino, U.; Coletta, M. pH dependence of the enzymatic processing of collagen I by MMP-1 (fibroblast collagenase), MMP-2 (gelatinase A), and MMP-14 ectodomain. J. Biol. Inorg. Chem. 2010, 15, 1219–1232. [Google Scholar] [CrossRef]
- Tao, C.; Huang, K.; Shi, J.; Hu, Q.; Li, K.; Zhu, X. Genomics and Prognosis Analysis of Epithelial-Mesenchymal Transition in Glioma. Front. Oncol. 2020, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Takata, K.; Ashihara, E. Inhibition of monocarboxylate transporter 1 suppresses the proliferation of glioblastoma stem cells. J. Physiol. Sci. 2016, 66, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.G.; Zhang, J.D.; Zhang, Y.F. [The effect of downregulation of MCT1 on the proliferation of glioma cells]. Zhonghua Zhong Liu Za Zhi 2019, 41, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Korkolopoulou, P.; Perdiki, M.; Thymara, I.; Boviatsis, E.; Agrogiannis, G.; Kotsiakis, X.; Angelidakis, D.; Rologis, D.; Diamantopoulou, K.; Thomas-Tsagli, E.; et al. Expression of hypoxia-related tissue factors in astrocytic gliomas. A multivariate survival study with emphasis upon carbonic anhydrase IX. Hum. Pathol. 2007, 38, 629–638. [Google Scholar] [CrossRef]
- Cetin, B.; Gonul, I.I.; Gumusay, O.; Bilgetekin, I.; Algin, E.; Ozet, A.; Uner, A. Carbonic anhydrase IX is a prognostic biomarker in glioblastoma multiforme. Neuropathology 2018, 38, 457–462. [Google Scholar] [CrossRef]
- Proescholdt, M.A.; Merrill, M.J.; Stoerr, E.M.; Lohmeier, A.; Pohl, F.; Brawanski, A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro Oncol. 2012, 14, 1357–1366. [Google Scholar] [CrossRef]
- Huang, B.R.; Liu, Y.S.; Lai, S.W.; Lin, H.J.; Shen, C.K.; Yang, L.Y.; Lu, D.Y. CAIX Regulates GBM Motility and TAM Adhesion and Polarization through EGFR/STAT3 under Hypoxic Conditions. Int. J. Mol. Sci. 2020, 21, 5838. [Google Scholar] [CrossRef]
- McIntyre, A.; Patiar, S.; Wigfield, S.; Li, J.L.; Ledaki, I.; Turley, H.; Leek, R.; Snell, C.; Gatter, K.; Sly, W.S.; et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. 2012, 18, 3100–3111. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef]
- Kumar, V.B.; Viji, R.I.; Kiran, M.S.; Sudhakaran, P.R. Endothelial cell response to lactate: Implication of PAR modification of VEGF. J. Cell Physiol. 2007, 211, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Fukumura, D.; Jain, R.K. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: Mechanism of low pH-induced VEGF. J. Biol. Chem. 2002, 277, 11368–11374. [Google Scholar] [CrossRef]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Bezerra, F.; Costa-Almeida, R.; Freitas-Cunha, M.; Soares, R.; Martinho, O.; Reis, R.M.; Pinheiro, C.; Baltazar, F. Monocarboxylate transporter 1 is a key player in glioma-endothelial cell crosstalk. Mol. Carcinog. 2017, 56, 2630–2642. [Google Scholar] [CrossRef]
- Thakur, A.; Qiu, G.; Xu, C.; Han, X.; Yang, T.; Ng, S.P.; Chan, K.W.Y.; Wu, C.M.L.; Lee, Y. Label-free sensing of exosomal MCT1 and CD147 for tracking metabolic reprogramming and malignant progression in glioma. Sci. Adv. 2020, 6, eaaz6119. [Google Scholar] [CrossRef]
- Talasila, K.M.; Røsland, G.V.; Hagland, H.R.; Eskilsson, E.; Flønes, I.H.; Fritah, S.; Azuaje, F.; Atai, N.; Harter, P.N.; Mittelbronn, M.; et al. The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro Oncol. 2017, 19, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Li, K.; Huang, Y.; Hu, Q.; Gao, X.; Jie, S. miR-495 mediates metabolic shift in glioma cells via targeting Glut1. J. Craniofac. Surg. 2015, 26, e155–e158. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; De Lay, M.; Van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, D.; Ansari, K.; Nowicki, M.O.; Salińska, E.; Bronisz, A.; Godlewski, J. MicroRNA-451 Inhibits Migration of Glioblastoma while Making It More Susceptible to Conventional Therapy. Noncoding RNA 2019, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Nan, Y.; Zhen, Y.; Zhang, Y.; Guo, L.; Yu, K.; Huang, Q.; Zhong, Y. miRNA-451 inhibits glioma cell proliferation and invasion by downregulating glucose transporter 1. Tumour. Biol. 2016, 37, 13751–13761. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, L.; Li, T.; Zhu, M.; Zhang, C.; Chen, L.; Zhao, P.; Zhou, H.; Yu, S.; Yang, X. The role of miR-451 in the switching between proliferation and migration in malignant glioma cells: AMPK signaling, mTOR modulation and Rac1 activation required. Int. J. Oncol. 2017, 50, 1989–1999. [Google Scholar] [CrossRef]
- Nan, Y.; Guo, H.; Guo, L.; Wang, L.; Ren, B.; Yu, K.; Huang, Q.; Zhong, Y. MiRNA-451 Inhibits Glioma Cell Proliferation and Invasion Through the mTOR/HIF-1α/VEGF Signaling Pathway by Targeting CAB39. Hum. Gene Clin. Dev. 2018, 29, 156–166. [Google Scholar] [CrossRef]
- Li, S.J.; Liu, H.L.; Tang, S.L.; Li, X.J.; Wang, X.Y. MicroRNA-150 regulates glycolysis by targeting von Hippel-Lindau in glioma cells. Am. J. Transl Res. 2017, 9, 1058–1066. [Google Scholar]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef]
- Tan, X.; Wang, S.; Yang, B.; Zhu, L.; Yin, B.; Chao, T.; Zhao, J.; Yuan, J.; Qiang, B.; Peng, X. The CREB-miR-9 negative feedback minicircuitry coordinates the migration and proliferation of glioma cells. PLoS ONE 2012, 7, e49570. [Google Scholar] [CrossRef]
- Yang, Y.; Dodbele, S.; Park, T.; Glass, R.; Bhat, K.; Sulman, E.P.; Zhang, Y.; Abounader, R. MicroRNA-29a inhibits glioblastoma stem cells and tumor growth by regulating the PDGF pathway. J. Neurooncol. 2019, 145, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Jiao, B.H.; Fan, F.S.; Lu, S.K.; Song, J.; Guo, C.Y.; Yang, J.K.; Yang, L. Downregulation of miR-95-3p inhibits proliferation, and invasion promoting apoptosis of glioma cells by targeting CELF2. Int. J. Oncol. 2015, 47, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cheung, W.K.C.; Ng, S.S.; Jiang, X.; Jiang, S.; Sze, J.; Leung, G.K.K.; Lu, G.; Chan, D.T.M.; Bian, X.W.; et al. Loss of brain-enriched miR-124 microRNA enhances stem-like traits and invasiveness of glioma cells. J. Biol. Chem. 2012, 287, 9962–9971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kim, J.; Mueller, A.C.; Dey, B.; Yang, Y.; Lee, D.H.; Hachmann, J.; Finderle, S.; Park, D.M.; Christensen, J.; et al. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ. 2014, 21, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, S.J.; Seo, H.H.; Shin, S.P.; Kim, D.; Park, C.S.; Kim, K.T.; Kim, Y.H.; Jeong, J.S.; Kim, I.H. Over-expression of miR-145 enhances the effectiveness of HSVtk gene therapy for malignant glioma. Cancer Lett. 2012, 320, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zhang, Y.; Skalski, M.; Hayes, J.; Kefas, B.; Schiff, D.; Purow, B.; Parsons, S.; Lawler, S.; Abounader, R. microRNA-148a is a prognostic oncomiR that targets MIG6 and BIM to regulate EGFR and apoptosis in glioblastoma. Cancer Res. 2014, 74, 1541–1553. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Shi, Z.; Wei, W.; Lu, C.; Wei, Y.; Yan, W.; Li, R.; Zhang, J.; You, Y.; Wang, X. MiR-181b suppress glioblastoma multiforme growth through inhibition of SP1-mediated glucose metabolism. Cancer Cell Int. 2020, 20, 69. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Shi, Z.M.; Wang, X.R.; Cao, L.; Wang, Y.Y.; Zhang, J.X.; Yin, Y.; Luo, H.; Kang, C.S.; Liu, N.; et al. MiR-181d acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2. J. Cancer Res. Clin. Oncol. 2012, 138, 573–584. [Google Scholar] [CrossRef]
- He, J.; Deng, Y.; Yang, G.; Xie, W. MicroRNA-203 down-regulation is associated with unfavorable prognosis in human glioma. J. Surg Oncol. 2013, 108, 121–125. [Google Scholar] [CrossRef]
- Xiong, Z.; Zhou, C.; Wang, L.; Zhu, R.; Zhong, L.; Wan, D.; Wang, Q. Circular RNA SMO sponges miR-338-3p to promote the growth of glioma by enhancing the expression of SMO. Aging (Albany NY) 2019, 11, 12345–12360. [Google Scholar] [CrossRef]
- Wu, X.; Hu, C.; Long, C.; Zhai, X.; Liang, P.; Yu, Z. MicroRNA-351 Promotes the Proliferation and Invasion of Glioma Cells through Downregulation of NAIF1. J. Mol. Neurosci. 2020, 70, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Wu, Z.; You, H.; Ge, H.; Zheng, S.; Lin, Y.; Wu, X.; Lin, Z.; Kang, D. CircNFIX promotes progression of glioma through regulating miR-378e/RPN2 axis. J. Exp. Clin. Cancer Res. 2019, 38, 506. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zeng, A.; Hu, Q.; Yan, W.; Liu, Y.; You, Y. miR-423-5p contributes to a malignant phenotype and temozolomide chemoresistance in glioblastomas. Neuro Oncol. 2017, 19, 55–65. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Lin, G.; Wang, D. Down-regulation of circ-PTN suppresses cell proliferation, invasion and glycolysis in glioma by regulating miR-432-5p/RAB10 axis. Neurosci. Lett. 2020, 735, 135153. [Google Scholar] [CrossRef]
- Nan, Y.; Han, L.; Zhang, A.; Wang, G.; Jia, Z.; Yang, Y.; Yue, X.; Pu, P.; Zhong, Y.; Kang, C. MiRNA-451 plays a role as tumor suppressor in human glioma cells. Brain Res. 2010, 1359, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Cao, W.; Ma, H. Knockdown of lncRNA LSINCT5 suppresses growth and metastasis of human glioma cells via up-regulating miR-451. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2507–2515. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chen, C.; Zhang, X.; Liu, Q.; Xu, J.L.; Zhang, H.R.; Yao, X.H.; Jiang, T.; He, Z.C.; Ren, Y.; et al. Primate-specific miR-663 functions as a tumor suppressor by targeting PIK3CD and predicts the prognosis of human glioblastoma. Clin. Cancer Res. 2014, 20, 1803–1813. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Xiao, M.T.; Liu, L.X.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chakhoyan, A.; Nathanson, D.A.; Yong, W.H.; Salamon, N.; Raymond, C.; Mareninov, S.; Lai, A.; Nghiemphu, P.L.; Prins, R.M.; et al. Metabolic characterization of human IDH mutant and wild type gliomas using simultaneous pH- and oxygen-sensitive molecular MRI. Neuro Oncol. 2019, 21, 1184–1196. [Google Scholar] [CrossRef]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef]
- Khurshed, M.; Molenaar, R.J.; Lenting, K.; Leenders, W.P.; van Noorden, C.J.F. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget 2017, 8, 49165–49177. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Chaumeil, M.M.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS ONE 2015, 10, e0118781. [Google Scholar] [CrossRef]
- Wenger, K.J.; Steinbach, J.P.; Bähr, O.; Pilatus, U.; Hattingen, E. Lower Lactate Levels and Lower Intracellular pH in Patients with IDH-Mutant versus Wild-Type Gliomas. AJNR Am. J. Neuroradiol. 2020, 41, 1414–1422. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Radoul, M.; Najac, C.; Eriksson, P.; Viswanath, P.; Blough, M.D.; Chesnelong, C.; Luchman, H.A.; Cairncross, J.G.; Ronen, S.M. Hyperpolarized (13)C MR imaging detects no lactate production in mutant IDH1 gliomas: Implications for diagnosis and response monitoring. Neuroimage Clin. 2016, 12, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, P.; Najac, C.; Izquierdo-Garcia, J.L.; Pankov, A.; Hong, C.; Eriksson, P.; Costello, J.F.; Pieper, R.O.; Ronen, S.M. Mutant IDH1 expression is associated with down-regulation of monocarboxylate transporters. Oncotarget 2016, 7, 34942–34955. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ruiz-Rodado, V.; Malta, T.M.; Seki, T.; Lita, A.; Dowdy, T.; Celiku, O.; Cavazos-Saldana, A.; Li, A.; Liu, Y.; Han, S.; et al. Metabolic reprogramming associated with aggressiveness occurs in the G-CIMP-high molecular subtypes of IDH1mut lower grade gliomas. Neuro Oncol 2020, 22, 480–492. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Ishida, C.T.; Bianchetti, E.; Zhang, Y.; Shu, C.; Tsujiuchi, T.; Banu, M.A.; Garcia, F.; Roth, K.A.; Bruce, J.N.; et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat. Commun. 2017, 8, 1067. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagné, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P. 2-Hydroxyglutarate in Cancer Cells. Antioxid. Redox Signal. 2020, 33, 903–926. [Google Scholar] [CrossRef]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour. Biol. 2015, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Liu, Y.; Neltner, J.; Pu, H.; Morris, A.; Sunkara, M.; Pittman, T.; Kyprianou, N.; Horbinski, C. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol. 2014, 127, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Mohrenz, I.V.; Antonietti, P.; Pusch, S.; Capper, D.; Balss, J.; Voigt, S.; Weissert, S.; Mukrowsky, A.; Frank, J.; Senft, C.; et al. Isocitrate dehydrogenase 1 mutant R132H sensitizes glioma cells to BCNU-induced oxidative stress and cell death. Apoptosis 2013, 18, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, L.; Valable, S.; Lechapt-Zalcman, E.; Ponte, K.; Collet, S.; Constans, J.M.; Levallet, G.; Bordji, K.; Petit, E.; Branger, P.; et al. [18F]-FMISO PET study of hypoxia in gliomas before surgery: Correlation with molecular markers of hypoxia and angiogenesis. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, P.; Callea, M.; Fallanca, F.; Castellano, A.; Bailo, M.; Scifo, P.; Bettinardi, V.; Conte, G.M.; Monterisi, C.; Rancoita, P.M.V.; et al. 18F-FAZA PET/CT in pretreatment assessment of hypoxic status in high-grade glioma: Correlation with hypoxia immunohistochemical biomarkers. Nucl. Med. Commun. 2021. [Google Scholar] [CrossRef]
- Sadeghzadeh, M.; Wenzel, B.; Gündel, D.; Deuther-Conrad, W.; Toussaint, M.; Moldovan, R.P.; Fischer, S.; Ludwig, F.A.; Teodoro, R.; Jonnalagadda, S.; et al. Development of Novel Analogs of the Monocarboxylate Transporter Ligand FACH and Biological Validation of One Potential Radiotracer for Positron Emission Tomography (PET) Imaging. Molecules 2020, 25, 2309. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, H.; Li, N.; Huang, H.; Liu, T.; Guo, X.; Xu, X.; Xia, L.; Deng, C.; Tian, X.; et al. Targeting CAIX with [64 Cu]XYIMSR-06 Small Molecular Radiotracer Enables Noninvasive PET Imaging of Malignant Glioma in U87 MG Tumor Cell Xenograft Mice. Mol. Pharm. 2019, 16, 1532–1540. [Google Scholar] [CrossRef]
- Radoul, M.; Najac, C.; Viswanath, P.; Mukherjee, J.; Kelly, M.; Gillespie, A.M.; Chaumeil, M.M.; Eriksson, P.; Delos Santos, R.; Pieper, R.O.; et al. HDAC inhibition in glioblastoma monitored by hyperpolarized 13C MRSI. Nmr. Biomed. 2019, 32, e4044. [Google Scholar] [CrossRef]
- Rafiki, A.; Boulland, J.L.; Halestrap, A.P.; Ottersen, O.P.; Bergersen, L. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience 2003, 122, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: An in vivo study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef]
- Voss, D.M.; Spina, R.; Carter, D.L.; Lim, K.S.; Jeffery, C.J.; Bar, E.E. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci. Rep. 2017, 7, 4292. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Cardoso-Carneiro, D.; Valbom, I.; Cury, F.P.; Silva, V.A.; Granja, S.; Reis, R.M.; Baltazar, F.; Martinho, O. Metabolic alterations underlying Bevacizumab therapy in glioblastoma cells. Oncotarget 2017, 8, 103657–103670. [Google Scholar] [CrossRef]
- Colen, C.B.; Seraji-Bozorgzad, N.; Marples, B.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: An in vitro study. Neurosurgery 2006, 59, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Amiri, A.; Le, P.U.; Moquin, A.; Machkalyan, G.; Petrecca, K.; Gillard, J.W.; Yoganathan, N.; Maysinger, D. Inhibition of carbonic anhydrase IX in glioblastoma multiforme. Eur. J. Pharm. Biopharm. 2016, 109, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.L.; Aguirre, M.T.; Ravichandran, S.; Leishman, L.L.; Berrondo, C.; Gamboa, J.T.; Wang, L.; King, R.; Wang, X.; Tan, M.; et al. RNA interference targeting hypoxia-inducible factor 1α via a novel multifunctional surfactant attenuates glioma growth in an intracranial mouse model. J. Neurosurg. 2015, 122, 331–341. [Google Scholar] [CrossRef]
- El Sayed, S.M.; El-Magd, R.M.; Shishido, Y.; Chung, S.P.; Diem, T.H.; Sakai, T.; Watanabe, H.; Kagami, S.; Fukui, K. 3-Bromopyruvate antagonizes effects of lactate and pyruvate, synergizes with citrate and exerts novel anti-glioma effects. J. Bioenerg. Biomembr. 2012, 44, 61–79. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Thiepold, A.L.; Lorenz, N.I.; Foltyn, M.; Engel, A.L.; Divé, I.; Urban, H.; Heller, S.; Bruns, I.; Hofmann, U.; Dröse, S.; et al. Mammalian target of rapamycin complex 1 activation sensitizes human glioma cells to hypoxia-induced cell death. Brain 2017, 140, 2623–2638. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Xue, S.; Song, G.; Yu, J. The prognostic significance of PD-L1 expression in patients with glioma: A meta-analysis. Sci. Rep. 2017, 7, 4231. [Google Scholar] [CrossRef] [PubMed]
- Givechian, K.B.; Garner, C.; Benz, S.; Rabizadeh, S.; Soon-Shiong, P. Glycolytic expression in lower-grade glioma reveals an epigenetic association between IDH mutation status and PDL1/2 expression. Neurooncol. Adv. 2021, 3, vdaa162. [Google Scholar] [CrossRef]
- Vallejo, F.A.; Vanni, S.; Graham, R.M. UCP2 as a Potential Biomarker for Adjunctive Metabolic Therapies in Tumor Management. Front. Oncol. 2021, 11, 640720. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef]
- O’Sullivan, D.; Pearce, E.L. Targeting T cell metabolism for therapy. Trends Immunol. 2015, 36, 71–80. [Google Scholar] [CrossRef]
- Singer, K.; Kastenberger, M.; Gottfried, E.; Hammerschmied, C.G.; Büttner, M.; Aigner, M.; Seliger, B.; Walter, B.; Schlösser, H.; Hartmann, A.; et al. Warburg phenotype in renal cell carcinoma: High expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. Int. J. Cancer 2011, 128, 2085–2095. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 2010, 32, 67–78. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Bristow, C.A.; Carugo, A.; Peoples, M.D.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, Q.; Song, Q.; Wang, H.; Dmitriev, P.; Sun, M.Y.; Cao, X.; Wang, Y.; Guo, L.; Indig, I.H.; et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR T-cell therapy against glioblastoma. Neuro. Oncol. 2019, 21, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.S.; Poschet, G.; Nonnenmacher, Y.; Becker, H.M.; Sapcariu, S.; Gaupel, A.C.; Schlotter, M.; Wu, Y.; Kneisel, N.; Seiffert, M.; et al. Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 2017, 18, 2172–2185. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xu, Z.; Duan, H.; Ji, H.; Zhen, Z.; Li, B.; Wang, H.; Tang, H.; Zhou, J.; Guo, T.; et al. Tumor-associated macrophage interleukin-β promotes glycerol-3-phosphate dehydrogenase activation, glycolysis and tumorigenesis in glioma cells. Cancer Sci. 2020, 111, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Meng, G.; Su, L.; Chen, A.; Xia, M.; Xu, C.; Yu, D.; Jiang, A.; Wei, J. Dichloroacetate blocks aerobic glycolytic adaptation to attenuated measles virus and promotes viral replication leading to enhanced oncolysis in glioblastoma. Oncotarget 2015, 6, 1544–1555. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | MCT1 | MCT2 | MCT4 |
|---|---|---|---|
| Formate | - | - | >500 |
| Bicarbonate | - | - | >500 |
| Oxamate | - | - | >500 |
| Glyoxylate | - | - | >500 |
| L-Lactate | 3.5 | 0.74 | 28 |
| D-Lactate | >60 | - | 519 |
| Pyruvate | 1.0 | 0.08 | 153 |
| S-Chloropropionate | - | - | 46 |
| R-Chloropropionate | - | - | 51 |
| D,L-α-Hydroxybutyrate | - | - | 56 |
| L-β-Hydroxybutyrate | - | 1.2 | 824 |
| D-β-Hydroxybutyrate | - | 1.2 | 130 |
| γ-Hydroxybutyrate | - | - | >500 |
| Acetoacetate | - | 0.8 | 216 |
| α-Ketobutyrate | - | - | 57 |
| α-Ketoisocaproate | 0.7 | 0.1 | 95 |
| α-Ketoisovalerate | 1.3 | 0.3 | 113 |
| β-Phenylpyruvate | - | - | >500 |
| miRNA | Expression in Glioma | Targeted by | Targets | Effects in Glioma | Literature |
|---|---|---|---|---|---|
| miR-1 | Downregulated | - | Annexin A2 | Decreases proliferation, invasion, and angiogenesis in glioma cells and xenografts | [89] |
| miR-9 | Overexpressed | CREB | CREB, NF1 | Decreases proliferation and increases migration in glioma cells | [90] |
| miR-29a | Downregulated | - | PDGFC, PDGFA | Decreases proliferation, cell viability, migration, and invasion in glioma cells, and tumor growth in xenografts | [91] |
| miR-95-3p | Downregulated | - | CELF2 | Decreases proliferation, cell viability, and invasion in glioma cells | [92] |
| miR-124 | Downregulated | - | SNAl2 | Decreases proliferation and invasion in glioma cells and tumor growth in xenografts | [93] |
| miR-134 | Downregulated | - | KRAS, STAT5B | Decreases proliferation and cell viability in glioma and glioma stem cells and tumor growth in xenografts | [94] |
| miR-145 | Downregulated | - | - | Decreases migration and invasion in glioma cells | [95] |
| miR-148a | Overexpressed | - | MIG6, BIM | Increases proliferation, cell viability, migration, and invasion in glioma cells, and tumor growth in xenografts | [96] |
| miR-150 | Overexpressed | - | VHL | Increases glucose uptake, lactate secretion, and proliferation in glioma cells, and tumor growth in xenografts | [88] |
| miR-181b | Downregulated | - | SP1 | Decreases glucose uptake and proliferation in glioma cells and tumor growth in xenografts | [97] |
| miR-181d | Downregulated | - | KRAS, Bcl-2 | Decreases proliferation and cell viability in glioma cells and tumor growth in xenografts | [98] |
| miR-203 | Downregulated | - | - | - | [99] |
| miR-338-3p | Downregulated | circSMO742 &SMO | - | Decreases proliferation, cell viability, migration, and invasion in glioma cells | [100] |
| miR-351 | Overexpressed | - | NAIF1 | Increases cell viability, migration, and invasion in glioma cells | [101] |
| miR-378e | Downregulated | circNFIX | RPN2 | Decreases glucose uptake, lactate secretion, cell viability, migration, and invasion in glioma cells | [102] |
| miR-423-5p | Overexpressed | - | ING-4 | Increases proliferation, invasion, angiogenesis, and temozolomide resistance in glioma cells and tumor growth and invasion in xenografts | [103] |
| miR-432-5p | Downregulated | - | RAB10 | Decreases glucose uptake, lactate secretion, invasion, and proliferation in glioma cells | [104] |
| miR-451 | Downregulated | - | - | Increases cell viability and decreases invasion in glioma cells | [86] |
| miR-451 | Downregulated | - | CAB39 | Decreases proliferation, invasion, and migration in glioma cells and tumor growth in xenografts | [87] |
| miR-451 | Downregulated in low glucose level glioma cells | - | - | Decreases migration in glioma cells andinvasion in xenografts, increases sensitivity to temozolomide treatment in glioma cells | [84] |
| miR-451 | Downregulated | - | - | Decreases proliferation, cell viability, and invasion in glioma cells | [105] |
| miR-451 | Downregulated | lncRNA LSINCT5 | CAB39 | Decreases glycolysis, cell viability, invasion, and migration in glioma cells | [85,106] |
| miR-495 | Downregulated | - | GLUT1 | Decreases glucose uptake and lactate secretion in glioma cells | [82] |
| miR-663 | Downregulated | - | PIK3CD | Decreases proliferation and invasion in glioma cells | [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reuss, A.M.; Groos, D.; Buchfelder, M.; Savaskan, N. The Acidic Brain—Glycolytic Switch in the Microenvironment of Malignant Glioma. Int. J. Mol. Sci. 2021, 22, 5518. https://doi.org/10.3390/ijms22115518
Reuss AM, Groos D, Buchfelder M, Savaskan N. The Acidic Brain—Glycolytic Switch in the Microenvironment of Malignant Glioma. International Journal of Molecular Sciences. 2021; 22(11):5518. https://doi.org/10.3390/ijms22115518
Chicago/Turabian StyleReuss, Anna Maria, Dominik Groos, Michael Buchfelder, and Nicolai Savaskan. 2021. "The Acidic Brain—Glycolytic Switch in the Microenvironment of Malignant Glioma" International Journal of Molecular Sciences 22, no. 11: 5518. https://doi.org/10.3390/ijms22115518
APA StyleReuss, A. M., Groos, D., Buchfelder, M., & Savaskan, N. (2021). The Acidic Brain—Glycolytic Switch in the Microenvironment of Malignant Glioma. International Journal of Molecular Sciences, 22(11), 5518. https://doi.org/10.3390/ijms22115518