
Insight into Bortezomib Focusing on Its Efficacy against P-gp-Positive MDR Leukemia Cells

 , , and
, , and
Abstract
1. Introduction
2. Results
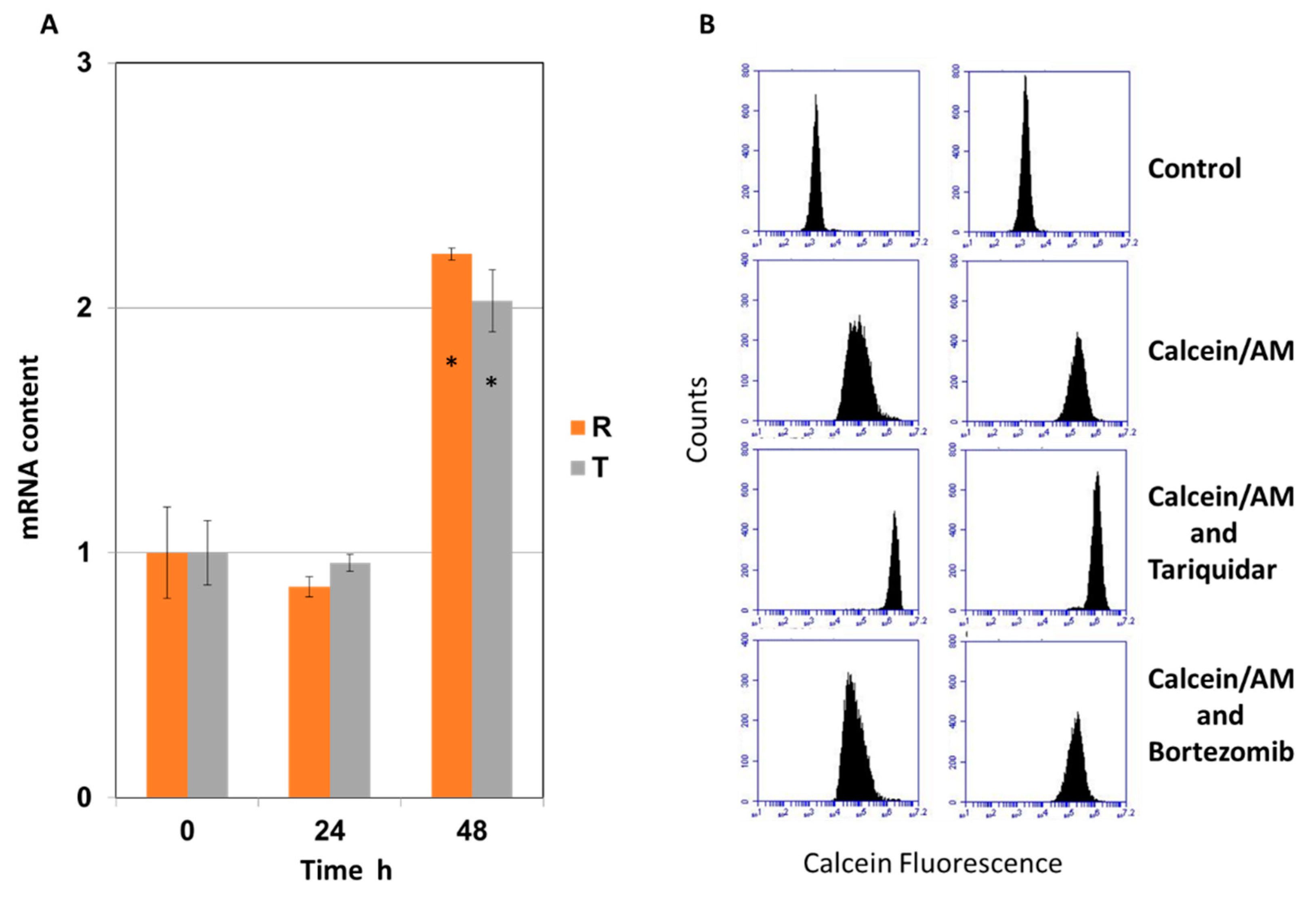
2.1. Effect of Bortezomib on the Expression and Activity of P-glycoprotein
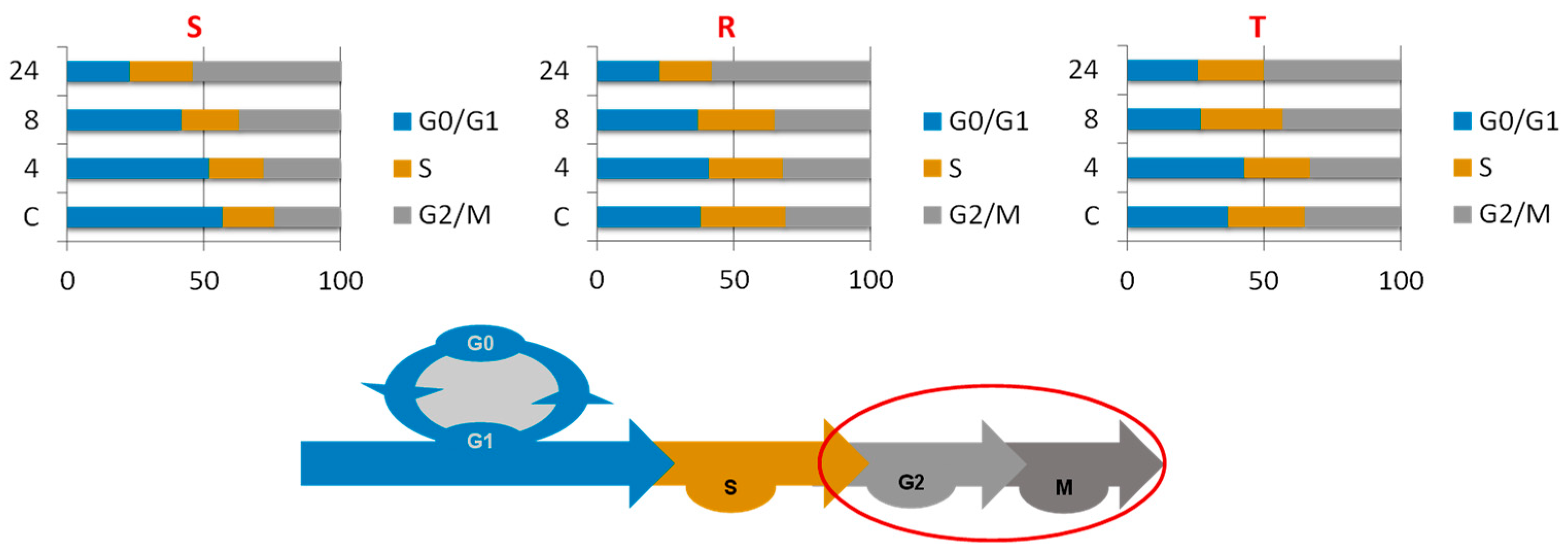
2.2. Effect of Bortezomib on the Cell Cycling of the S, R, and T Cell Variants
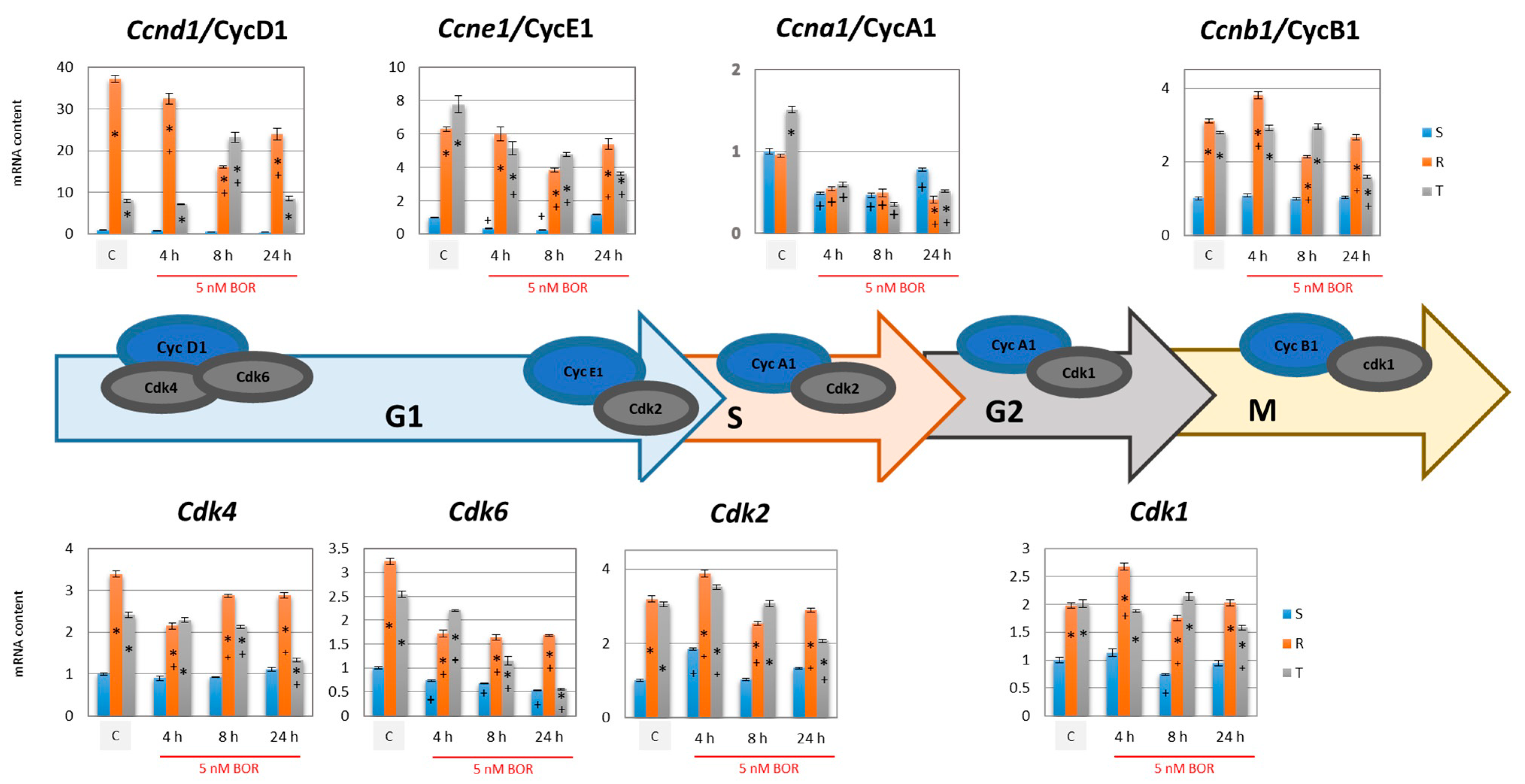
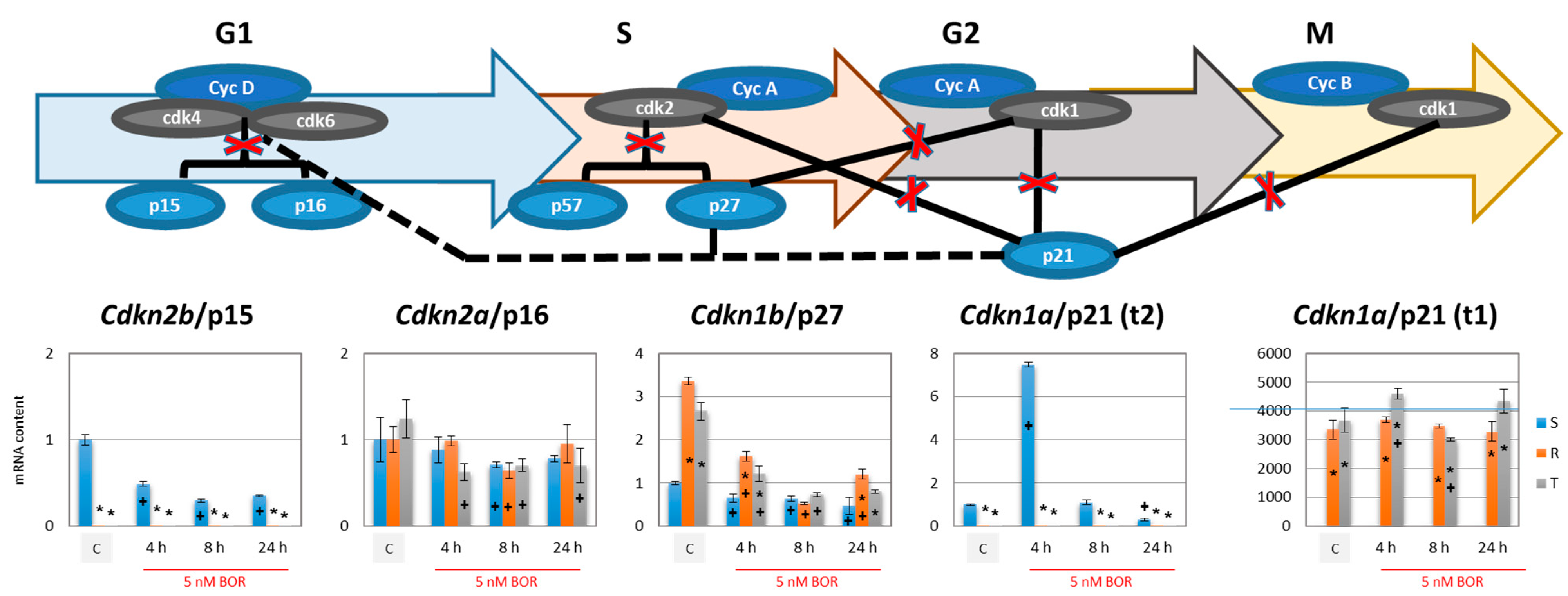
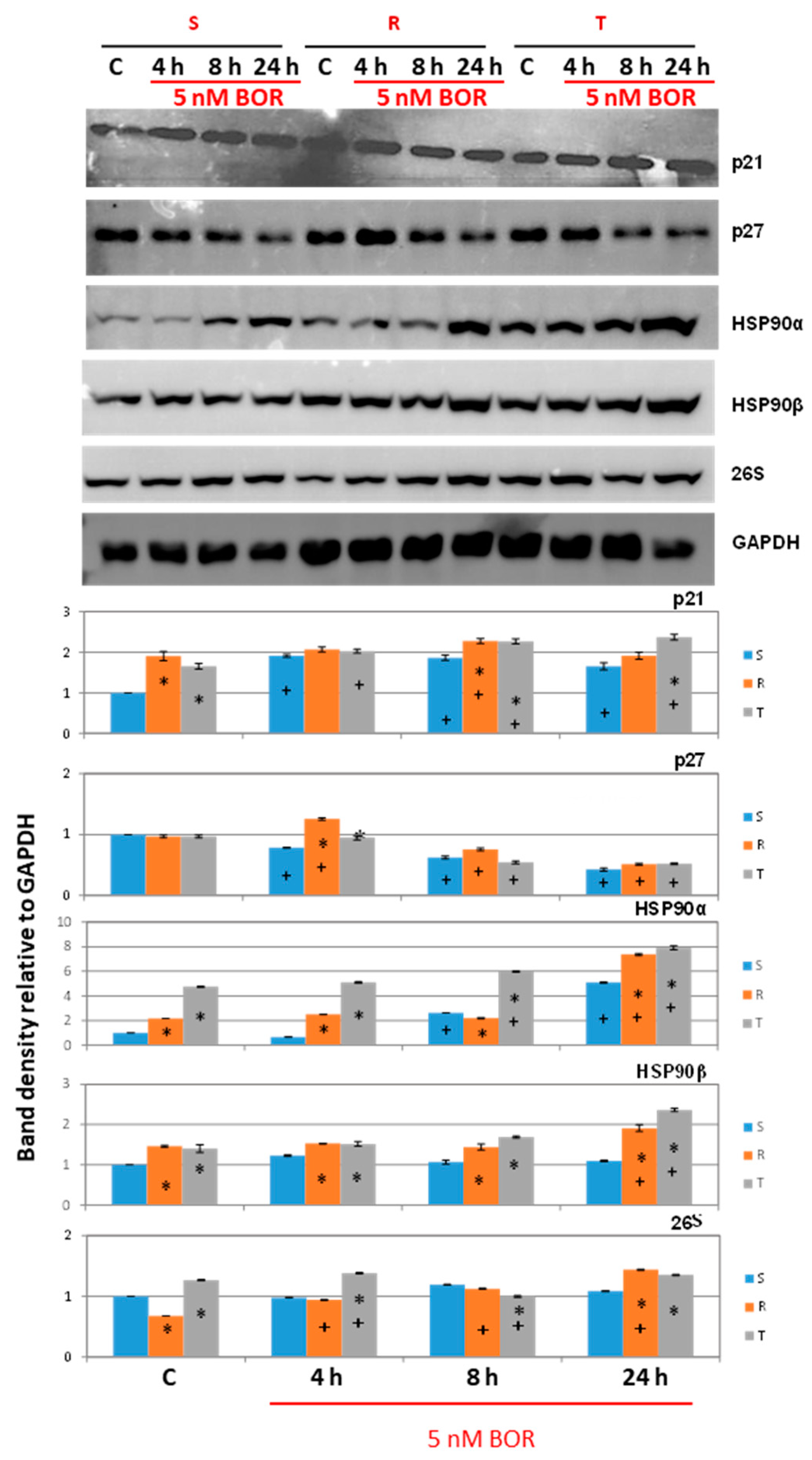
2.3. Effects of Bortezomib on the Expression of Proteins Associated with CC Progression in S, R, and T Cells
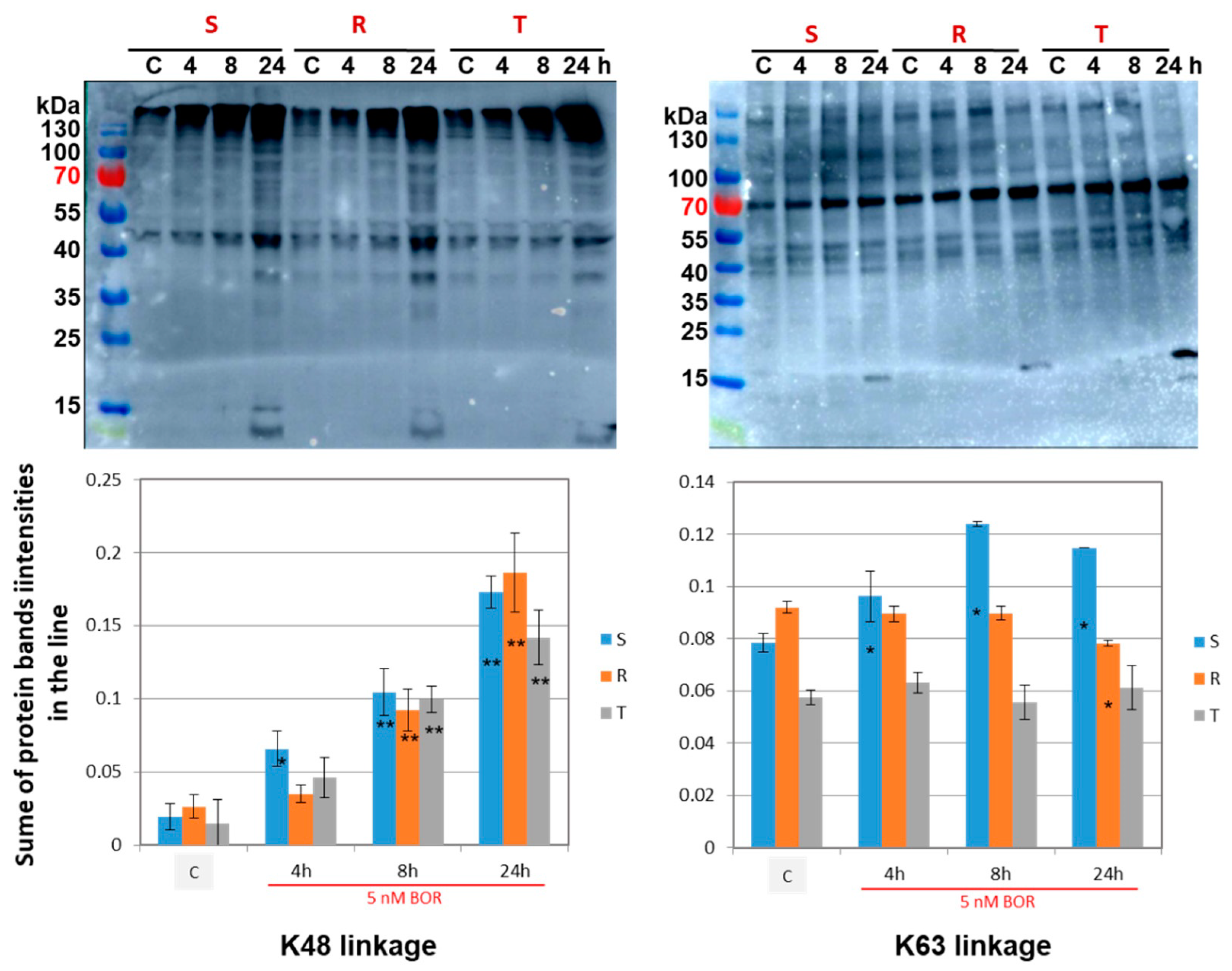
2.4. Effects of Bortezomib on the Quantity and Linkage of Ubiquitination
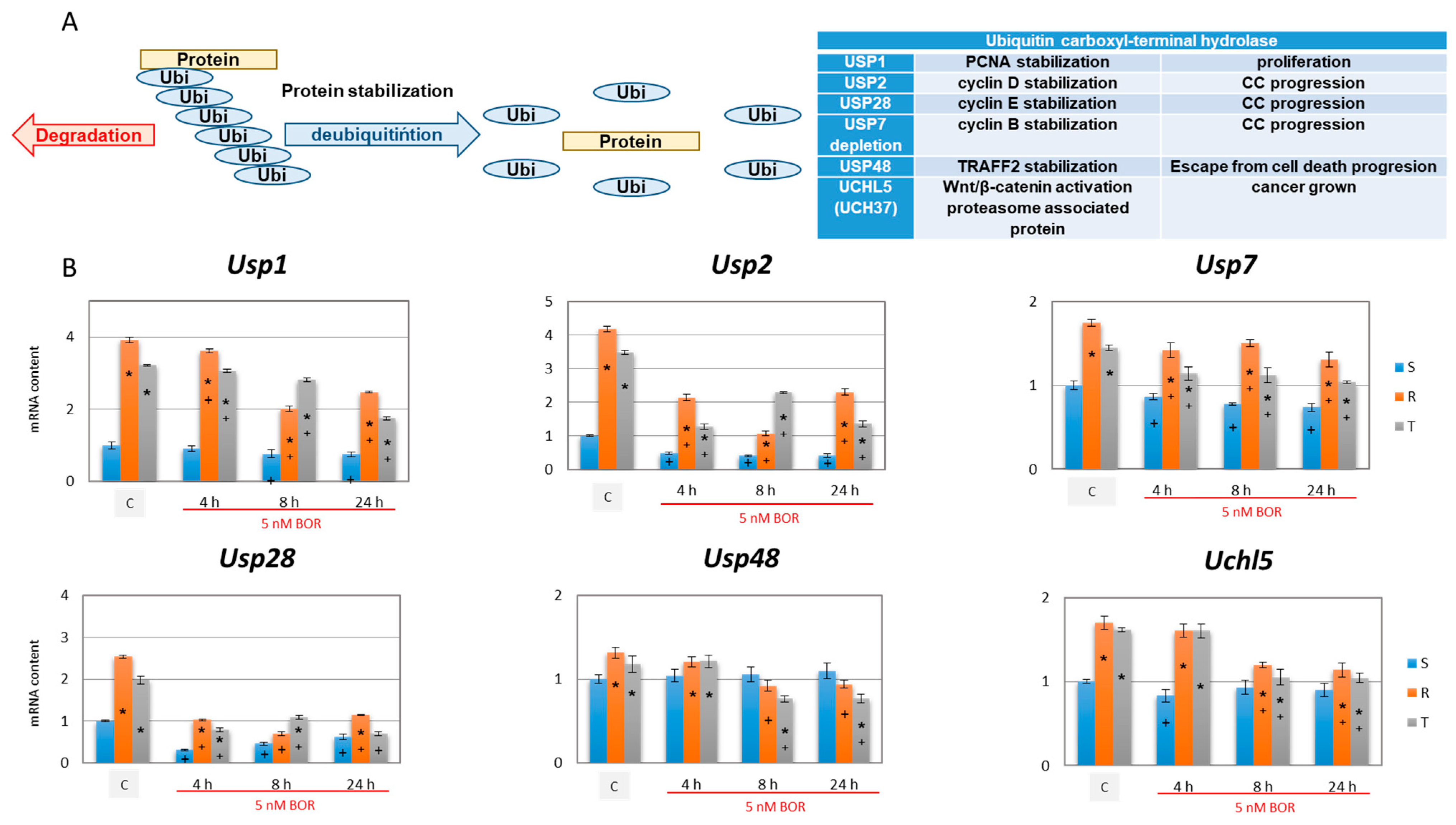
2.5. Effects of Bortezomib on the Expression of Deubiquitinating Enzymes
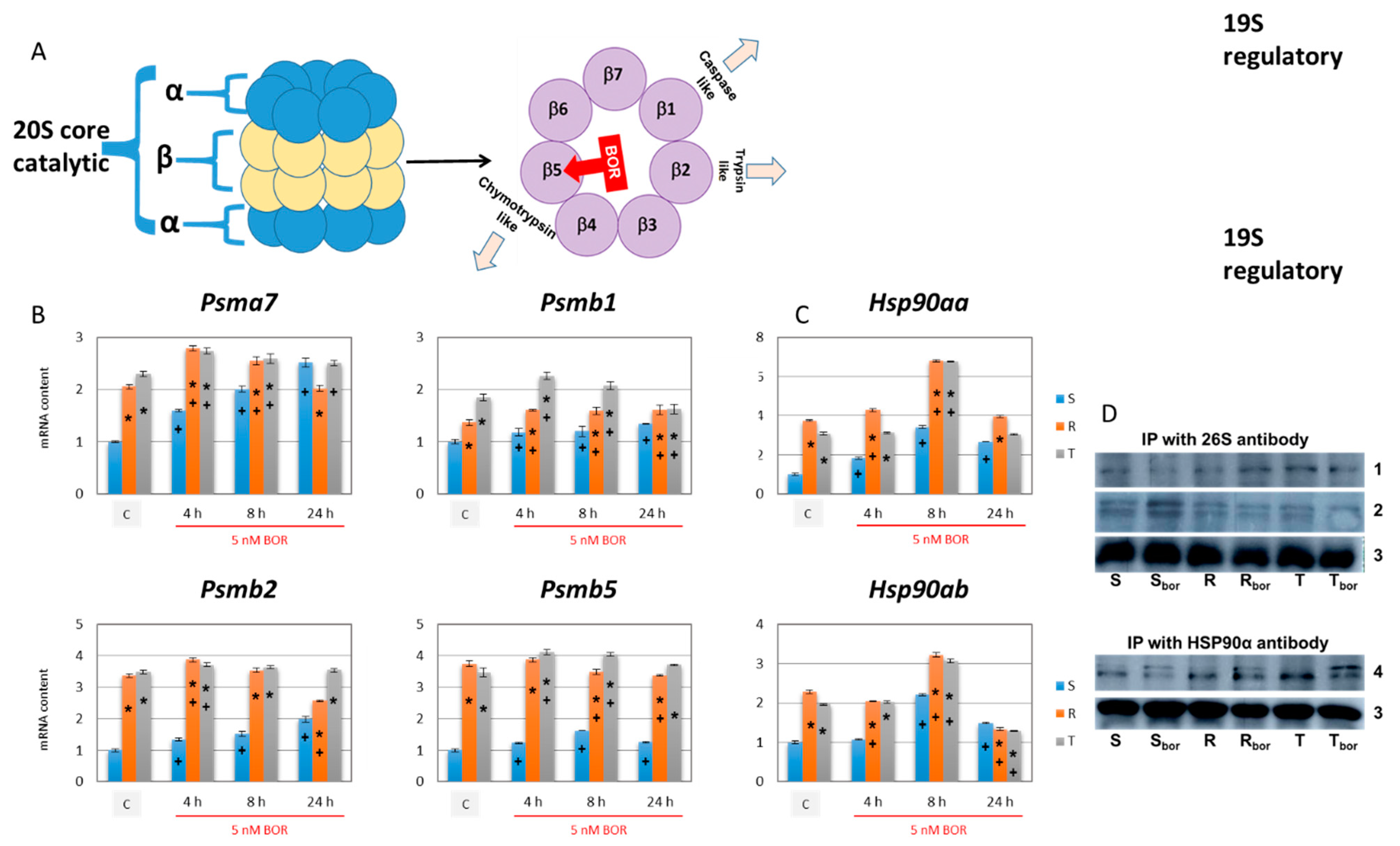
2.6. Effect of Bortezomib on the Expression of Proteins Required for Proteasome Formation
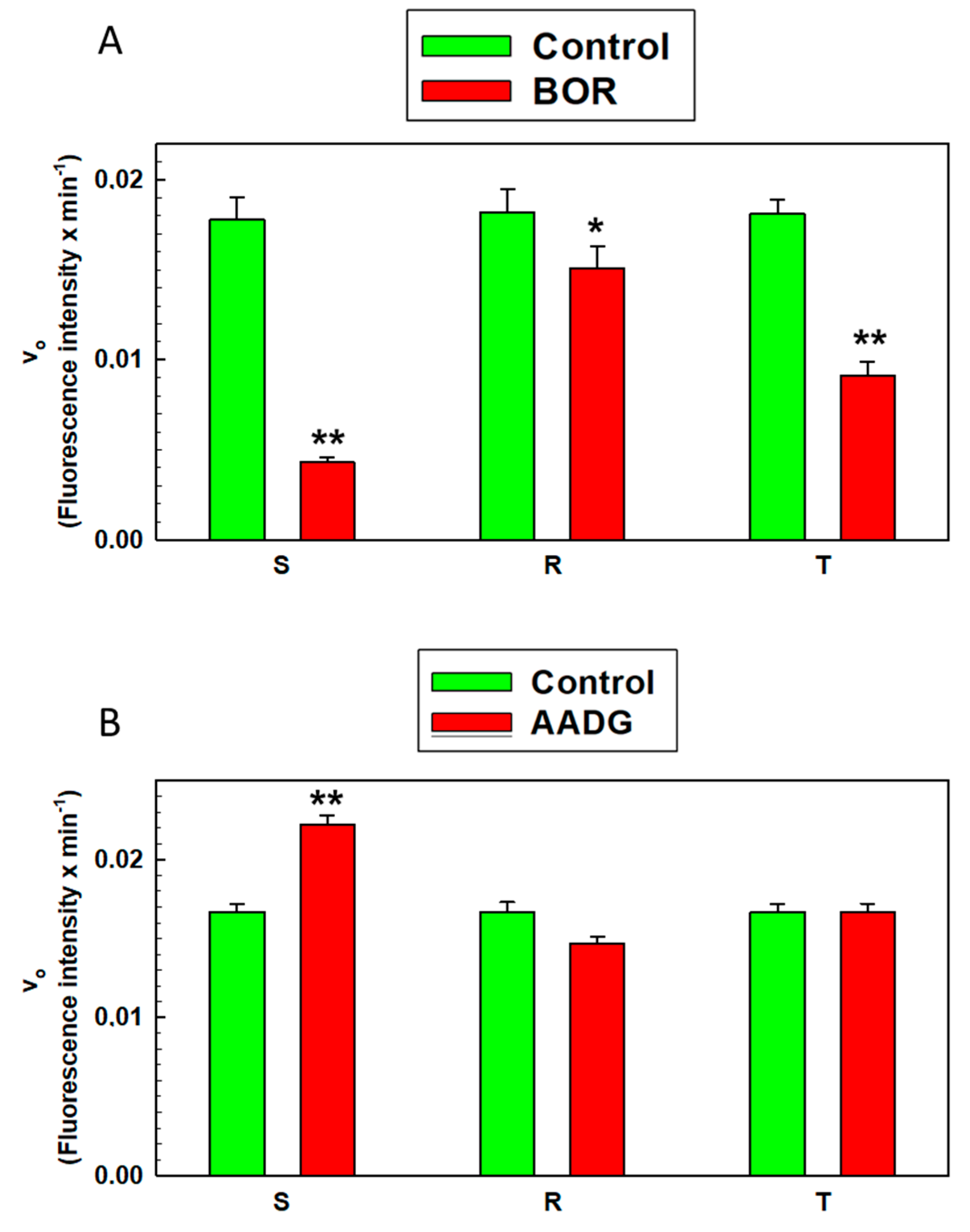
2.7. Measurement of Proteasome Activity in S, R, and T Cells
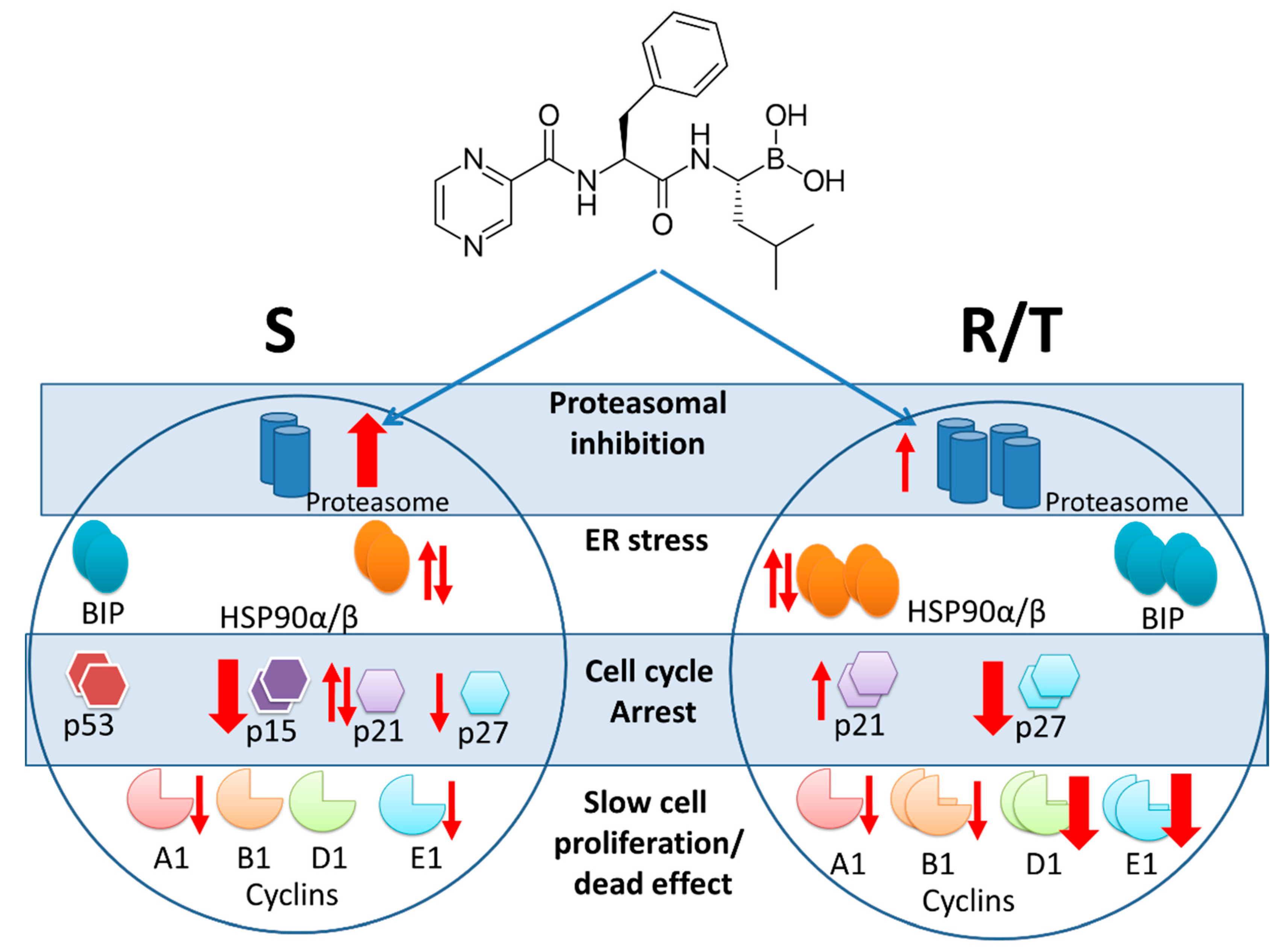
3. Discussion
- S cells showed a greater tendency to enter CC arrest in the G2/M phase than R or T cells, which manifests as S cells arrested within 4 h of treatment (Figure 2).
- The gene expression of cyclins B1, D1, and E1 and all four CDKs was higher in the R and T cells than in the S cells (Figure 4). This expression level was maintained for CDK1 and CDK2 (active in S and G2/M phase [35,36]) and cyclins B1 (activating CDK1 in the M phase [36]), D1, and E1 (both activating CDK4 and CDK6 in the G1 phase [33,34]) even 24 h after the addition of 5 nM BOR. In the T cells but not in the R cells, 24 h after the addition of BOR, the expression levels of the CDK4- and CDK6-encoding genes approached the levels in the S cells. Cyclin A1 (activating CDK2 in the S phase [35] and CDK1 in the G2 phase [36]) gene expression levels decreased after treatment of all three cell variants with BOR (Figure 4).
- Cdkn2b (encoding the p15 inhibitors CDK4 and CDK6 [38]) was expressed at a lower level in the R and T cells than in the S cells and persisted at this level even after BOR treatment, although the expression of this gene in the S cells decreased (Figure 4). Higher expression levels of the Cdkn1b gene (the p27 inhibitor CDK1 and CDK2 and stabilizer of the CDK4 and CDK6 complexes formed with cyclin D1 [39]) was observed in the R and T cells than in the S cells, a trend that persisted even after BOR treatment, although the gene expression levels for this protein decreased in all three cell variants (Figure 4). Consistent with its gene expression, the p27 protein level also decreased in all three cell variants after treatment with BOR (Figure 5). Expression of a multimodal modulator of CDKs encoded by the Cdkn1a gene (p21, an inhibitor of CDK1 and CDK2 and stabilizer of complexes formed by CDK4 or CDK6 with cyclin D1 [39,60]) is expressed from a common t2 transcript variant in S cells and from an alternative t1 transcript variant in R and T cells (Figure S5 in supplementary files).
4. Materials and Methods
4.1. Cells and Cultivation Conditions
4.2. Cell Viability Tested by MTT Assay
4.3. Measurement of P-gp Transport Activity by Calcein/AM Retention Assay
4.4. Monitoring of Cell Cycle Progression
4.5. Detection of Gene Expression by qRT-PCR
4.6. Western Blotting
4.7. Visualization of HSP90α-and β-Immunoreactive Proteins in S, R, and T Cells by Immunofluorescence Confocal Microscopy
4.8. Immunoprecipitation of the 26S Proteasome and HSP90α or HSP90β
4.9. Proteasome Activity Assay
4.10. Statistical Analysis and Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 19S | Regulatory cap of proteasome |
| 20S | Catalytic core of proteasome |
| 26S | Completed proteasome |
| AADG | 17-(allylamino)-17-demethoxygeldanamycin |
| ABC | ATP-binding cassette |
| ABCB1 | B1 member of ABC transporter family, P-gp |
| ABCC1 | C1 member of ABC transporter family, MRP1 |
| ABCC/ABCG | C and G subfamily of ABC transporter |
| Akt | Serine/threonine specific protein kinase B (PKB) |
| ALL | Acute lymphoblastic leukemia |
| AMC | 7-amino-4-methylcoumarin |
| ATP | Adenosine triphosphate |
| BiP | Binding immunoglobulin protein |
| BOR | Bortezomib |
| BSA | Bovine serum albumin |
| Calcein/AM | Acetoxymethyl-calcein |
| CC | Cell cycle |
| Ccna1, b1,d1,e1 | Mouse gene encoding cyclin A1, B1, D1, and E1, respectively |
| CDK(s) | Cyclin-dependent kinase(s) |
| Cdk1, 2, 4, 6 | Mouse gene encoding CDK1, 2, 4, and 6, respectively |
| CDK1, 2, 4, 6 | Cyclin-dependent kinase 1, 2, 4, and 6, respectively |
| CIP | CDK-interacting protein, inhibitor of CDK |
| CKIs | Cyclin-dependent kinase inhibitors |
| DAPI | 4′,6-diamidino-2-phenylindole, fluorescent stain |
| DSMZ | German Collection of Microorganisms and Cell Culture |
| ECL | Enhanced chemiluminescence |
| EMA | European medicines agency |
| ER | Endoplasmic reticulum |
| FAV | Fluorescein isothiocyanate labeled annexin V |
| FDA | U.S. Food and Drug Administration |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GRP78 | Glucose-regulated protein 78-kda (bip) |
| HSP90 | Heat shock protein 90 |
| Hsp90aa, ab | Mouse gene encoding HSP90α and β, respectively |
| HSP90α, β | Heat shock protein 90α and β, respectively |
| IC50 | Half maximal inhibitory concentration |
| IgG | Immunoglobulin G |
| INK4 | Inhibitor of CDK4 |
| K48 | Lysine 48 |
| K63 | Lysine 63 |
| KIP | Kinase inhibitory protein, inhibitor of CDK |
| L1210 | Mouse lymphocytic leukemia cell line |
| MDR | Multidrug resistance |
| MG-132 | Inhibitor of proteasome |
| MTT | Methylthiazolyldiphenyl-tetrazolium bromide |
| p15, 16 | CDK4/6-inhibitors known also as INK4 |
| p21, 27, 57 | CDK-interacting protein/kinase inhibitory protein (CIP/KIP) |
| PAGE | Polyacrylamide gel electrophoresis |
| PBS | Phosphate buffered saline |
| PCR | Polymerase chain reaction |
| PFD | Paraformaldehyde |
| P-gp | P-glycoprotein |
| PI | Propidium iodide |
| PNCA | Proliferating cell nuclear antigen |
| Psma7 | Mouse gene encoding proteasome subunit α7 |
| Psmb1, b2, b5 | Mouse gene encoding proteasome β1, β2, and β5, respectively |
| PVDF | Polyvinylidene fluoride, special plastic |
| qRT-PCR | Quantitative reverse transcription-polymerase chain reaction |
| R | P-gp positive L1210 cells obtained by selectin with VCR |
| RPMI 1640 | Roswell Park Memorial Institute 1640 medium |
| S | Parental L1210cells |
| SDS | Sodium dodecyl sulfate |
| T | P-gp positive L1210 cells obtained by transfection with P-gp |
| TRAF2 | Tumor necrosis factor receptor-associated factor 2 |
| Ubi | Ubiquitin |
| UPR | Unfolded protein response |
| UCHL5 | Ubiquitin C-Terminal Hydrolase L5 |
| USP1 | Ubiquitin-specific protease 1 |
| USP2 | Ubiquitin-specific protease 2 |
| USP28 | Ubiquitin-specific protease 28 |
| USP48 | Ubiquitin-specific protease 48 |
| USP7 | Ubiquitin-specific protease 7 |
| VCR | Vincristine |
| YB-1 | Y box binding protein 1 |
References
- Kazandjian, D.; Blumenthal, G.M.; Yuan, W.; He, K.; Keegan, P.; Pazdur, R. Fda approval of gefitinib for the treatment of patients with metastatic egfr mutation-positive non-small cell lung cancer. Clin. Cancer Res. 2016, 22, 1307–1312. [Google Scholar] [CrossRef]
- Ema Bortezomib Drug Label (Epar Poduct Information). Available online: Http://www.Ema.Europa.Eu/docs/en_gb/document_library/epar_-_product_information/human/000539/wc500048471.Pdf (accessed on 21 March 2021).
- Terpos, E.; Katodritou, E.; de la Rubia, J.; Hungria, V.; Hulin, C.; Roussou, M.; Delforge, M.; Bries, G.; Stoppa, A.M.; Aagesen, J.; et al. Bortezomib-based therapy for relapsed/refractory multiple myeloma in real-world medical practice. Eur. J. Haematol. 2018, 101, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Robak, P.; Robak, T. Bortezomib for the treatment of hematologic malignancies: 15 years later. Drugs R&D 2019, 19, 73–92. [Google Scholar]
- Bonvini, P.; Zorzi, E.; Basso, G.; Rosolen, A. Bortezomib-mediated 26s proteasome inhibition causes cell-cycle arrest and induces apoptosis in cd-30+ anaplastic large cell lymphoma. Leukemia 2007, 21, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Bolognese, A.; Esposito, A.; Manfra, M.; Catalano, L.; Petruzziello, F.; Martorelli, M.C.; Pagliuca, R.; Mazzarelli, V.; Ottiero, M.; Scalfaro, M.; et al. An nmr study of the bortezomib degradation under clinical use conditions. Adv. Hematol. 2009, 2009, 704928. [Google Scholar] [CrossRef][Green Version]
- Bruning, A.; Burger, P.; Vogel, M.; Rahmeh, M.; Friese, K.; Lenhard, M.; Burges, A. Bortezomib treatment of ovarian cancer cells mediates endoplasmic reticulum stress, cell cycle arrest, and apoptosis. Investig. New Drugs 2009, 27, 543–551. [Google Scholar] [CrossRef]
- Bertaina, A.; Vinti, L.; Strocchio, L.; Gaspari, S.; Caruso, R.; Algeri, M.; Coletti, V.; Gurnari, C.; Romano, M.; Cefalo, M.G.; et al. The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br. J. Haematol. 2017, 176, 629–636. [Google Scholar] [CrossRef]
- Messinger, Y.H.; Gaynon, P.S.; Sposto, R.; van der Giessen, J.; Eckroth, E.; Malvar, J.; Bostrom, B.C.; Therapeutic Advances in Childhood, L.; Lymphoma, C. Bortezomib with chemotherapy is highly active in advanced b-precursor acute lymphoblastic leukemia: Therapeutic advances in childhood leukemia & lymphoma (tacl) study. Blood 2012, 120, 285–290. [Google Scholar] [PubMed]
- O’Connor, R.; Ooi, M.G.; Meiller, J.; Jakubikova, J.; Klippel, S.; Delmore, J.; Richardson, P.; Anderson, K.; Clynes, M.; Mitsiades, C.S.; et al. The interaction of bortezomib with multidrug transporters: Implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 2013, 71, 1357. [Google Scholar] [CrossRef]
- Panischeva, L.A.; Kakpakova, E.S.; Rybalkina, E.Y.; Stavrovskaya, A.A. Influence of proteasome inhibitor bortezomib on the expression of multidrug resistance genes and akt kinase activity. Biochemistry 2011, 76, 1009–1016. [Google Scholar] [CrossRef]
- Mynott, R.L.; Wallington-Beddoe, C.T. Inhibition of p-glycoprotein does not increase the efficacy of proteasome inhibitors in multiple myeloma cells. ACS Pharmacol. Transl. Sci. 2021, 4, 713–729. [Google Scholar] [CrossRef]
- Seres, M.; Pavlikova, L.; Bohacova, V.; Kyca, T.; Borovska, I.; Lakatos, B.; Breier, A.; Sulova, Z. Overexpression of grp78/bip in p-glycoprotein-positive l1210 cells is responsible for altered response of cells to tunicamycin as a stressor of the endoplasmic reticulum. Cells 2020, 9, 890. [Google Scholar] [CrossRef]
- Takahashi, K.; Inukai, T.; Imamura, T.; Yano, M.; Tomoyasu, C.; Lucas, D.M.; Nemoto, A.; Sato, H.; Huang, M.; Abe, M.; et al. Anti-leukemic activity of bortezomib and carfilzomib on b-cell precursor all cell lines. PLoS ONE 2017, 12, e0188680. [Google Scholar]
- Uhrik, B.; Tribulova, N.; Klobusicka, M.; Barancik, M.; Breier, A. Characterization of morphological and histochemical changes induced by overexpression of p-glycoprotein in mouse leukemic cell line l1210. Neoplasma 1994, 41, 83–88. [Google Scholar]
- Polekova, L.; Barancik, M.; Mrazova, T.; Pirker, R.; Wallner, J.; Sulova, Z.; Breier, A. Adaptation of mouse leukemia cells l1210 to vincristine. Evidence for expression of p-glycoprotein. Neoplasma 1992, 39, 73–77. [Google Scholar]
- Sulova, Z.; Ditte, P.; Kurucova, T.; Polakova, E.; Rogozanova, K.; Gibalova, L.; Seres, M.; Skvarkova, L.; Sedlak, J.; Pastorek, J.; et al. The presence of p-glycoprotein in l1210 cells directly induces down-regulation of cell surface saccharide targets of concanavalin A. Anticancer Res. 2010, 30, 3661–3668. [Google Scholar]
- Gibalova, L.; Seres, M.; Rusnak, A.; Ditte, P.; Labudova, M.; Uhrik, B.; Pastorek, J.; Sedlak, J.; Breier, A.; Sulova, Z. P-glycoprotein depresses cisplatin sensitivity in l1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol. In Vitro 2012, 26, 435–444. [Google Scholar] [CrossRef]
- Pavlikova, L.; Seres, M.; Hano, M.; Bohacova, V.; Sevcikova, I.; Kyca, T.; Breier, A.; Sulova, Z. L1210 cells overexpressing abcb1 drug transporters are resistant to inhibitors of the n- and o-glycosylation of proteins. Molecules 2017, 22, 1104. [Google Scholar] [CrossRef]
- Seres, M.; Cholujova, D.; Bubencikova, T.; Breier, A.; Sulova, Z. Tunicamycin depresses p-glycoprotein glycosylation without an effect on its membrane localization and drug efflux activity in l1210 cells. Int. J. Mol. Sci. 2011, 12, 7772–7784. [Google Scholar] [CrossRef]
- Seres, M.; Polakova, E.; Krizanova, O.; Hudecova, S.; Klymenko, S.V.; Breier, A.; Sulova, Z. Overexpression of p-glycoprotein in l1210/vcr cells is associated with changes in several endoplasmic reticulum proteins that may be partially responsible for the lack of thapsigargin sensitivity. Gen. Physiol. Biophys. 2008, 27, 211–221. [Google Scholar]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med. Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef]
- Hano, M.; Tomasova, L.; Seres, M.; Pavlikova, L.; Breier, A.; Sulova, Z. Interplay between p-glycoprotein expression and resistance to endoplasmic reticulum stressors. Molecules 2018, 23, 337. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Cagala, M.; Pavlikova, L.; Seres, M.; Kadlecikova, K.; Breier, A.; Sulova, Z. Development of resistance to endoplasmic reticulum stress-inducing agents in mouse leukemic l1210 cells. Molecules 2020, 25, 2517. [Google Scholar] [CrossRef]
- Minderman, H.; Zhou, Y.; O’Loughlin, K.L.; Baer, M.R. Bortezomib activity and in vitro interactions with anthracyclines and cytarabine in acute myeloid leukemia cells are independent of multidrug resistance mechanisms and p53 status. Cancer Chemother. Pharmacol. 2007, 60, 245–255. [Google Scholar] [CrossRef]
- Volk, E.L.; Farley, K.M.; Wu, Y.; Li, F.; Robey, R.W.; Schneider, E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002, 62, 5035–5040. [Google Scholar]
- Winter, S.S.; Ricci, J.; Luo, L.; Lovato, D.M.; Khawaja, H.M.; Serna-Gallegos, T.; Debassige, N.; Larson, R.S. Atp binding cassette c1 (abcc1/mrp1)-mediated drug efflux contributes to disease progression in t-lineage acute lymphoblastic leukemia. Health 2013, 5, 41. [Google Scholar] [CrossRef]
- Elefantova, K.; Lakatos, B.; Kubickova, J.; Sulova, Z.; Breier, A. Detection of the mitochondrial membrane potential by the cationic dye jc-1 in l1210 cells with massive overexpression of the plasma membrane abcb1 drug transporter. Int. J. Mol. Sci. 2018, 19, 1985. [Google Scholar] [CrossRef]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of cell cycle by flow cytometry. Methods Mol. Biol. 2004, 281, 301–311. [Google Scholar]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef]
- Wood, D.J.; Endicott, J.A. Structural insights into the functional diversity of the cdk-cyclin family. Open Biol 2018, 8, 180112. [Google Scholar] [CrossRef]
- Dong, P.; Zhang, C.; Parker, B.T.; You, L.; Mathey-Prevot, B. Cyclin d/cdk4/6 activity controls g1 length in mammalian cells. PLoS ONE 2018, 13, e0185637. [Google Scholar] [CrossRef]
- Aleem, E.; Berthet, C.; Kaldis, P. Cdk2 as a master of s phase entry: Fact or fake? Cell Cycle 2004, 3, 35–37. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.; Oakes, V.; Beamish, H.; Giles, N.; Stevens, F.; Somodevilla-Torres, M.; Desouza, C.; Gabrielli, B. Cyclin a/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 2008, 27, 4261–4268. [Google Scholar] [CrossRef]
- Huang, Y.; Sramkoski, R.M.; Jacobberger, J.W. The kinetics of g2 and m transitions regulated by b cyclins. PLoS ONE 2013, 8, e80861. [Google Scholar] [CrossRef]
- Francesconi, A.; Kasai, M.; Petraitiene, R.; Petraitis, V.; Kelaher, A.M.; Schaufele, R.; Hope, W.W.; Shea, Y.R.; Bacher, J.; Walsh, T.J. Characterization and comparison of galactomannan enzyme immunoassay and quantitative real-time pcr assay for detection of aspergillus fumigatus in bronchoalveolar lavage fluid from experimental invasive pulmonary aspergillosis. J. Clin. Microbiol. 2006, 44, 2475–2480. [Google Scholar] [CrossRef]
- Rodriguez-Diez, E.; Quereda, V.; Bellutti, F.; Prchal-Murphy, M.; Partida, D.; Eguren, M.; Kollmann, K.; Gomez de Cedron, M.; Dubus, P.; Canamero, M.; et al. Cdk4 and cdk6 cooperate in counteracting the ink4 family of inhibitors during murine leukemogenesis. Blood 2014, 124, 2380–2390. [Google Scholar] [CrossRef]
- Cerqueira, A.; Martin, A.; Symonds, C.E.; Odajima, J.; Dubus, P.; Barbacid, M.; Santamaria, D. Genetic characterization of the role of the cip/kip family of proteins as cyclin-dependent kinase inhibitors and assembly factors. Mol. Cell. Biol. 2014, 34, 1452–1459. [Google Scholar] [CrossRef]
- Bayrak, A.; Oktay, K. The expression of cyclin-dependent kinase inhibitors p15, p16, p21, and p27 during ovarian follicle growth initiation in the mouse. Reprod. Biol. Endocrinol. 2003, 1, 41. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Gierut, J.; Gartel, A.L. Multiple alternate p21 transcripts are regulated by p53 in human cells. Oncogene 2006, 25, 1812–1815. [Google Scholar] [CrossRef]
- Wirth, M.; Schick, M.; Keller, U.; Kronke, J. Ubiquitination and ubiquitin-like modifications in multiple myeloma: Biology and therapy. Cancers 2020, 12, 3764. [Google Scholar] [CrossRef]
- Lub, S.; Maes, K.; Menu, E.; De Bruyne, E.; Vanderkerken, K.; Van Valckenborgh, E. Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget 2016, 7, 6521–6537. [Google Scholar] [CrossRef]
- Lim, K.S.; Li, H.; Roberts, E.A.; Gaudiano, E.F.; Clairmont, C.; Sambel, L.A.; Ponnienselvan, K.; Liu, J.C.; Yang, C.; Kozono, D.; et al. Usp1 is required for replication fork protection in brca1-deficient tumors. Mol. Cell 2018, 72, 925–941 e924. [Google Scholar] [CrossRef]
- Shan, J.; Zhao, W.; Gu, W. Suppression of cancer cell growth by promoting cyclin d1 degradation. Mol Cell 2009, 36, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease usp28 is required for myc stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef]
- Giovinazzi, S.; Morozov, V.M.; Summers, M.K.; Reinhold, W.C.; Ishov, A.M. Usp7 and daxx regulate mitosis progression and taxane sensitivity by affecting stability of aurora-a kinase. Cell Death Differ. 2013, 20, 721–731. [Google Scholar] [CrossRef]
- Li, S.; Wang, D.; Zhao, J.; Weathington, N.M.; Shang, D.; Zhao, Y. The deubiquitinating enzyme usp48 stabilizes traf2 and reduces e-cadherin-mediated adherens junctions. FASEB J. 2018, 32, 230–242. [Google Scholar] [CrossRef]
- Tada, K.; Okazaki, T.; Sakon, S.; Kobarai, T.; Kurosawa, K.; Yamaoka, S.; Hashimoto, H.; Mak, T.W.; Yagita, H.; Okumura, K.; et al. Critical roles of traf2 and traf5 in tumor necrosis factor-induced nf-kappa b activation and protection from cell death. J. Biol. Chem. 2001, 276, 36530–36534. [Google Scholar] [CrossRef]
- Liu, D.; Song, Z.; Wang, X.; Ouyang, L. Ubiquitin c-terminal hydrolase l5 (uchl5) accelerates the growth of endometrial cancer via activating the wnt/beta-catenin signaling pathway. Front. Oncol. 2020, 10, 865. [Google Scholar] [CrossRef]
- Liu, C.W.; Jacobson, A.D. Functions of the 19s complex in proteasomal degradation. Trends Biochem. Sci. 2013, 38, 103–110. [Google Scholar] [CrossRef]
- Ben-Nissan, G.; Sharon, M. Regulating the 20s proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef]
- Eleuteri, A.M.; Cuccioloni, M.; Bellesi, J.; Lupidi, G.; Fioretti, E.; Angeletti, M. Interaction of hsp90 with 20s proteasome: Thermodynamic and kinetic characterization. Proteins 2002, 48, 169–177. [Google Scholar] [CrossRef]
- Whittier, J.E.; Xiong, Y.; Rechsteiner, M.C.; Squier, T.C. Hsp90 enhances degradation of oxidized calmodulin by the 20 s proteasome. J. Biol. Chem. 2004, 279, 46135–46142. [Google Scholar] [CrossRef]
- Makhnevych, T.; Houry, W.A. The role of hsp90 in protein complex assembly. Biochim. Biophys. Acta 2012, 1823, 674–682. [Google Scholar] [CrossRef]
- Prodromou, C. Mechanisms of hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef]
- Wayne, N.; Mishra, P.; Bolon, D.N. Hsp90 and client protein maturation. Methods Mol. Biol. 2011, 787, 33–44. [Google Scholar] [PubMed]
- Proteasome Activity Assay Kit (ab107921). Available online: https://www.abcam.com/proteasome-activity-assay-kit-ab107921.html (accessed on 19 December 2020).
- Messingerova, L.; Imrichova, D.; Coculova, M.; Zelina, M.; Pavlikova, L.; Kavcova, H.; Seres, M.; Bohacova, V.; Lakatos, B.; Sulova, Z.; et al. Different mechanisms of drug resistance in myelodysplastic syndromes and acute myeloid leukemia. In Myelodysplastic Syndromes; Fusch, O., Ed.; Intech: Rijeka, Croatia, 2016; pp. 181–200. [Google Scholar]
- Chen, J.Y.; Lin, J.R.; Tsai, F.C.; Meyer, T. Dosage of dyrk1a shifts cells within a p21-cyclin d1 signaling map to control the decision to enter the cell cycle. Mol. Cell 2013, 52, 87–100. [Google Scholar] [CrossRef]
- Pavlíková, L.; Kyca, T.; Sulová, Z.; Boháčová, V.; Breier, A.; Šereš, M. Different Effect of Tunicamycin on Expression of Proteins Ivolved in Cell Cycle Regulation in Relation to p-Glycoprotein Presence in l1210 Cells. In Proceedings of the World Congress on Cancer Science and Therapy, Bangkok, Thailand, 2–3 December 2019. Int. J. Cancer Treat. 2019. Available online: https://www.innovationinfo.org/conferenceproceedings/7/1_Lucia_Pavlikova_abstrakt_rim_Lucia.pdf (accessed on 15 May 2021).
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef]
- Gartel, A.L.; Radhakrishnan, S.K.; Serfas, M.S.; Kwon, Y.H.; Tyner, A.L. A novel p21waf1/cip1 transcript is highly dependent on p53 for its basal expression in mouse tissues. Oncogene 2004, 23, 8154–8157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lehman, S.L.; Cerniglia, G.J.; Johannes, G.J.; Ye, J.; Ryeom, S.; Koumenis, C. Translational upregulation of an individual p21cip1 transcript variant by gcn2 regulates cell proliferation and survival under nutrient stress. PLoS Genet. 2015, 11, e1005212. [Google Scholar] [CrossRef]
- Takenokuchi, M.; Miyamoto, K.; Saigo, K.; Taniguchi, T. Bortezomib causes ER stress-related death of acute promyelocytic leukemia cells through excessive accumulation of PML-RARA. Anticancer Res 2015, 35, 3307–3316. [Google Scholar]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Antao, A.M.; Tyagi, A.; Kim, K.S.; Ramakrishna, S. Advances in deubiquitinating enzyme inhibition and applications in cancer therapeutics. Cancers 2020, 12, 1579. [Google Scholar] [CrossRef]
- Davis, C.; Spaller, B.L.; Matouschek, A. Mechanisms of substrate recognition by the 26s proteasome. Curr. Opin. Struct. Biol. 2020, 67, 161–169. [Google Scholar] [CrossRef]
- Sakata, E.; Eisele, M.R.; Baumeister, W. Molecular and cellular dynamics of the 26s proteasome. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140583. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The hsp90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Sanchez, E.R. Chaperoning steroidal physiology: Lessons from mouse genetic models of hsp90 and its cochaperones. Biochim. Biophys. Acta 2012, 1823, 722–729. [Google Scholar] [CrossRef]
- Wang, D.; Zong, C.; Koag, M.C.; Wang, Y.; Drews, O.; Fang, C.; Scruggs, S.B.; Ping, P. Proteome dynamics and proteome function of cardiac 19s proteasomes. Mol. Cell. Proteom. 2011, 10, M110.006122. [Google Scholar] [CrossRef]
- Pastan, I.; Gottesman, M.M.; Ueda, K.; Lovelace, E.; Rutherford, A.V.; Willingham, M.C. A retrovirus carrying an mdr1 cdna confers multidrug resistance and polarized expression of p-glycoprotein in mdck cells. Proc. Natl. Acad. Sci. USA 1988, 85, 4486–4490. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Revers Primer | bp |
|---|---|---|---|
| Actb | 5′-TCG CCA TGG ATG ACG ATA-3′ | 5′-CAC GAT GGA GGG GAA TAC AG-3′ | 110 |
| Human ABCB1 | 5′-GCA ATG GAG GAG CAA AGA AG-3′ | 5′-CCA AAG TTC CCA CCA CCA TA-3′ | 150 |
| Mouse Abcb1 | 5′-TGG GAA CTC TGG CTG CTA TT-3′ | 5′-GGC GTA CGT GGT CAT TTC TT-3′ | 179 |
| Abcc1 | 5′-ACC AGC AAC CCC GAC TTT AC-3′ | 5′-TGG TTT TGT TGA GGT GTG TCA-3′ | 151 |
| Abcg2 | 5′-CCA CGT GTT AGT ACC AAT GTC G-3′ | 5′-TTT CCG GAC TAG AAA CCC ACT-3′ | 151 |
| Psmb1 | 5′-CGA GAT AGC CCC AAA TGC TA-3′ | 5′-AAG AAG CGC CGT GAG TAC AG-3′ | 194 |
| Psmb2 | 5′-GTC ACC CCC AGC TCA GTC-3′ | 5′-GGA AGC GAC GAG GAC ATA GT-3′ | 153 |
| Psmb5 | 5′-GCC ATC TAC CAA GCC ACC TA-3′ | 5′-TAG CCA TGG AGA CCC ACC TA-3′ | 193 |
| Psma7 | 5′-GTT GGT GTT CGA GGA AAG GA-3′ | 5′-GTC ACT GGG TCC TCC ACT GT-3′ | 212 |
| Usp7 | 5′-AGC AAC GCA GAA GAG GAC AT-3′ | 5′-TTT GGT GTG GTC TGT CTG GA-3′ | 181 |
| Usp28 | 5′-CCA GTC ACG ACA CAA CTG CT-3′ | 5′-TCC AGG AGA CTC AAG GCA AT-3′ | 139 |
| Usp48 | 5′-GAT CCA TGG GGG AAA AGA TT-3′ | 5′-TGC TGC TGC CCA GTA TCT AA-3′ | 144 |
| Usp1 | 5′-AAG GGA AGC TGC AAA GAA GA-3′ | 5′-GCC TTG GCT GTG TAG CAA GT-3′ | 145 |
| Usp2 | 5′-GTG GTG AGC CCA TCT GAG TT-3′ | 5′-GTG GAG ACC ATC CAG AAG GA-3′ | 114 |
| Uchl5 | 5′-TGG TCC AGG ACT CCA GAC TT-3′ | 5′-TGC ACA TCT TGA TGC GTA CA-3′ | 115 |
| p15 | 5′-AGG ACG CTC ACC GAA GCT A-3′ | 5′-CTG TGG CAG AAA TGG TCC TT-3′ | 100 |
| p16 | 5′-CAA CGT TCA CGT AGC AGC TC-3′ | 5′-ACC AGC GTG TCC AGG AAG-3′ | 120 |
| p27 | 5′-AGT CAG CGC AAG TGG AAT TT-3′ | 5′-AGT AGA ACT CGG GCA AGC TG-3′ | 100 |
| p21 t1 | 5′-TCC ACA GCG ATA TCC AGA CA-3′ | 5′-ATG AGC GCA TCG CAA TCA C-3′ | 148 |
| p21 t2 | 5′-TTA AGG ACG TCC CAC TTT GC-3′ | 5′-AGA CAA CGG CAC ACT TTG CT-3′ | 107 |
| CycB1 | 5′-GGT GAC TTC GCC TTT GTG AC-3′ | 5′-CTA CGG AGG AAG TGC AGA GG-3′ | 125 |
| CycD1 | 5′-AGC AGA AGT GCG AAG AGG AG-3′ | 5′-CAA GGG AAT GGT CTC CTT CA-3′ | 149 |
| CycE1 | 5′-GGA AAA TCA GAC CAC CCA GA-3′ | 5′-AGG ATG ACG CTG CAG AAA GT-3′ | 131 |
| CycA1 | 5′-ACA CAG ACC CAA GGC TCA CT-3′ | 5′-ACA GGG TCT CTG TGC GAA GT-3′ | 122 |
| Cdk2 | 5′-CAT TCC TCT TCC CCT CAT CA-3′ | 5′-TAA GCA GGT TCT GGG GCT TA-3′ | 110 |
| Cdk4 | 5′-TAT GAA CCC GTG GCT GAA AT-3′ | 5′-TCC AGC TGC TCC TCC ATT AG-3′ | 123 |
| Cdk6 | 5′-AGA AGG TCG GTC CGT CTA GC-3′ | 5′-ACT CAG GCT GTC CTT CTC CA-3′ | 132 |
| Cdk1 | 5′-TTG AAA GCG AGG AAG AAG GA-3′ | 5′-TCC ATG GAC AGG AAC TCA AA-3′ | 148 |
| Hsp90aa | 5′-GGG AGC TCA TCT CCA ATT CA-3′ | 5′-ATT GAT GTG CAG CTC CTT CC-3′ | 101 |
| Hsp90ab | 5′-GAT TGT CAC CTT TTC AAC CTT CTT-3′ | 5′-TGA CCT CCT TGT CAG AGT ATG TGT-3′ | 344 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyca, T.; Pavlíková, L.; Boháčová, V.; Mišák, A.; Poturnayová, A.; Breier, A.; Sulová, Z.; Šereš, M. Insight into Bortezomib Focusing on Its Efficacy against P-gp-Positive MDR Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 5504. https://doi.org/10.3390/ijms22115504
Kyca T, Pavlíková L, Boháčová V, Mišák A, Poturnayová A, Breier A, Sulová Z, Šereš M. Insight into Bortezomib Focusing on Its Efficacy against P-gp-Positive MDR Leukemia Cells. International Journal of Molecular Sciences. 2021; 22(11):5504. https://doi.org/10.3390/ijms22115504
Chicago/Turabian StyleKyca, Tomáš, Lucia Pavlíková, Viera Boháčová, Anton Mišák, Alexandra Poturnayová, Albert Breier, Zdena Sulová, and Mário Šereš. 2021. "Insight into Bortezomib Focusing on Its Efficacy against P-gp-Positive MDR Leukemia Cells" International Journal of Molecular Sciences 22, no. 11: 5504. https://doi.org/10.3390/ijms22115504
APA StyleKyca, T., Pavlíková, L., Boháčová, V., Mišák, A., Poturnayová, A., Breier, A., Sulová, Z., & Šereš, M. (2021). Insight into Bortezomib Focusing on Its Efficacy against P-gp-Positive MDR Leukemia Cells. International Journal of Molecular Sciences, 22(11), 5504. https://doi.org/10.3390/ijms22115504