
TFEB Overexpression, Not mTOR Inhibition, Ameliorates RagCS75Y Cardiomyopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
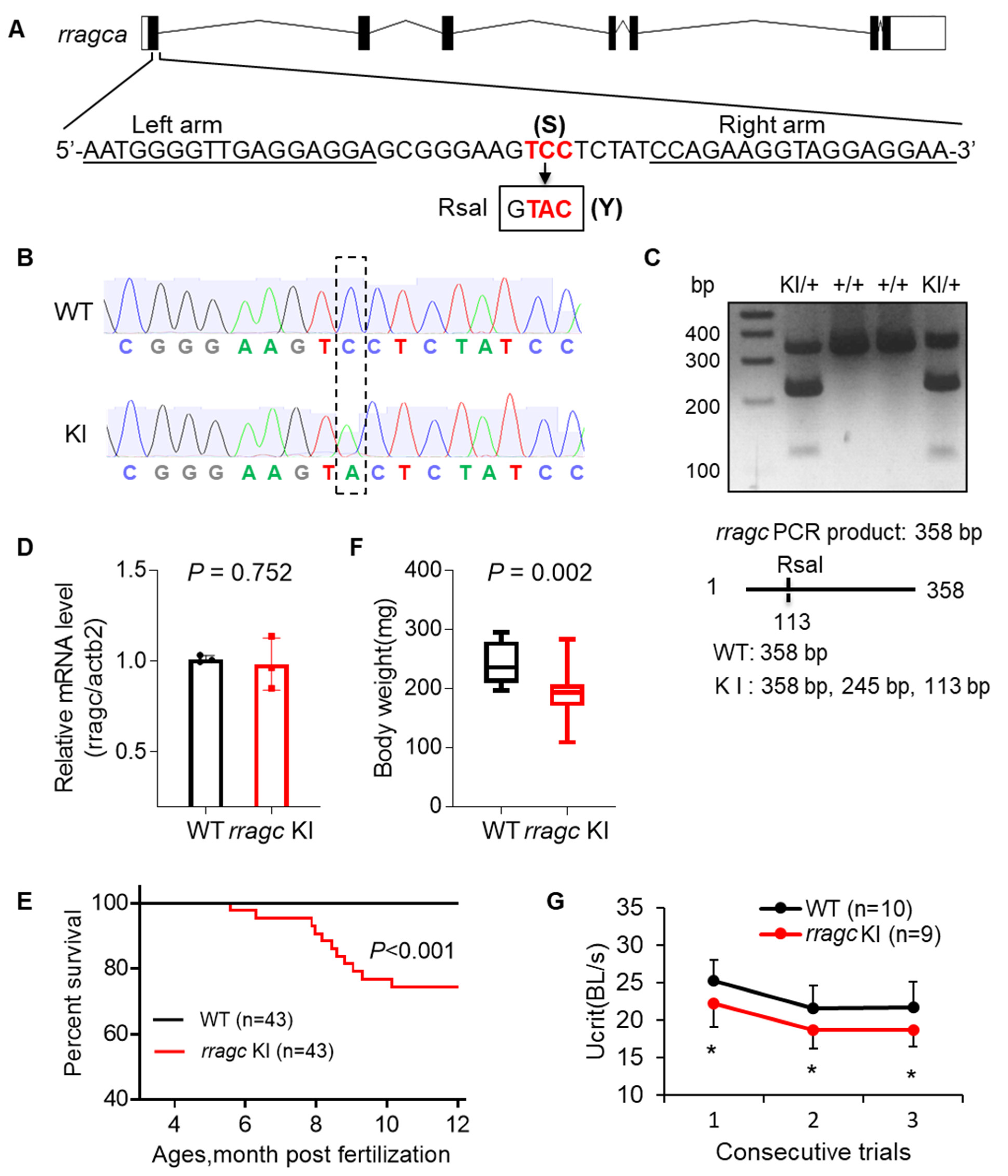
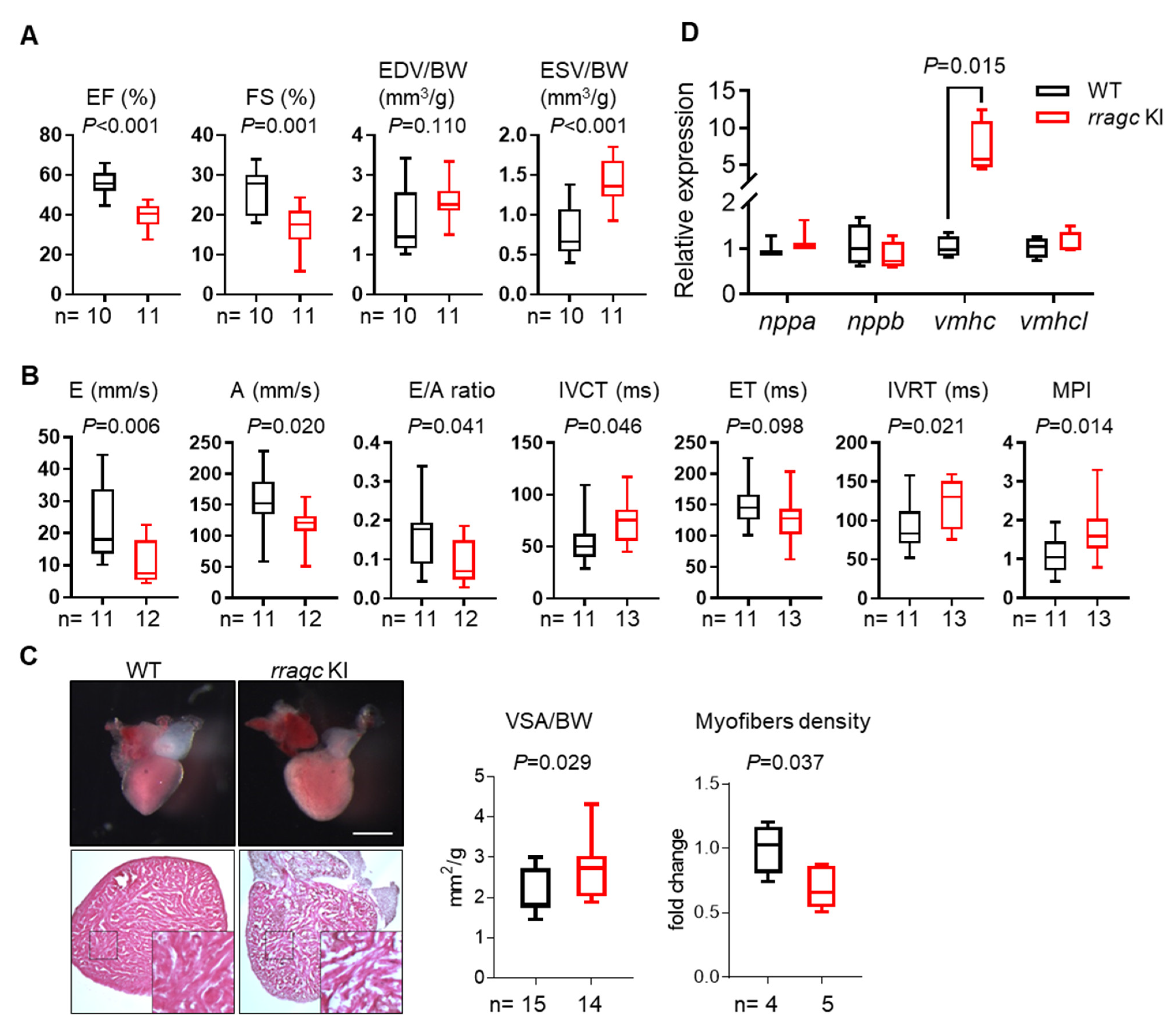
2.1. RragcS56Y KI Zebrafish Manifest Cardiomyopathy-Like Phenotypes
2.2. Rragc Knockout (KO) Zebrafish Does Not Manifest Cardiomyopathy-Like Phenotypes
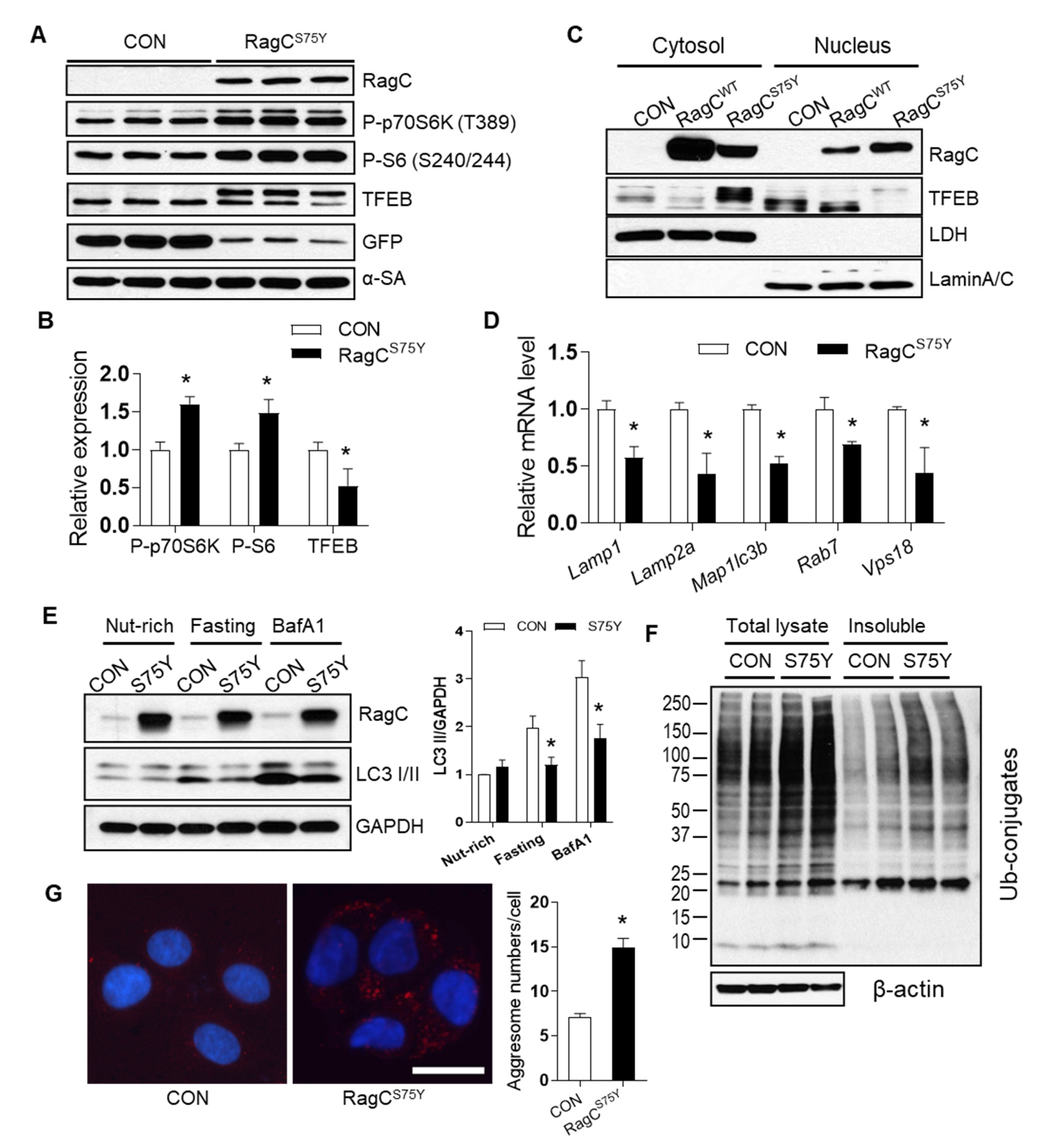
2.3. RagCS75Y Cardiomyopathy Models Exhibit Altered mTORC1–TFEB Signaling
2.4. mTOR Inhibition Does Not Attenuate RagCS75Y Cardiomyopathy
2.5. TFEB Activation Attenuates RagCS75Y Cardiomyopathy
3. Discussion
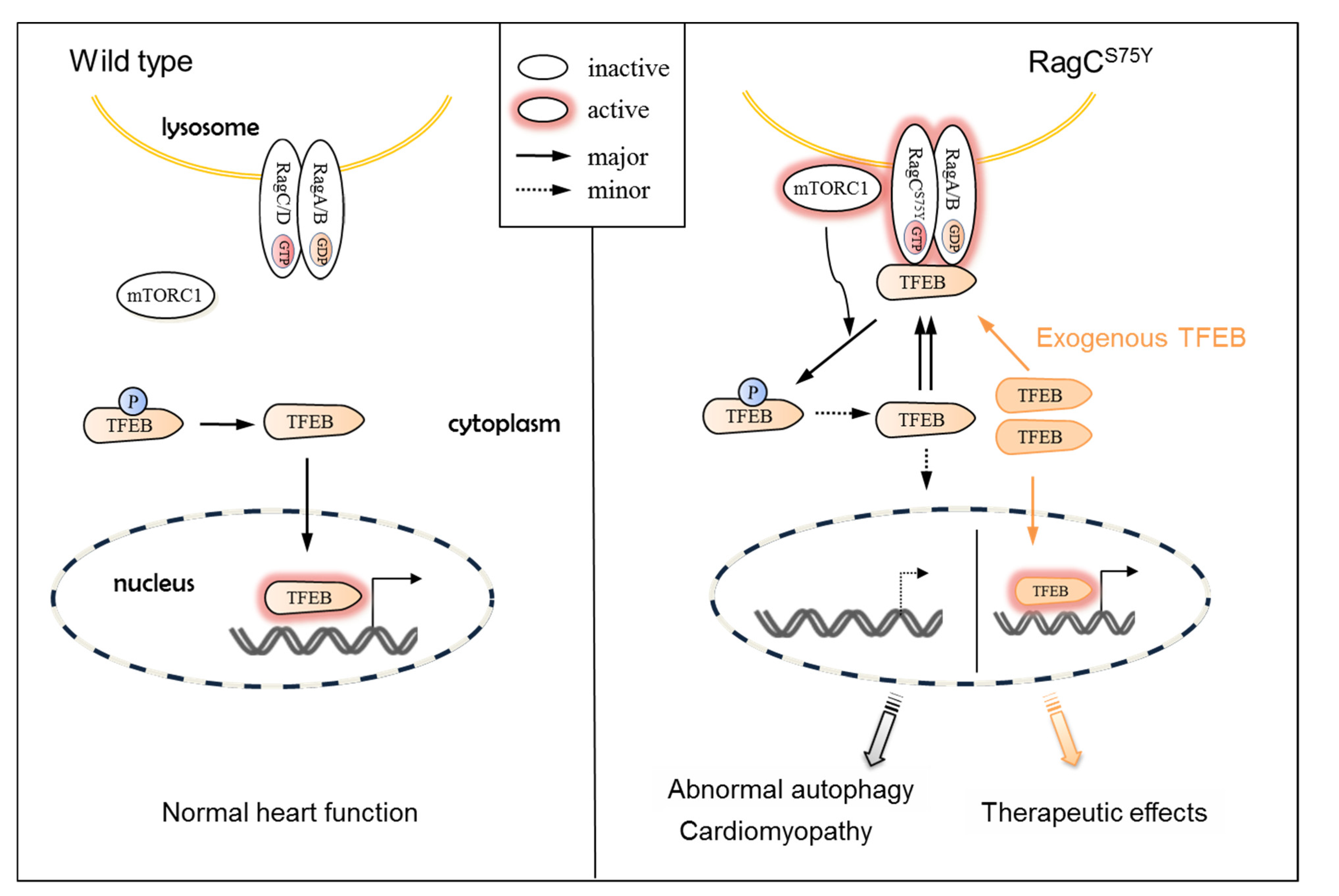
3.1. Molecular Mechanisms of RagCS75Y Cardiomyopathy
3.2. TFEB, Not mTOR, Could Be the Therapeutic Target for RagCS75Y Cardiomyopathy
3.3. Zebrafish Can Be Used for Studying Genetic Variants Identified in Human Cardiomyopathy
4. Materials and Methods
4.1. Production of Rragc Knock-in Fish
4.2. Production of Transgenic Fish Lines
4.3. Cell Culture
4.4. Production of Recombinant Adenovirus
4.5. Measurement of Cell Size
4.6. Preparation of Protein Extracts and Immunoblotting
4.7. Coimmunoprecipitation
4.8. Aggresome Staining
4.9. Autophagy Flux
4.10. RNA Isolation and RT-PCR
4.11. Zebrafish Echocardiography
4.12. Measurement of Ventricular Surface Area to Body Weight Index
4.13. Histology
4.14. Swimming Tunnel Assay
4.15. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Long, P.A.; Zimmermann, M.T.; Kim, M.; Evans, J.M.; Xu, X.; Olson, T.M. De novo RRAGC mutation activates mTORC1 signaling in syndromic fetal dilated cardiomyopathy. Hum. Genet. 2016, 135, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, R.; Sardu, A.; Panchaud, N.; De Virgilio, C. The Architecture of the Rag GTPase Signaling Network. Biomolecules 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Fromm, S.A.; Fu, Y.; Yokom, A.L.; Kim, D.J.; Thelen, A.M.; Young, L.N.; Lim, C.Y.; Samelson, A.J.; Hurley, J.H.; et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 2019, 366, 971–977. [Google Scholar] [CrossRef]
- Shen, K.; Rogala, K.B.; Chou, H.T.; Huang, R.K.; Yu, Z.; Sabatini, D.M. Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell 2019, 179, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Anandapadamanaban, M.; Masson, G.R.; Perisic, O.; Berndt, A.; Kaufman, J.; Johnson, C.M.; Santhanam, B.; Rogala, K.B.; Sabatini, D.M.; Williams, R.L. Architecture of human Rag GTPase heterodimers and their complex with mTORC1. Science 2019, 366, 203–210. [Google Scholar] [CrossRef]
- Rogala, K.B.; Gu, X.; Kedir, J.F.; Abu-Remaileh, M.; Bianchi, L.F.; Bottino, A.M.S.; Dueholm, R.; Niehaus, A.; Overwijn, D.; Fils, A.P.; et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 2019, 366, 468–475. [Google Scholar] [CrossRef]
- Martina, J.A.; Puertollano, R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 2013, 200, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef] [PubMed]
- Saraste, M.; Sibbald, P.R.; Wittinghofer, A. The P-loop—A common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 1990, 15, 430–434. [Google Scholar] [CrossRef]
- Okosun, J.; Wolfson, R.L.; Wang, J.; Araf, S.; Wilkins, L.; Castellano, B.M.; Escudero-Ibarz, L.; Al Seraihi, A.F.; Richter, J.; Bernhart, S.H.; et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat. Genet. 2016, 48, 183–188. [Google Scholar] [CrossRef]
- Ortega-Molina, A.; Deleyto-Seldas, N.; Carreras, J.; Sanz, A.; Lebrero-Fernández, C.; Menéndez, C.; Vandenberg, A.; Fernández-Ruiz, B.; Marín-Arraiza, L.; de la Calle Arregui, C.; et al. Oncogenic Rag GTPase signaling enhances B cell activation and drives follicular lymphoma sensitive to pharmacological inhibition of mTOR. Nat. Metab. 2019, 1, 775–789. [Google Scholar] [CrossRef]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- Ma, X.; Mani, K.; Liu, H.; Kovacs, A.; Murphy, J.T.; Foroughi, L.; French, B.A.; Weinheimer, C.J.; Kraja, A.; Benjamin, I.J.; et al. Transcription Factor EB Activation Rescues Advanced alphaB-Crystallin Mutation-Induced Cardiomyopathy by Normalizing Desmin Localization. J. Am. Heart Assoc. 2019, 8, e010866. [Google Scholar] [CrossRef]
- Godar, R.J.; Ma, X.; Liu, H.; Murphy, J.T.; Weinheimer, C.J.; Kovacs, A.; Crosby, S.D.; Saftig, P.; Diwan, A. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy 2015, 11, 1537–1560. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Shih, Y.H.; Zhang, Y.; Ding, Y.; Ross, C.A.; Li, H.; Olson, T.M.; Xu, X. Cardiac transcriptome and dilated cardiomyopathy genes in zebrafish. Circ. Cardiovasc. Genet. 2015, 8, 261–269. [Google Scholar] [CrossRef]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef]
- Shen, K.; Choe, A.; Sabatini, D.M. Intersubunit Crosstalk in the Rag GTPase Heterodimer Enables mTORC1 to Respond Rapidly to Amino Acid Availability. Mol. Cell 2017, 68, 552–565. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; McMahon, C.J.; Smith, L.R.; Bersola, J.; Adesina, A.M.; Breinholt, J.P.; Kearney, D.L.; Dreyer, W.J.; Denfield, S.W.; Price, J.F.; et al. Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation 2005, 112, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Sun, X.; Huang, W.; Hoage, T.; Redfield, M.; Kushwaha, S.; Sivasubbu, S.; Lin, X.; Ekker, S.; Xu, X. Haploinsufficiency of target of rapamycin attenuates cardiomyopathies in adult zebrafish. Circ. Res. 2011, 109, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.X.; Jin, M.; Peterson, L.F.; Bernard, D.; Saiya-Cork, K.; Yildiz, M.; Wang, S.; Kaminski, M.S.; Chang, A.E.; Klionsky, D.J.; et al. Recurrent Mutations in the MTOR Regulator RRAGC in Follicular Lymphoma. Clin. Cancer Res. 2016, 22, 5383–5393. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Park, H.W.; Sciarretta, S.; Mo, J.S.; Jewell, J.L.; Russell, R.C.; Wu, X.; Sadoshima, J.; Guan, K.L. Rag GTPases are cardioprotective by regulating lysosomal function. Nat. Commun. 2014, 5, 4241. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Weber, T.; Hajjar, R.J. Human Cardiac Gene Therapy. Circ. Res. 2018, 123, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Dvornikov, A.V.; Ma, X.; Zhang, H.; Wang, Y.; Lowerison, M.; Packard, R.R.; Wang, L.; Chen, J.; Zhang, Y.; et al. Haploinsufficiency of mechanistic target of rapamycin ameliorates bag3 cardiomyopathy in adult zebrafish. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Dvornikov, A.V.; Wang, M.; Yang, J.; Zhu, P.; Le, T.; Lin, X.; Cao, H.; Xu, X. Phenotyping an adult zebrafish lamp2 cardiomyopathy model identifies mTOR inhibition as a candidate therapy. J. Mol. Cell. Cardiol. 2019, 133, 199–208. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, W.; Deng, Y.; Jomok, B.; Yang, J.; Huang, W.; Clark, K.J.; Zhong, T.P.; Lin, X.; Ekker, S.C.; et al. Trapping cardiac recessive mutants via expression-based insertional mutagenesis screening. Circ. Res. 2013, 112, 606–617. [Google Scholar] [CrossRef]
- Dvornikov, A.V.; de Tombe, P.P.; Xu, X. Phenotyping cardiomyopathy in adult zebrafish. Prog. Biophys. Mol. Biol. 2018, 138, 116–125. [Google Scholar] [CrossRef]
- Prykhozhij, S.V.; Fuller, C.; Steele, S.L.; Veinotte, C.J.; Razaghi, B.; Robitaille, J.M.; McMaster, C.R.; Shlien, A.; Malkin, D.; Berman, J.N. Optimized knock-in of point mutations in zebrafish using CRISPR/Cas9. Nucleic Acids Res. 2018, 46, e102. [Google Scholar] [CrossRef]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug II, R.G.; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.H.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef]
- Tessadori, F.; Roessler, H.I.; Savelberg, S.M.C.; Chocron, S.; Kamel, S.M.; Duran, K.J.; van Haelst, M.M.; van Haaften, G.; Bakkers, J. Effective CRISPR/Cas9-based nucleotide editing in zebrafish to model human genetic cardiovascular disorders. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef]
- Shih, Y.H.; Dvornikov, A.V.; Zhu, P.; Ma, X.; Kim, M.; Ding, Y.; Xu, X. Exon- and contraction-dependent functions of titin in sarcomere assembly. Development 2016, 143, 4713–4722. [Google Scholar] [CrossRef]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef]
- Tamayo Caro, M.; Palomo Irigoyen, M.; Perez Andres, E.; Barreira Manrique, A.; Varela Rey, M.; Woodhoo, A. Analyzing Autophagic Flux in Nerve Cultures. Methods Mol. Biol. 2018, 1791, 193–206. [Google Scholar] [CrossRef]
- Wang, L.W.; Huttner, I.G.; Santiago, C.F.; Kesteven, S.H.; Yu, Z.Y.; Feneley, M.P.; Fatkin, D. Standardized echocardiographic assessment of cardiac function in normal adult zebrafish and heart disease models. Dis. Models Mech. 2017, 10, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Packard, R.R.S.; Baek, K.I.; Beebe, T.; Jen, N.; Ding, Y.; Shi, F.; Fei, P.; Kang, B.J.; Chen, P.H.; Gau, J.; et al. Automated Segmentation of Light-Sheet Fluorescent Imaging to Characterize Experimental Doxorubicin-Induced Cardiac Injury and Repair. Sci. Rep. 2017, 7, 8603. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Panakova, D.; Kikuchi, K.; Holdway, J.E.; Gemberling, M.; Burris, J.S.; Singh, S.P.; Dickson, A.L.; Lin, Y.F.; Sabeh, M.K.; et al. The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development 2011, 138, 3421–3430. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Lu, L.; Dvornikov, A.V.; Ma, X.; Ding, Y.; Zhu, P.; Olson, T.M.; Lin, X.; Xu, X. TFEB Overexpression, Not mTOR Inhibition, Ameliorates RagCS75Y Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 5494. https://doi.org/10.3390/ijms22115494
Kim M, Lu L, Dvornikov AV, Ma X, Ding Y, Zhu P, Olson TM, Lin X, Xu X. TFEB Overexpression, Not mTOR Inhibition, Ameliorates RagCS75Y Cardiomyopathy. International Journal of Molecular Sciences. 2021; 22(11):5494. https://doi.org/10.3390/ijms22115494
Chicago/Turabian StyleKim, Maengjo, Linghui Lu, Alexey V. Dvornikov, Xiao Ma, Yonghe Ding, Ping Zhu, Timothy M. Olson, Xueying Lin, and Xiaolei Xu. 2021. "TFEB Overexpression, Not mTOR Inhibition, Ameliorates RagCS75Y Cardiomyopathy" International Journal of Molecular Sciences 22, no. 11: 5494. https://doi.org/10.3390/ijms22115494
APA StyleKim, M., Lu, L., Dvornikov, A. V., Ma, X., Ding, Y., Zhu, P., Olson, T. M., Lin, X., & Xu, X. (2021). TFEB Overexpression, Not mTOR Inhibition, Ameliorates RagCS75Y Cardiomyopathy. International Journal of Molecular Sciences, 22(11), 5494. https://doi.org/10.3390/ijms22115494