
Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
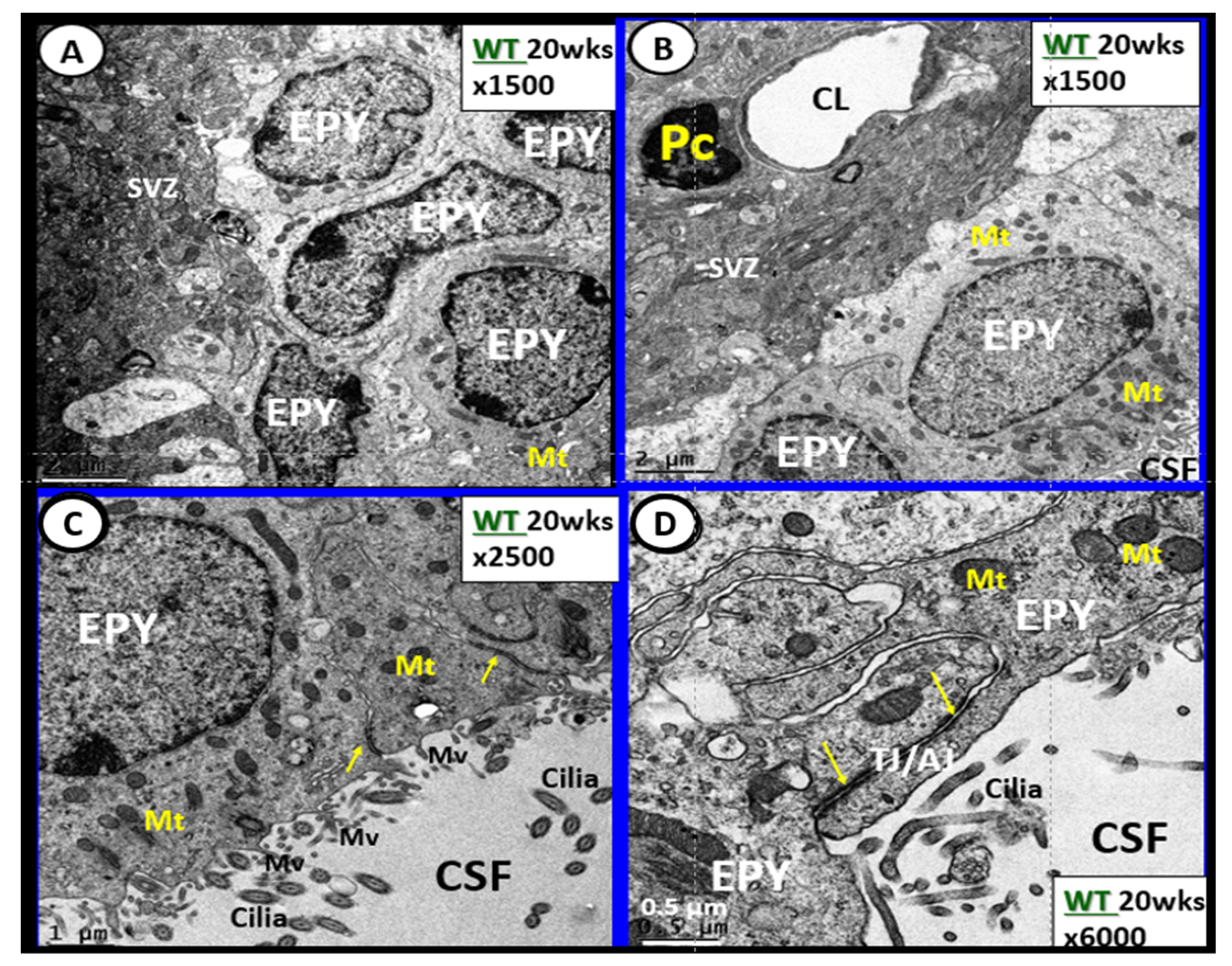{kind=link}
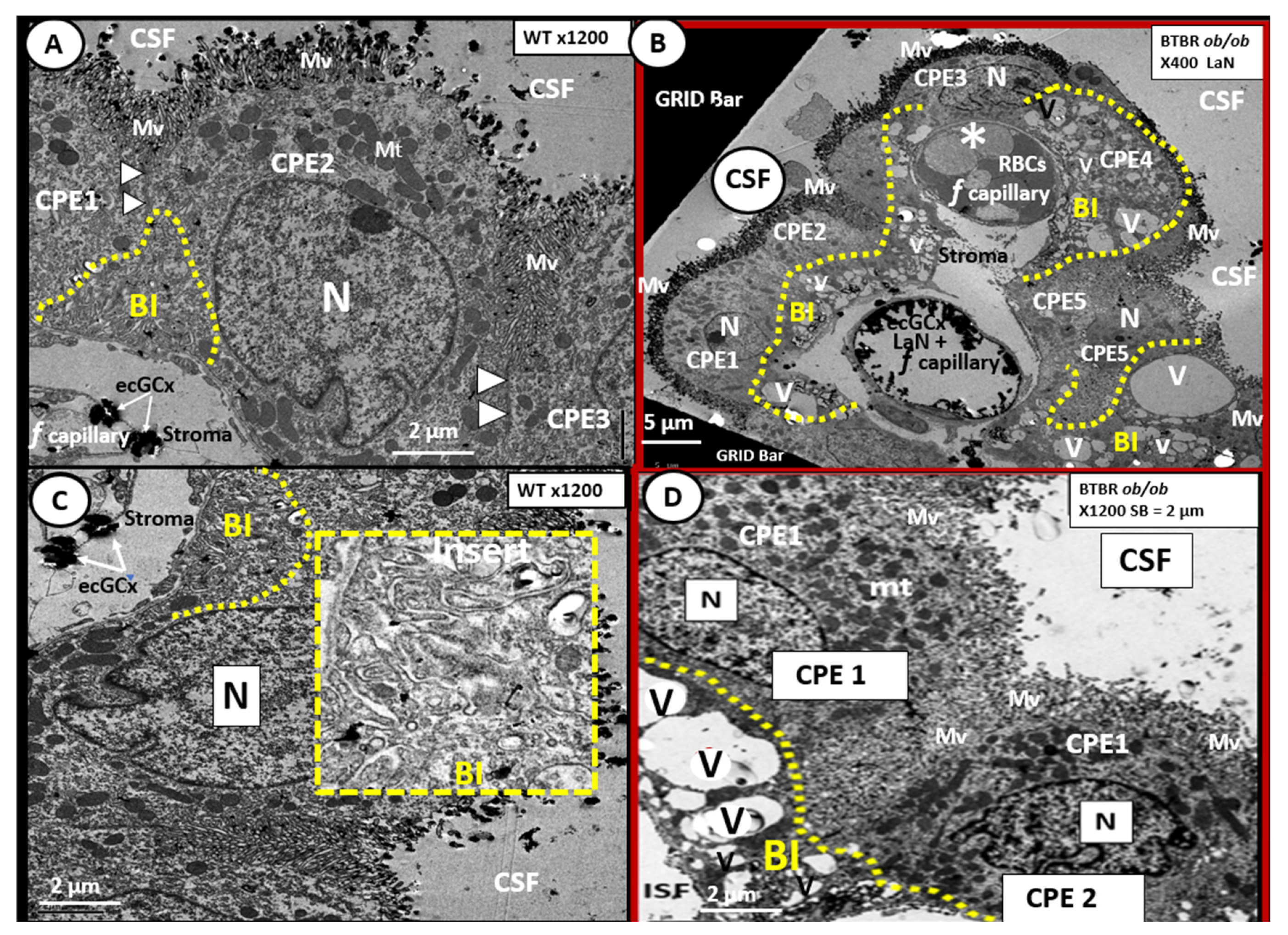{kind=link}
Abstract
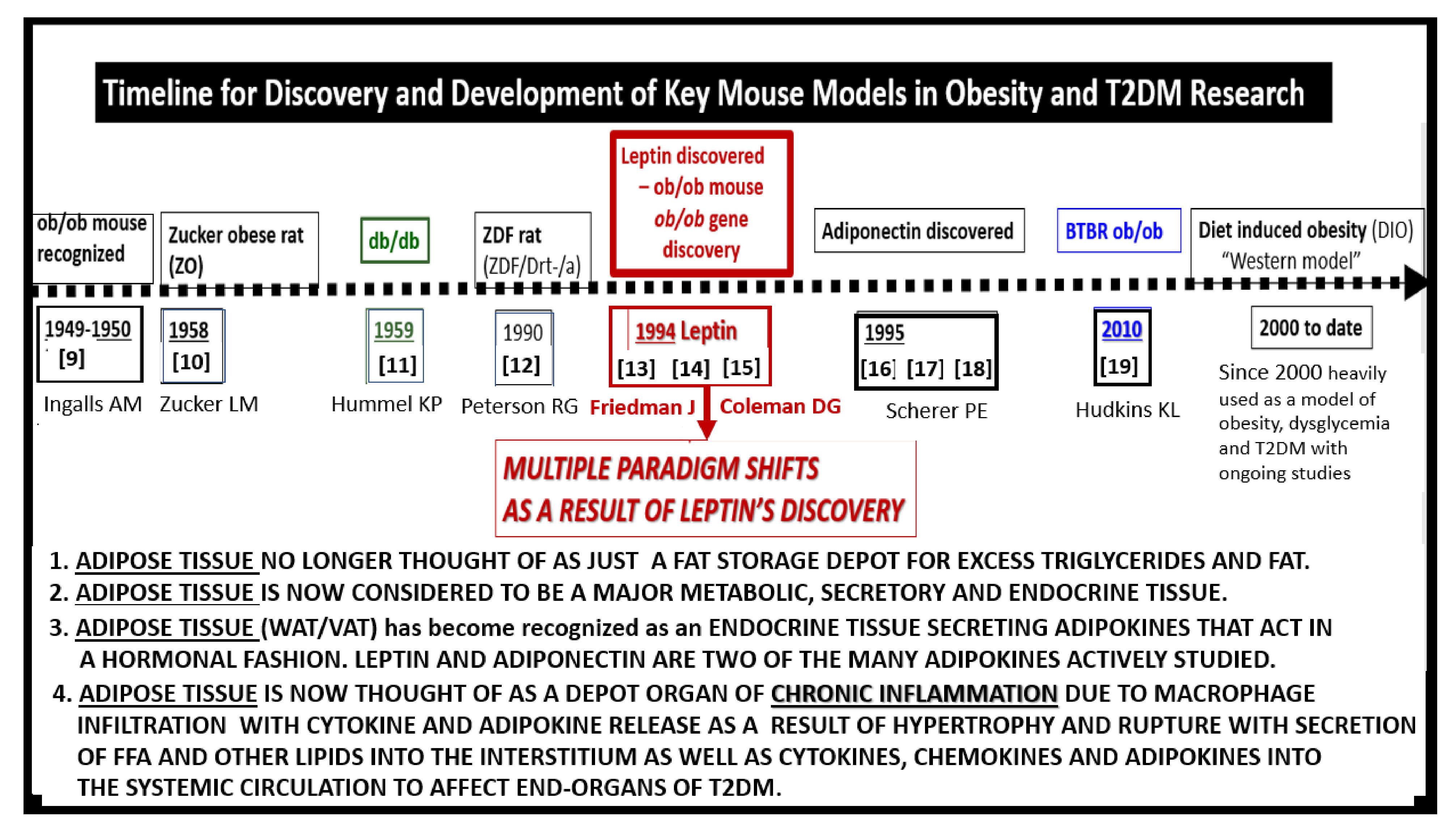
1. Introduction
2. Leptin
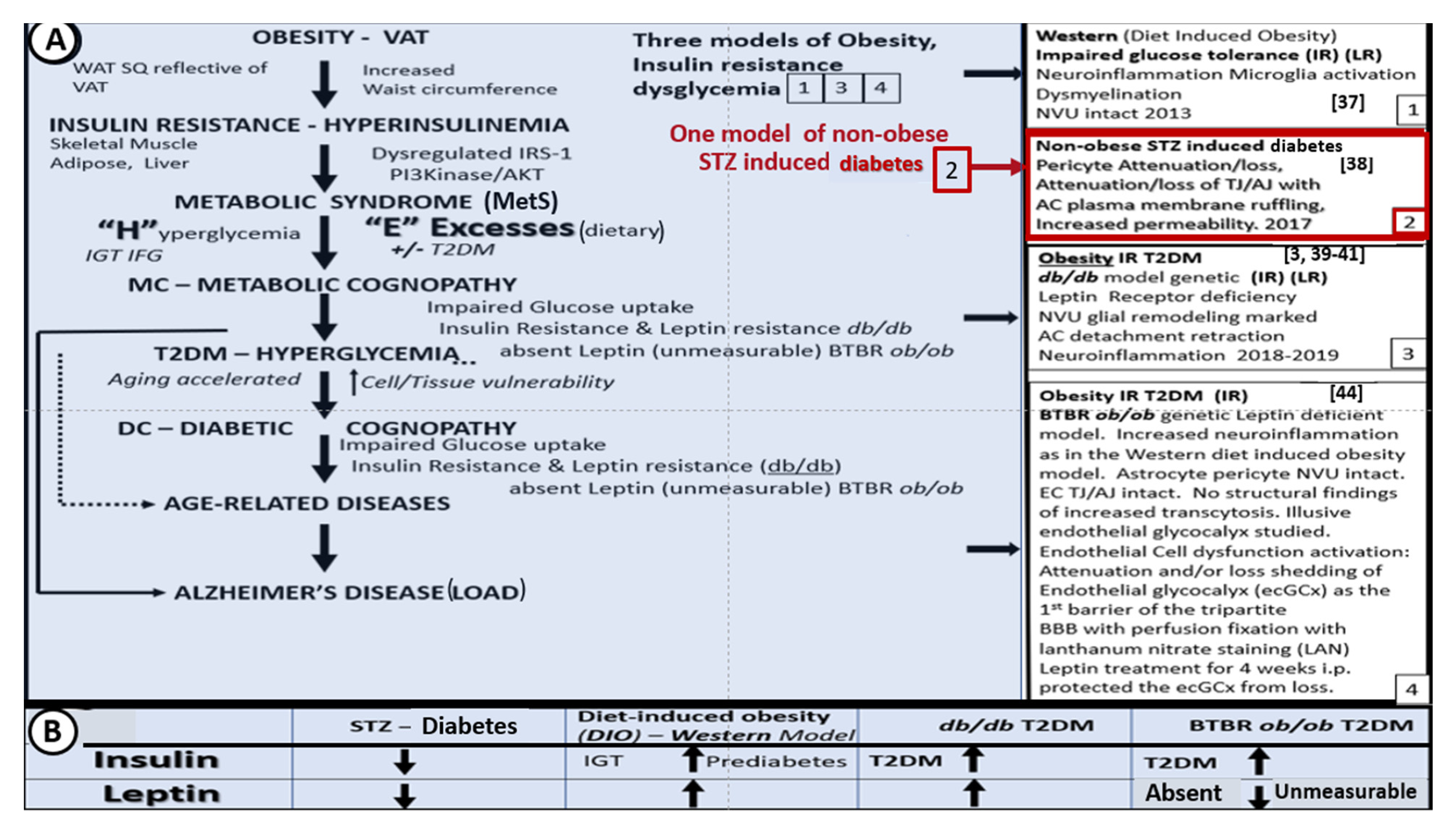
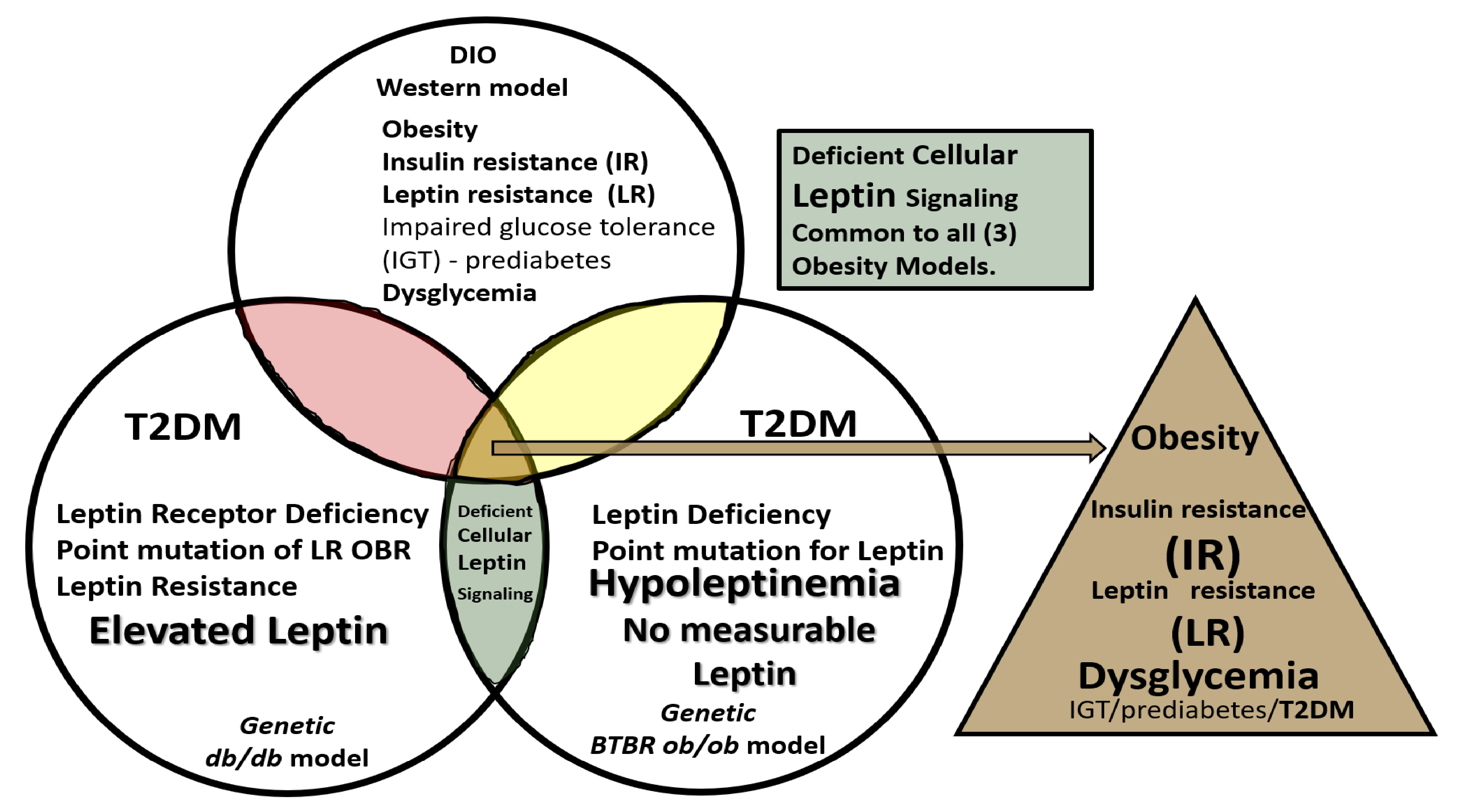
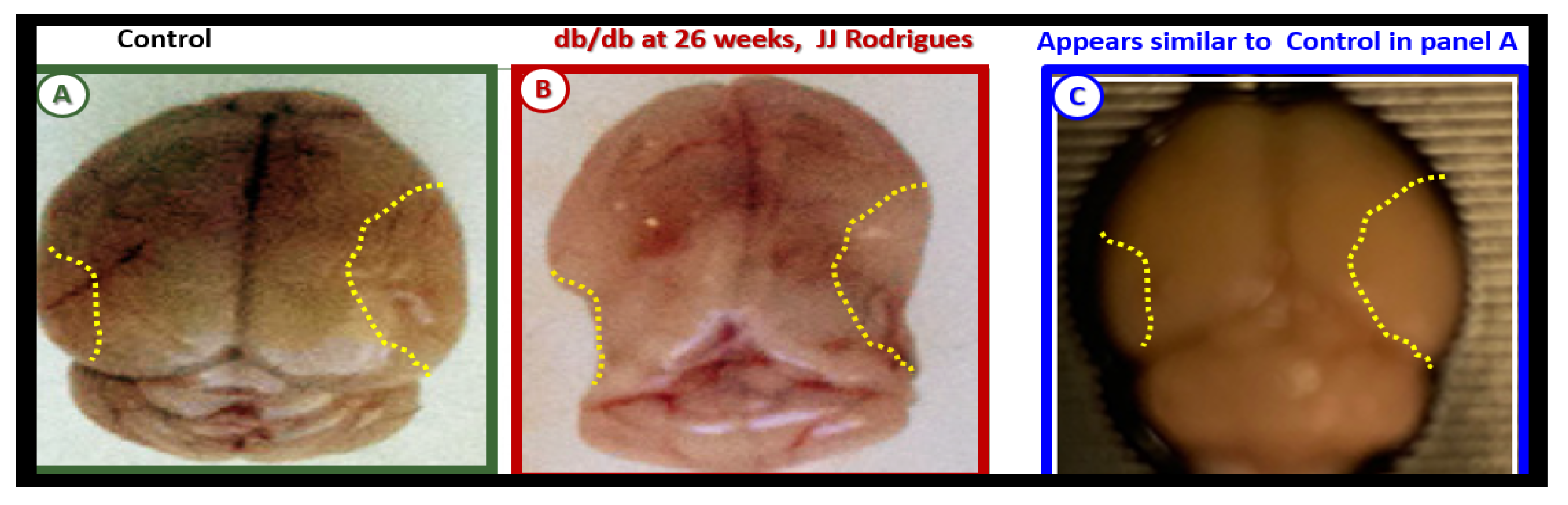
3. Central Nervous System (CNS) Roles of Leptin in Diet induced Obesity (DIO), db/db and BTBR ob/ob: Genetic Preclinical Models
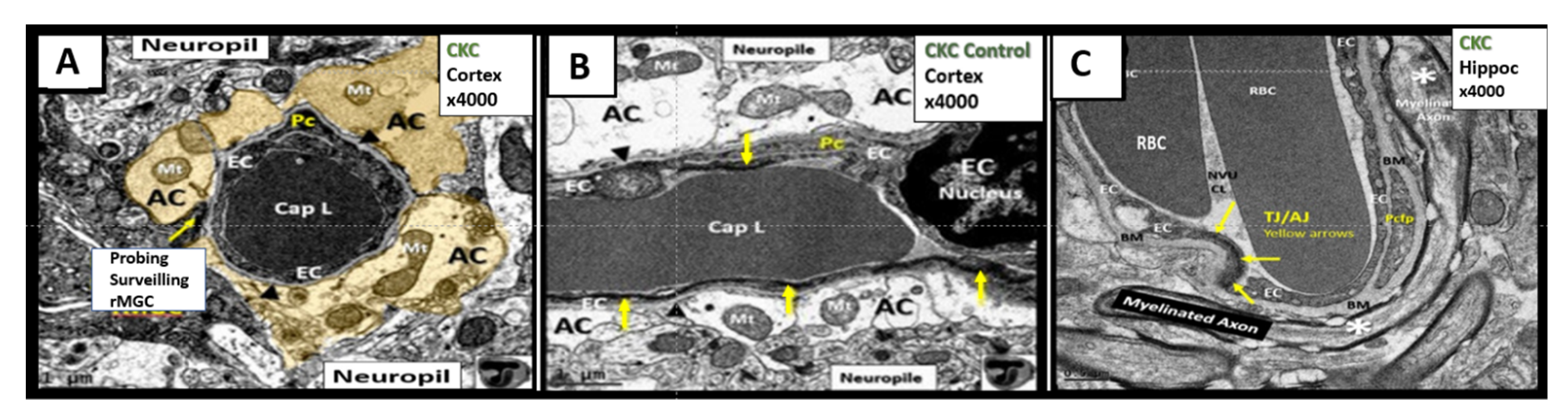
4. Neurovascular Unit (NVU) as an Anatomical Ultrastructural and Functional Unit
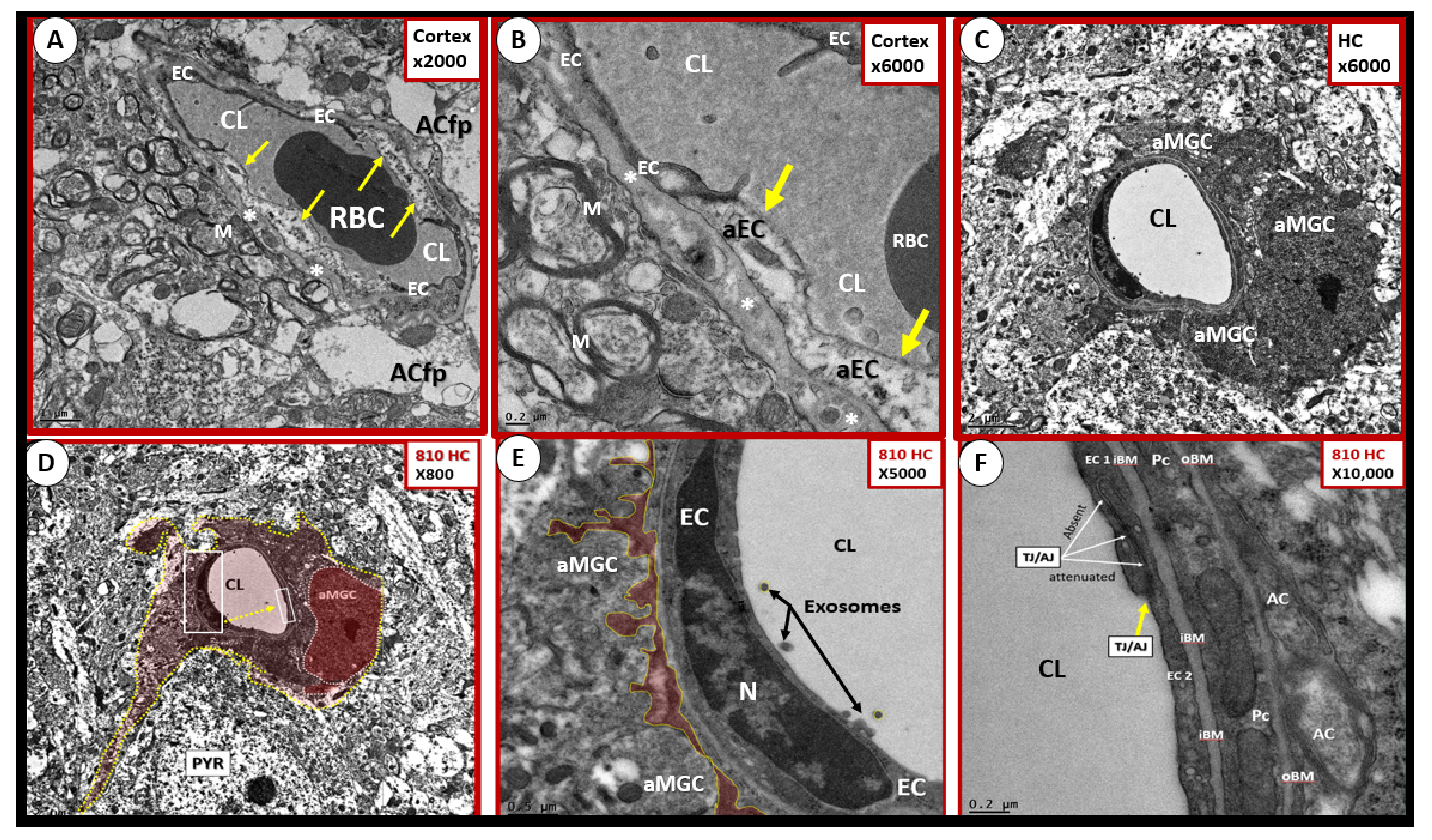
4.1. The Brain Endothelial Cell (BEC)
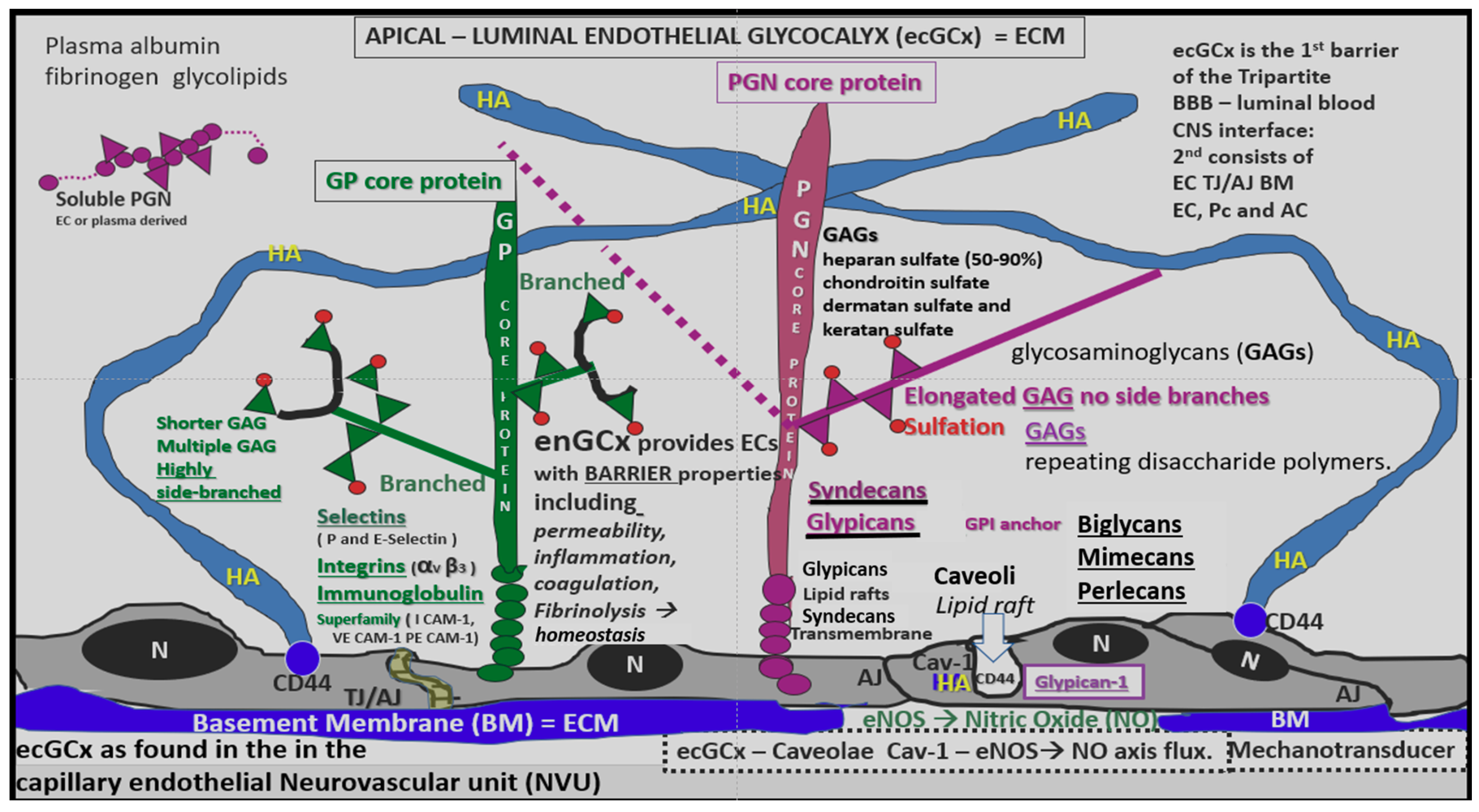
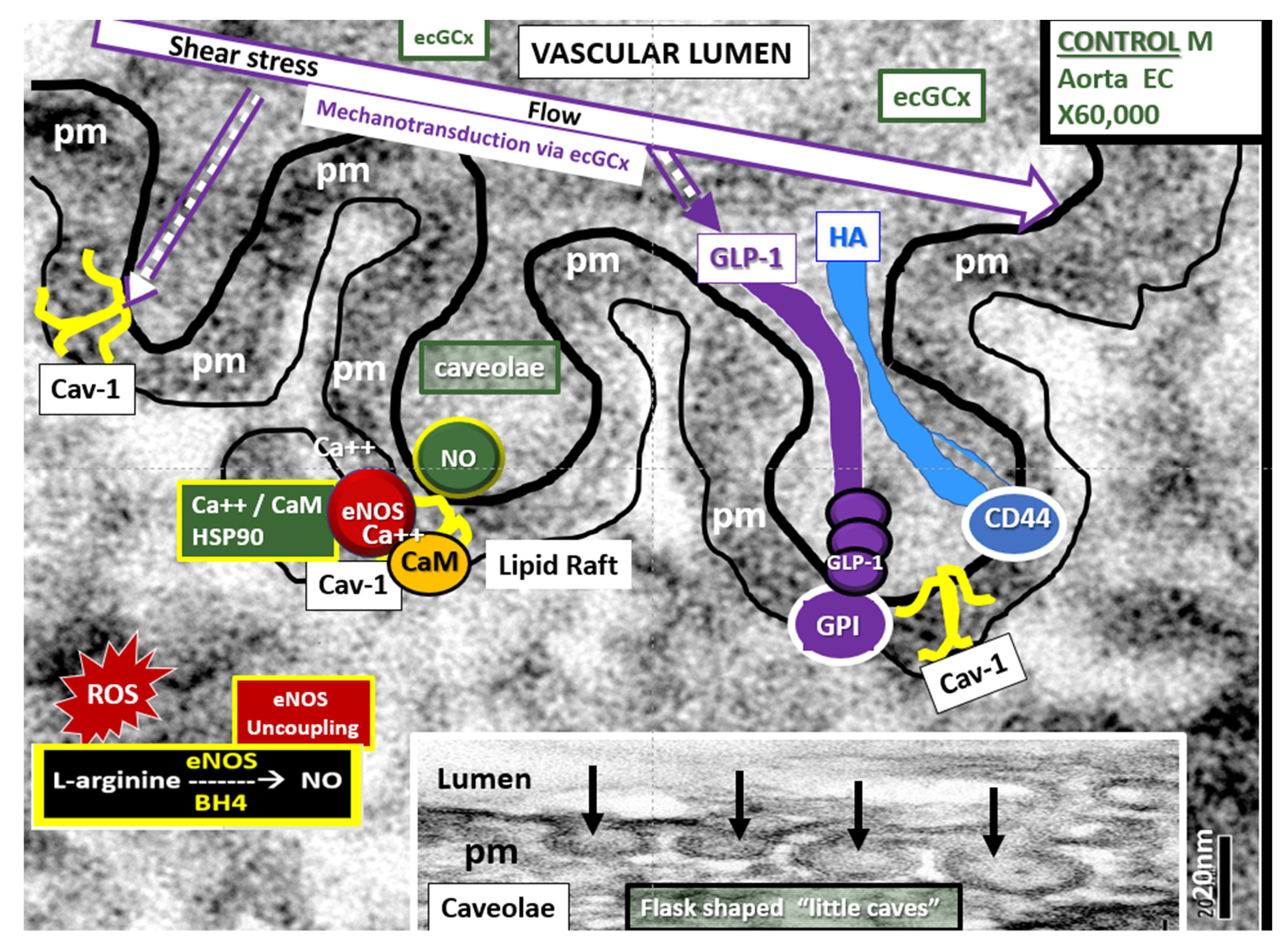
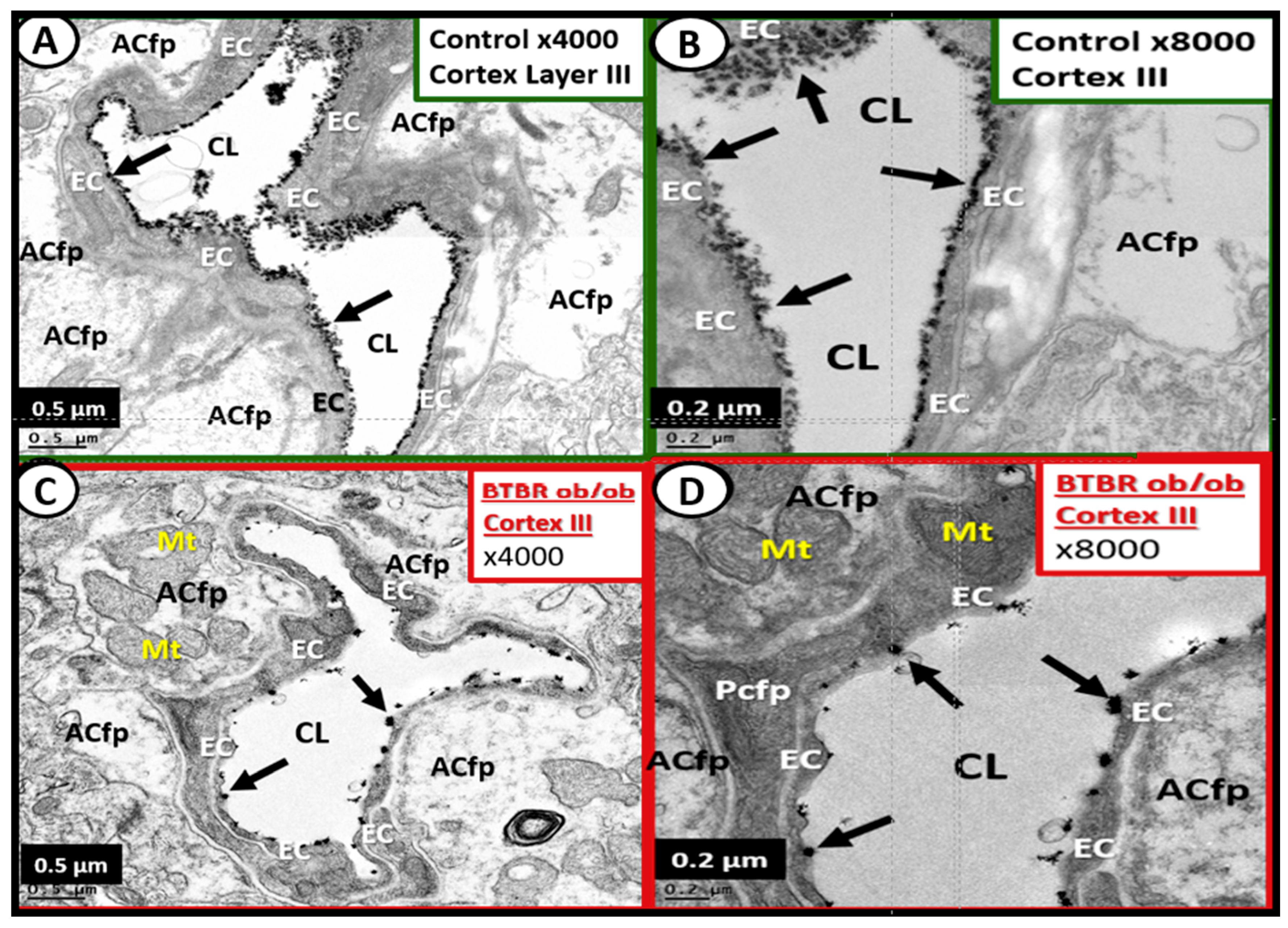
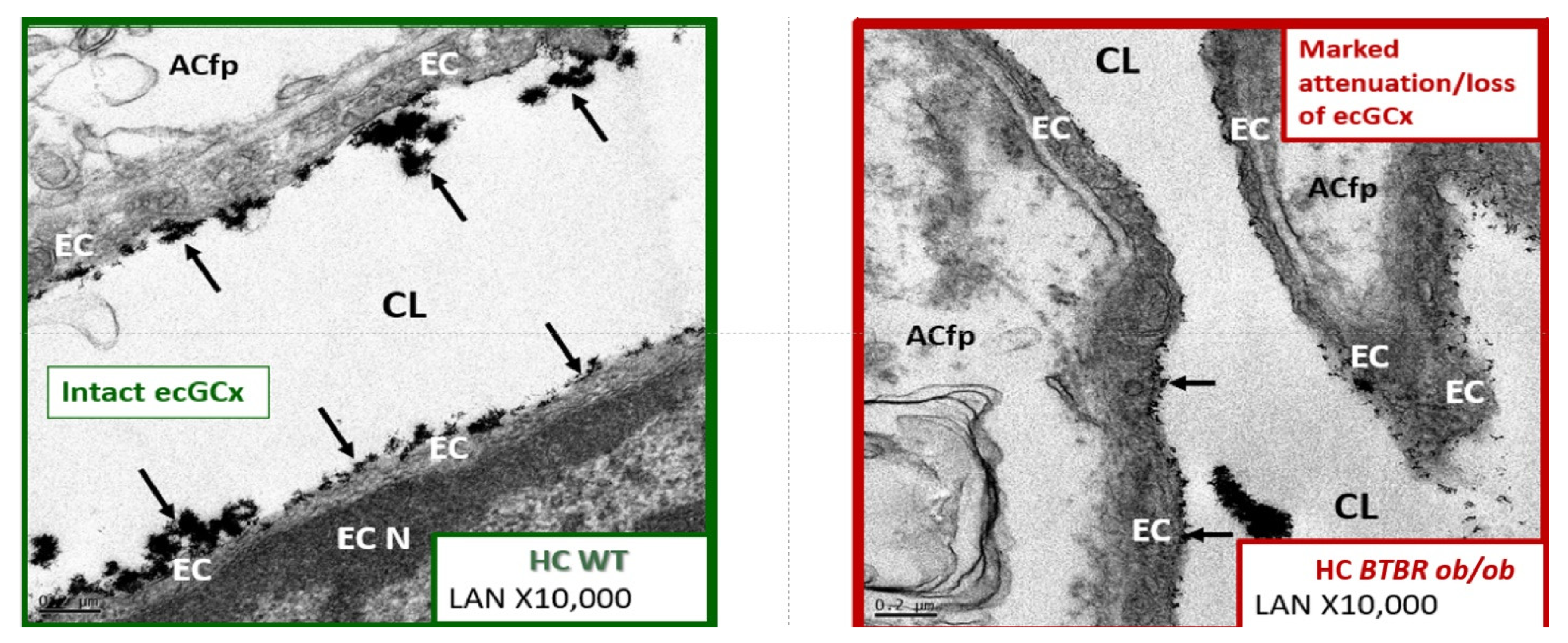
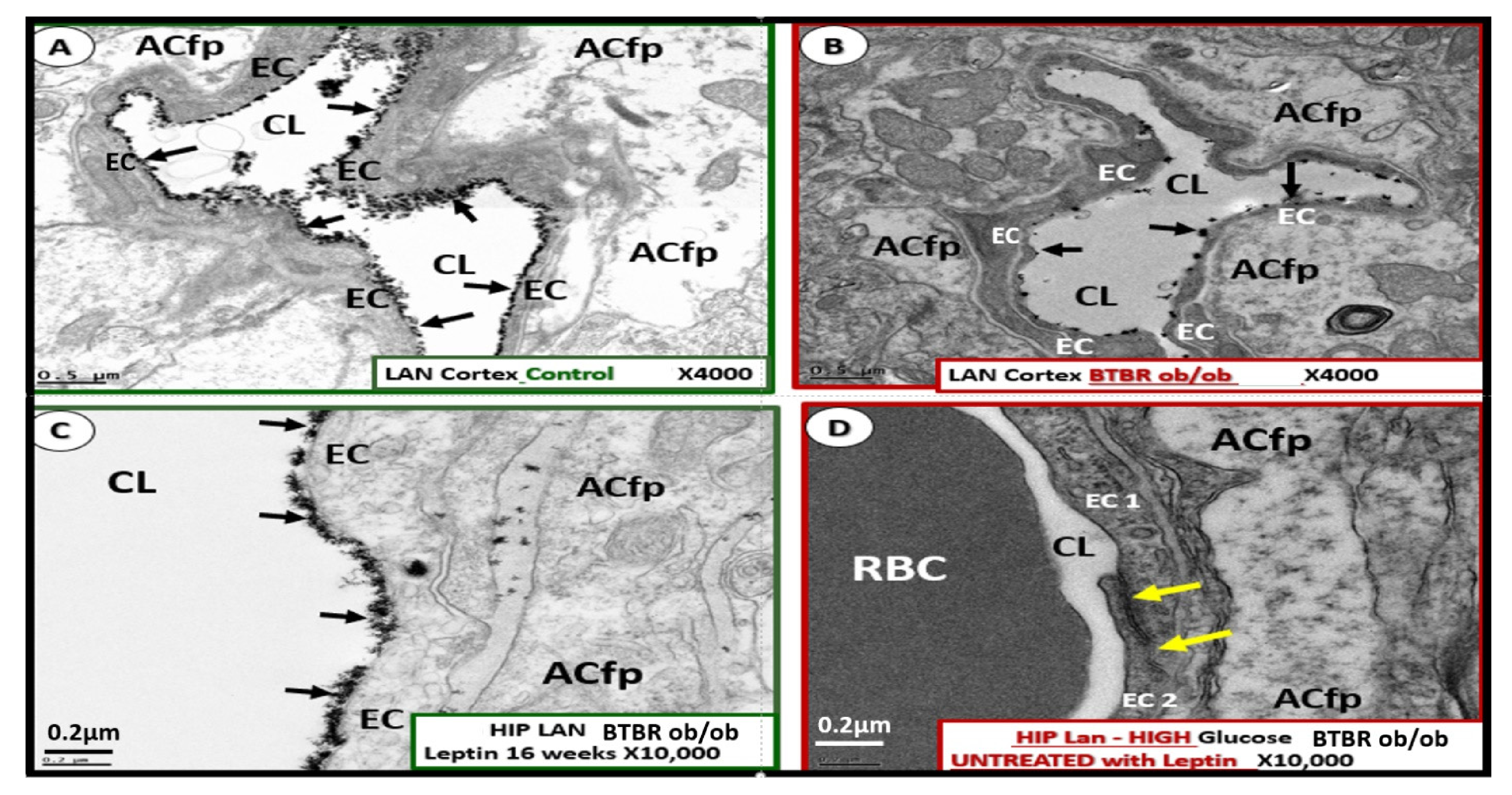
4.1.1. The BECs and Endothelial Glycocalyx (ecGCx)
4.1.2. The BEC Basement Membrane
4.1.3. BEC Activation-Dysfunction of the NVU
4.2. The Neurovascular Unit Pericyte (Pc)
4.3. Neurovascular Unit Astrocyte (AC) and Astrocyte Foot Process (ACfp)
4.4. NVU Oligodendrocytes and Myelinated Neurons
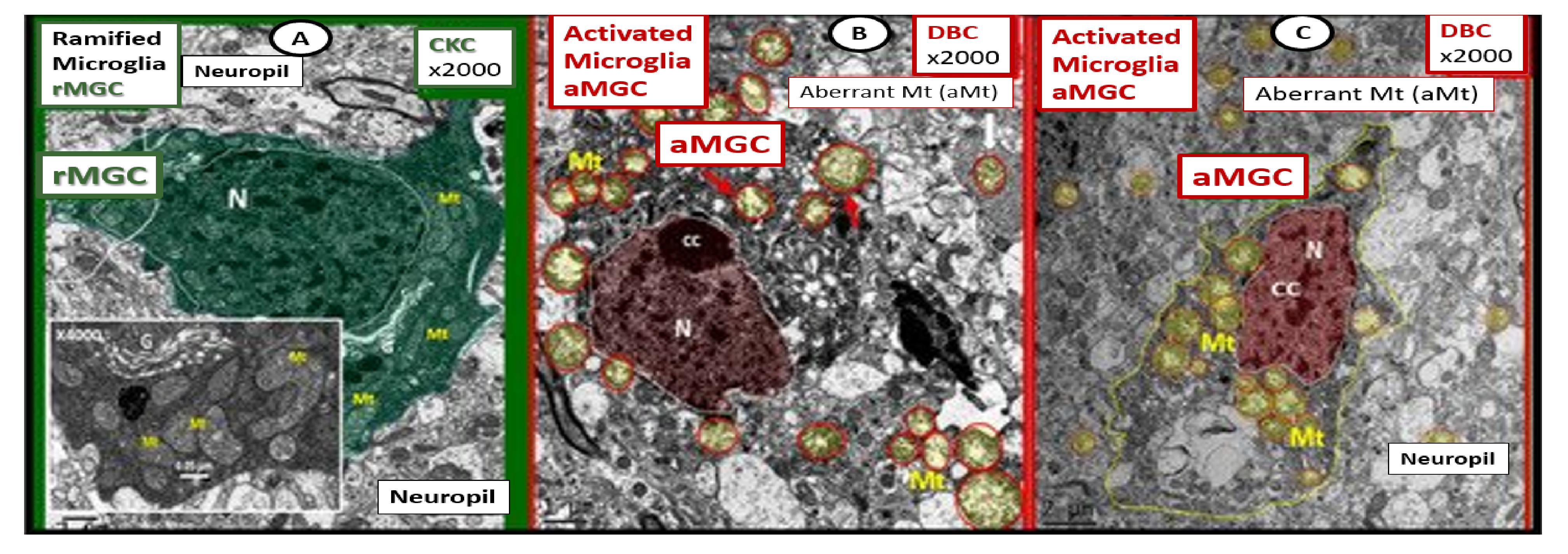
5. Microglia (MGC)
6. Blood-Cerebrospinal Fluid Barrier (BCSFB), Choroid Plexus, Median Eminence of Third Ventricle, Tanycytes, Circumventricular organ(s) (CVOs) and Hypothalamic Nuclei
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rippe, J.M.; Crossley, S.; Ringer, R. Obesity as a chronic disease: Modern medical and lifestyle management. J. Am. Diet. Assoc. 1998, 98 (Suppl. 2), S15–S19. [Google Scholar] [CrossRef]
- Aroor, A.R.; Jia, G.; Sowers, J.R. Cellular mechanisms underlying obesity-induced arterial stiffness. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R387–R398. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. T2DM increases the risk of late-onset Alzheimer’s disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.Z.; Lu, W.; Zong, X.F.; Ruan, H.Y.; Liu, Y. Obesity and hypertension. Exp. Ther. Med. 2016, 12, 2395–2399. [Google Scholar] [CrossRef]
- Hayden, M.R. An immediate and Long-Term Complication of COVID-19 May Be Type 2 Diabetes Mellitus: The Central Role of beta-Cell Dysfunction, Apoptosis and Exploration of Possible Mechanisms. Cells 2020, 9, 2475. [Google Scholar] [CrossRef]
- Mechanick, J.I.; Garber, A.J.; Grunberger, G.; Handelsman, Y.; Garvey, W.T. Dysglycemia-based chronic disease: An American Association of Clinical Endocrinologists position statement. Endocr. Pract. 2018, 24, 995–1011. [Google Scholar] [CrossRef]
- Mechanick, J.I.; Hurley, D.L.; Garvey, W.T. Adiposity-based chronic disease as a new diagnostic term: The American Association of Clinical Endocrinologists and American College of Endocrinology position statement. Endocr. Pract. 2017, 23, 372–378. [Google Scholar] [CrossRef]
- Sims, E.A.; Danforth, E.; Horton, E.S., Jr.; Bray, G.A.; Glennon, J.A.; Salans, L.B. Endocrine and metabolic effects of experimental obesity in man. Recent. Prog. Horm. Res. 1973, 29, 457–496. [Google Scholar] [CrossRef]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef]
- Zucker, L.M.; Zucker, T.F. Fatty, a new mutation in the rat. J. Hered. 1961, 52, 275–278. [Google Scholar] [CrossRef]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef]
- Peterson, R.G.; Shaw, W.N.; Neel, M.A.; Little, L.A.; Eichberg, J. Zucker Diabetic Fatty Rat as a Model for Non-insulin-dependent Diabetes Mellitus. Ilar J. 1990, 32, 16–19. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Friedman, J. Leptin at 20: An overview. J. Endocrinol. 2014, 223, 1. [Google Scholar] [CrossRef]
- Friedman, J. The long road to leptin. J. Clin. Investig. 2016, 126, 4727–4734. [Google Scholar] [CrossRef]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995, 170, 26746–26749. [Google Scholar] [CrossRef]
- Trujillo, M.E.; Scherer, P.E. Adiponectin—Journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J. Int. Med. 2005, 257, 167–175. [Google Scholar] [CrossRef]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Hudkins, K.L.; Pichaiwong, W.; Wietecha, T.; Kowalewska, J.; Banas, M.C.; Spencer, M.W.; Mühlfeld, A.; Koelling, M.; Pippin, J.W.; Shankland, S.J.; et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 1533–1542. [Google Scholar] [CrossRef]
- Flier, J.S.; Maratos-Flier, E. Lasker lauds leptin. Cell Metab. 2010, 143, 9–12. [Google Scholar] [CrossRef]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [PubMed]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Lollmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef]
- Vázquez-Vela, M.E.; Torres, N.; Tovar, A.R. White adipose tissue as endocrine organ and its role in obesity. Arch. Med. Res. 2008, 39, 715–728. [Google Scholar] [CrossRef]
- Park, H.K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34. [Google Scholar] [CrossRef]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef]
- Gruzdeva, O.; Borodkina, D.; Uchasova, E.; Dyleva, Y.; Barbarash, O. Leptin resistance: Underlying mechanisms and diagnosis. Diabetes Metab. Syndr. Obes. 2019, 12, 191–198. [Google Scholar] [CrossRef]
- Tang, B.L. Leptin as a neuroprotective agent. Biochem. Biophys. Res. Commun. 2008, 368, 181–185. [Google Scholar] [CrossRef]
- Winick, J.D.; Stoffel, M.; Friedman, J.M. Identification of microsatellite markers linked to the human leptin receptor gene on chromosome 1. Genomics 1966, 36, 221–222. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1955, 83, 1263–1271. [Google Scholar] [CrossRef]
- Kelesidis, T.; Kelesidis, I.; Chou, S.; Mantzoros, C.S. Narrative review: The role of leptin in human physiology: Emerging clinical applications. Ann. Intern. Med. 2010, 152, 93–100. [Google Scholar] [CrossRef]
- Banks, W.A.; Niehoff, M.L.; Martin, D.; Farrell, C.L. Leptin transport across the blood-brain barrier of the Koletsky rat is not mediated by a product of the leptin receptor gene. Brain Res. 2002, 950, 130–136. [Google Scholar] [CrossRef]
- Di Spiezio, A.; Sandin, E.S.; Dore, R.; Müller-Fielitz, H.; Storck, S.E.; Bernau, M.; Mier, W.; Oster, H.; Jöhren, O.; Pietrzik, C.U.; et al. The LepR-mediated leptin transport across brain barriers controls food reward. Mol. Metab. 2018, 8, 13–22. [Google Scholar] [CrossRef]
- Bjørbæk, C.; Elmquist, J.K.; Michl, P.; Ahima, R.S.; Van Bueren, A.; McCall, A.L.; Flier, J.S. Expression of leptin receptor isoforms in rat brain microvessels. Endocrinology 1998, 139, 3485–3491. [Google Scholar] [CrossRef]
- Ramos-Lobo, A.M.; Donato, J., Jr. The role of leptin in health and disease. Temperature 2017, 4, 258–291. [Google Scholar] [CrossRef]
- Hayden, M.R.; Banks, W.A.; Shah, G.N.; Gu, Z.; Sowers, J.R. Cardiorenal metabolic syndrome and diabetic cognopathy. Cardiorenal Med. 2013, 3, 265–282. [Google Scholar] [CrossRef]
- Salameh, T.S.; Shah, G.N.; Price, T.O.; Hayden, M.R.; Banks, W.A. Blood-Brain Barrier Disruption and Neurovascular Unit Dysfunction in Diabetic Mice: Protection with the Mitochondrial Carbonic Anhydrase Inhibitor Topiramate. J. Pharmacol Exp. Ther. 2016, 359, 452–459. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.; Demarco, V.G. Ultrastructural Remodeling of The Neurovascular Unit in The Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia 2018, 1, 15. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.; Aroor, A.; Demarco, V.G. Ultrastructural Remodeling of The Neurovascular Unit in The Female Diabetic db/db Model—Part II: Microglia and Mitochondria. Neuroglia 2018, 1, 21. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.; Demarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part III: Oligodendrocyte and Myelin. Neuroglia 2018, 1, 24. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female db/db Mouse. Brain Sci. 2019, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Hypothesis: Astrocyte Foot Processes Detachment from the Neurovascular Unit in Female Diabetic Mice May Impair Modulation of Information Processing-Six Degrees of Separation. Brain Sci. 2019, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Mortell, W.; Hayden, M.R.; Banks, W.A. Role of Leptin in Blood-Brain Barrier Dysfunction. In Poster Session Presented at: American Diabetes Association. Poster Number 1958-P 2019 June 7–10; American Diabetes Association: San Francisco, CA, USA, 2019; Available online: https://ada.scientificposters.com/epsAbstractADA.cfm?id=12 (accessed on 19 April 2021).
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Serlin, Y.; Shelef, I.; Knyazer, B.; Friedman, A. Anatomy and Physiology of the Blood-Brain Barrier. Semin. Cell Dev. Biol. 2015, 38, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Prevot, V.; Dehouck, B.; Sharif, A.; Ciofi, P.; Giacobini, P.; Clasadonte, J. The Versatile Tanycyte: A Hypothalamic Integrator of Reproduction and Energy Metabolism. Endocr. Rev. 2018, 39, 333–368. [Google Scholar] [CrossRef] [PubMed]
- Johanson, C.E.; Johanson, N.L. Choroid Plexus Blood-CSF Barrier: Major Player in Brain Disease Modeling and Neuromedicine. J. Neurol. Neuromed. 2018, 3, 39–58. [Google Scholar] [CrossRef]
- Luft, J.H. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed. Proc. 1966, 25, 1773–1783. [Google Scholar] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflügers Arch.-Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Haeren, R.H.L.; Rijkers, K.; Schijns, O.E.M.G.; Dings, J.; Hoogland, G.; van Zandvoort, M.A.M.J.; Vink, H.; van Overbeeke, J.J. In vivo assessment of the human cerebral microcirculation and its glycocalyx: A technical report. J. Neurosci. Methods 2018, 303, 114–125. [Google Scholar] [CrossRef]
- Yoon, J.H.; Jeong, Y. In vivo imaging for neurovascular disease research. Arch. Pharm. Res. 2019, 42, 263–273. [Google Scholar] [CrossRef]
- Reed, M.J.; Damodarasamy, M.; Banks, W.A. The extracellular matrix of the blood-brain barrier: Structural and functional roles in health, aging, and Alzheimer’s disease. Tissue Barriers 2019, 7, 1651157. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R.; Tyagi, S.C. The central role of vascular extracellular matrix and basement membrane remodeling in metabolic syndrome and type 2 diabetes: The matrix preloaded. Cardiovasc. Diabetol. 2005, 4, 9. [Google Scholar] [CrossRef]
- Pahakis, M.Y.; Kosky, J.R.; Dull, R.O.; Tarbell, J.M. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 2007, 355, 228–233. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Pahakis, M.Y. Mechanotransduction and the glycocalyx. J. Int. Med. 2006, 259, 339–350. [Google Scholar] [CrossRef]
- Ando, Y.; Okada, H.; Takemura, G.; Suzuki, K.; Takada, C.; Tomita, H.; Zaikokuji, R.; Hotta, Y.; Miyazaki, N.; Yano, H.; et al. Brain-Specific Ultrastructure of Capillary Endothelial Glycocalyx and Its Possible Contribution for Blood Brain Barrier. Sci. Rep. 2018, 8, 17523. [Google Scholar] [CrossRef]
- Okada, H.; Takemura, G.; Suzuki, K.; Oda, K.; Takada, C.; Hotta, Y.; Miyazaki, N.; Tsujimoto, A.; Muraki, I.; Ando, Y.; et al. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic condition. Crit. Care 2017, 21, 261. [Google Scholar] [CrossRef]
- Kutuzov, N.; Flyvbjerg, H.; Lauritzen, M. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood–brain barrier. Proc. Natl. Acad. Sci. USA 2018, 115, E9429–E9438. [Google Scholar] [CrossRef]
- Haeren, R.; Vink, H.; Staals, J.; van Zandvoort, M.; Dings, J.; van Overbeeke, J.; Hoogland, G.; Rijkers, K.; Schijns, O. Protocol for intraoperative assessment of the human cerebrovascular glycocalyx. BMJ Open 2017, 7, e013954. [Google Scholar] [CrossRef][Green Version]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Tambuyzer, B.R.; Ponsaerts, P.; Nouwen, E.J. Microglia: Gatekeepers of the central nervous system immunology. J. Leukoc. Biol. 2009, 85, 352–370. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Crotti, A.; Ransohoff, R.M. Microglial physiology and pathophysiology: Insights from genome-wide transcriptional profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Liu, Y.; Li, M.; Zhang, Z.; Ye, Y.; Zhou, J. Role of microglia-neuron interactions in diabetic encephalopathy. Ageing Res. Rev. 2017, 42, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Koellhoffer, E.C.; McCullough, L.D.; Ritzel, R.M. Old maids: Aging and its impact on microglia function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Pósfai, B.; Cserép, C.; Orsolits, B.; Dénes, Á. New Insights into microglia-neuron interactions: A neuron’s perspective. Neuroscience 2019, 405, 103–117. [Google Scholar] [CrossRef]
- Tang, Y. Editorial: Microglial polarization in the pathogenesis and therapeutics of neurodegenerative diseases. Front. Aging Neurosci. 2019, 13, 542. [Google Scholar] [CrossRef]
- Sousa, C.; Biber, K.; Micheluccie, A. Cellular and molecular characterization of microglia: A unique immune cell population. Front. Immunol. 2017, 8, 198. [Google Scholar] [CrossRef]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef]
- Habibi, J.; Aroor, A.R.; Sowers, J.R.; Jia, G.; Hayden, M.R.; Garro, M.; Barron, B.; Mayoux, E.; Rector, R.S.; Whaley-Connell, A.; et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc. Diabetol. 2017, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Rodriguez, J.J.; Ortiz, O.; Jimenez-Palomares, M.; Kay, K.R.; Berrocoso, E.; Murillo-Carretero, M.I.; Perdomo, G.; Spires-Jones, T.; Cozar-Castellano, I.; Lechuga-Sancho, A.M.; et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 2013, 38, 2462–2475. [Google Scholar] [CrossRef]
- Jiménez, A.J.; Domínguez-Pinos, M.D.; Guerra, M.M.; Fernández-Llebrez, P.F.; Pérez-Fígares, J.M. Structure and function of the ependymal barrier and diseases associated with ependyma disruption. Tissue Barriers 2014, 2, e28426. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Iliff, J.J.; Nedergaard, M. Is there a cerebral lymphatic system? Stroke 2013, 44, S93–S95. [Google Scholar] [CrossRef]
- Jessenm, N.A.; Munk, A.S.; Lundgarrd, I.; Nedergaard, M. The glymphatic system—A beginner’s guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Kida, S.; Pantazis, A.; Weller, R.O. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol. Appl. Neurobiol. 1993, 19, 480–488. [Google Scholar] [CrossRef]
- Weller, R.O.; Sharp, M.M.; Christodoulides, M.; Carare, R.O.; Mollgard, K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. 2018, 135, 363–385. [Google Scholar] [CrossRef]
- Bacyinski, A.; Maosheng, X.U.; Wang, W.; Hu, J. The paravascular pathway for brain current understanding, significance and controversy. Front. Neroanat. 2017, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A. Development of the choroid plexus and blood-CSF barrier. Front. Neurosci. 2015, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Mazucanti, C.H.; Liu, Q.R.; Lang, D.; Huang, N.; O’Connell, J.F.; Camandola, S.; Egan, J.M. Release of insulin produced by the choroid plexis is regulated by serotonergic signaling. JCI Insight 2019, 4, e131682. [Google Scholar] [CrossRef] [PubMed]
- Horstmann, E. Die Faserglia des Selachiergehirns. Z. Zellforsch. Mikrosk. Anat. 1954, 39, 588–617. [Google Scholar] [CrossRef]
- Rodríguez, E.M.; Blázquez, J.L.; Pastor, F.E.; Peláez, B.; Peña, P.; Peruzzo, B.; Amat, P. Hypothalamic tanycytes: A key component of brain-endocrine interaction. Int. Rev. Cytol. 2005, 247, 89–164. [Google Scholar] [CrossRef]
- Gao, Y.; Tschöp, M.H.; Luquet, S. Hypothalamic tanycytes: Gatekeepers to metabolic control. Cell Metab. 2014, 19, 173–175. [Google Scholar] [CrossRef]
- Balland, E.; Dam, J.; Langlet, F.; Caron, E.; Steculorum, S.; Messina, A.; Rasika, S.; Falluel-Morel, A.; Anouar, Y.; Dehouck, B.; et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014, 19, 293–301. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef]
- Harrison, L.; Schriever, S.C.; Feuchtinger, A.; Kyriakou, E.; Baumann, P.; Pfuhlmann, K.; Messias, A.C.; Walch, A.; Tschöp, M.H.; Pfluger, P.T. Fluorescent blood-brain barrier tracing shows intact leptin transport in obese mice. Int. J. Obes. 2019, 43, 1305–1318. [Google Scholar] [CrossRef]
- Elizondo-Vega, R.; Cortes-Campos, C.; Barahona, M.J.; Oyarce, K.A.; Carril, C.A.; García-Robles, M.A. The role of tanycytes in hypothalamic glucosensing. J. Cell. Mol. Med. 2015, 19, 1471–1482. [Google Scholar] [CrossRef]
- Raikwar, S.P.; Bhagavan, S.M.; Ramaswamy, S.B.; Thangavel, R.; Dubova, I.; Selvakumar, G.P.; Ahmed, M.E.; Kempuraj, D.; Zaheer, S.; Iyer, S.; et al. Are Tanycytes the Missing Link Between Type 2 Diabetes and Alzheimer’s Disease? Mol. Neurobiol. 2019, 56, 833–843. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Jovanovic, S.; Miao, W.; Samara, S.; Verma, S.; Farrell, C.L. Differential regulation of leptin transport by the choroid plexus and blood-brain barrier and high affinity transport systems for entry into hypothalamus and across the blood-cerebrospinal fluid barrier. Endocrinology 2000, 141, 1434–1441. [Google Scholar] [CrossRef]
- Yoo, S.; Cha, D.; Kim, D.W.; Hoang, T.V.; Blackshaw, S. Tanycyte-Independent Control of Hypothalamic Leptin Signaling. Front. Neurosci. 2019, 13, 240. [Google Scholar] [CrossRef]
- Huang, L.; Li, C. Leptin: A multifunctional hormone. Cell Res. 2000, 10, 81–92. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayden, M.R.; Banks, W.A. Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 5427. https://doi.org/10.3390/ijms22115427
Hayden MR, Banks WA. Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus. International Journal of Molecular Sciences. 2021; 22(11):5427. https://doi.org/10.3390/ijms22115427
Chicago/Turabian StyleHayden, Melvin R., and William A. Banks. 2021. "Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus" International Journal of Molecular Sciences 22, no. 11: 5427. https://doi.org/10.3390/ijms22115427
APA StyleHayden, M. R., & Banks, W. A. (2021). Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus. International Journal of Molecular Sciences, 22(11), 5427. https://doi.org/10.3390/ijms22115427