
α-Helices in the Type III Secretion Effectors: A Prevalent Feature with Versatile Roles



Abstract
1. Introduction
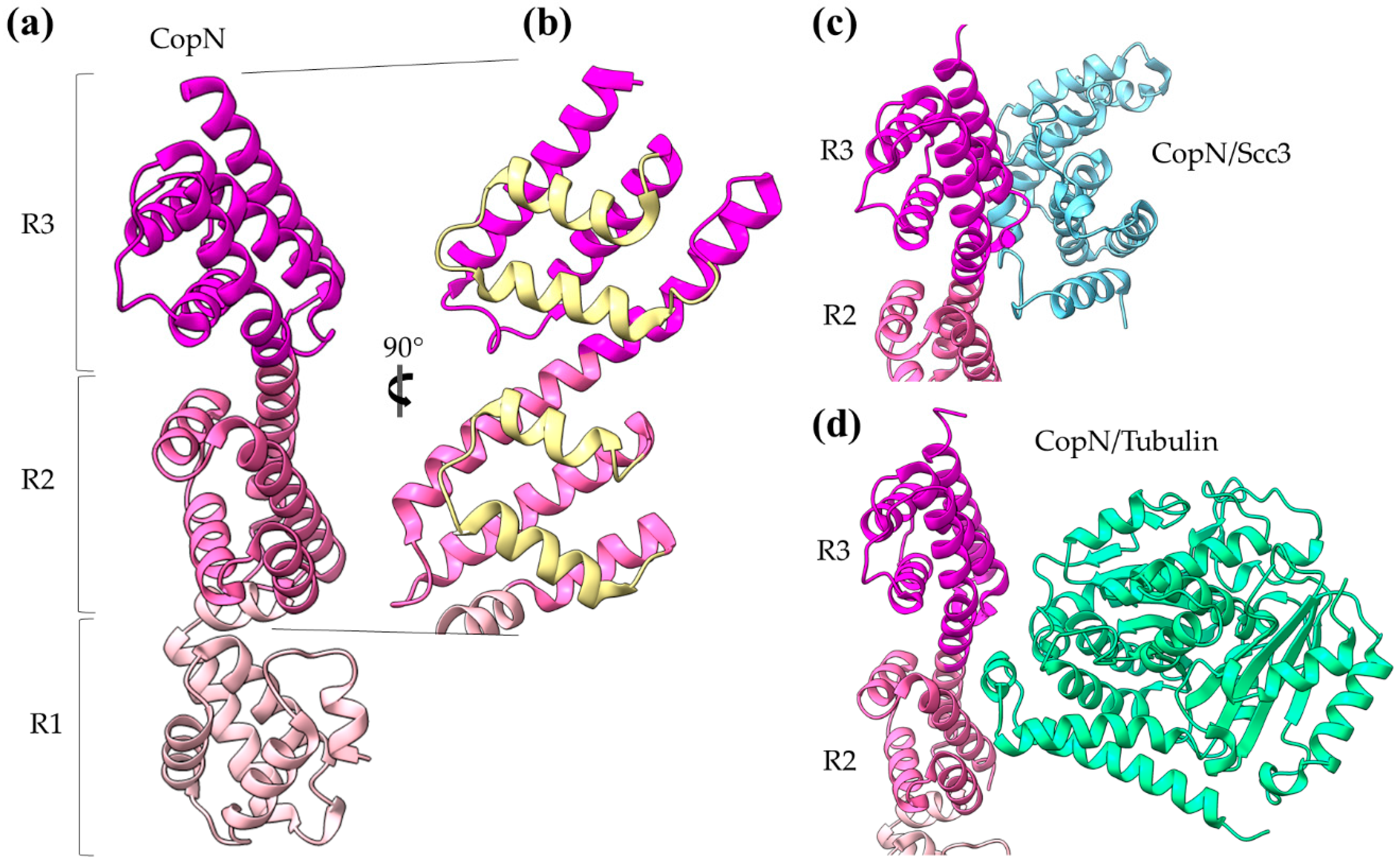
2. The T3SS Gatekeepers Are α-Helical Proteins
3. The Diverse Roles of α-Helical Domains in Animal T3S Effectors
3.1. Membrane Docking
3.2. Subverting G Proteins: GAP Activity
3.3. Subverting G Proteins: GEF and GDI Activities
3.4. Acting as Post-Translational Modification Domains
3.4.1. Adenylylation/AMPylation with Fido Domains
3.4.2. Arg-GlcNAcylation
4. The Importance of α-Helical Domains in the (a) Virulence of Bacterial Plant Pathogens
4.1. The Helix–Loop–Helix Transcription Activator-Like (TAL) Effectors
4.2. Coiled-Coils Avirulent Proteins
4.3. Bacterial Avirulence Based on 3- and 4-helix Bundles
4.4. Effector Activation Sites Are Formed by α-Helices
4.5. The Multiple Facets of Enzymatic Regulation by α-Helices
5. Marginal Resemblance of T3SE Folds to Functionally Related Proteins
5.1. The NleC/GtgA Family of T3S Zinc Metalloproteases
5.2. The NleH Family of T3S Kinases
5.3. The LRR-Containing T3S Effectors
6. Discussion
7. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Portaliou, A.G.; Tsolis, K.C.; Loos, M.S.; Zorzini, V.; Economou, A. Type III Secretion: Building and Operating a Remarkable Nanomachine. Trends Biochem. Sci. 2016, 41, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Tampakaki, A.P.; Skandalis, N.; Gazi, A.D.; Bastaki, M.N.; Sarris, P.F.; Charova, S.N.; Kokkinidis, M.; Panopoulos, N.J. Playing the “Harp”: Evolution of our understanding of hrp/hrc genes. Annu. Rev. Phytopathol. 2010, 48, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Abby, S.S.; Rocha, E.P.C. The Non-Flagellar Type III Secretion System Evolved from the Bacterial Flagellum and Diversified into Host-Cell Adapted Systems. PLoS Genet. 2012, 8, e1002983. [Google Scholar] [CrossRef]
- Lunelli, M.; Kamprad, A.; Bürger, J.; Mielke, T.; Spahn, C.M.T.; Kolbe, M. Cryo-EM structure of the Shigella type III needle complex. PLoS Pathog. 2020, 16, e1008263. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Morado, D.R.; Margolin, W.; Rohde, J.R.; Arizmendi, O.; Picking, W.L.; Picking, W.D.; Liu, J. Visualization of the type III secretion sorting platform of Shigella flexneri. Proc. Natl. Acad. Sci. USA 2015, 112, 1047–1052. [Google Scholar] [CrossRef]
- Charova, S.N.; Gazi, A.D.; Mylonas, E.; Pozidis, C.; Sabarit, B.; Anagnostou, D.; Psatha, K.; Aivaliotis, M.; Beuzon, C.R.; Panopoulos, N.J.; et al. Migration of type III secretion system transcriptional regulators links gene expression to secretion. mBio 2018, 9. [Google Scholar] [CrossRef]
- Deng, W.; Marshall, N.C.; Rowland, J.L.; McCoy, J.M.; Worrall, L.J.; Santos, A.S.; Strynadka, N.C.J.; Finlay, B.B. Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 2017, 15, 323–337. [Google Scholar] [CrossRef]
- Li, G.; Froehlich, J.E.; Elowsky, C.; Msanne, J.; Ostosh, A.C.; Zhang, C.; Awada, T.; Alfano, J.R. Distinct Pseudomonas type-III effectors use a cleavable transit peptide to target chloroplasts. Plant J. 2014, 77, 310–321. [Google Scholar] [CrossRef]
- Parsot, C. Shigella type III secretion effectors: How, where, when, for what purposes? Curr. Opin. Microbiol. 2009, 12, 110–116. [Google Scholar] [CrossRef]
- De Groot, N.S.; Burgas, M.T. Bacteria use structural imperfect mimicry to hijack the host interactome. bioRxiv 2020, 16, e1008395. [Google Scholar]
- Ruano-Gallego, D.; Sanchez-Garrido, J.; Kozik, Z.; Núñez-Berrueco, E.; Cepeda-Molero, M.; Mullineaux-Sanders, C.; Clark, J.N.B.; Slater, S.L.; Wagner, N.; Glegola-Madejska, I.; et al. Type III secretion system effectors form robust and flexible intracellular virulence networks. Science 2021, 371. [Google Scholar] [CrossRef]
- McCann, H.C.; Guttman, D.S. Evolution of the type III secretion system and its effectors in plant-microbe interactions. New Phytol. 2007, 177, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.L.; Charova, S.N.; Sansonetti, P.J.; Mylonas, E.; Gazi, A.D. Structural Insights of Shigella Translocator IpaB and Its Chaperone IpgC in Solution. Front. Cell. Infect. Microbiol. 2021, 11, 353. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.S.; Jobichen, C.; Tan, K.W.; Tan, Y.W.; Chan, S.L.; Ramesh, K.; Yuan, Y.; Hong, Y.; Seetharaman, J.; Leung, K.Y.; et al. Structure of AcrH-AopB Chaperone-Translocator Complex Reveals a Role for Membrane Hairpins in Type III Secretion System Translocon Assembly. Structure 2015, 23, 2022–2031. [Google Scholar] [CrossRef]
- Cunnac, S.; Lindeberg, M.; Collmer, A. Pseudomonas syringae type III secretion system effectors: Repertoires in search of functions. Curr. Opin. Microbiol. 2009, 12, 53–60. [Google Scholar] [CrossRef]
- Akeda, Y.; Galán, J.E. Chaperone release and unfolding of substrates in type III secretion. Nature 2005, 437, 911–915. [Google Scholar] [CrossRef]
- Radics, J.; Königsmaier, L.; Marlovits, T.C. Structure of a pathogenic type 3 secretion system in action. Nat. Struct. Mol. Biol. 2014, 21, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Rietsch, A. Fueling type III secretion. Trends Microbiol. 2015, 23, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Minamino, T. Flagella-Driven Motility of Bacteria. Biomolecules 2019, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Ohgita, T.; Saito, H. Biophysical mechanism of protein export by bacterial type III secretion system. Chem. Pharm. Bull. 2019, 67, 341–344. [Google Scholar] [CrossRef]
- Miletic, S.; Fahrenkamp, D.; Goessweiner-Mohr, N.; Wald, J.; Pantel, M.; Vesper, O.; Kotov, V.; Marlovits, T.C. Substrate-engaged type III secretion system structures reveal gating mechanism for unfolded protein translocation. Nat. Commun. 2021, 12, 1546. [Google Scholar] [CrossRef]
- Eisenberg, D. The discovery of the α-helix and β-sheet, the principal structural features of proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 11207–11210. [Google Scholar] [CrossRef]
- Gazi, A.D.; Charova, S.N.; Panopoulos, N.J.; Kokkinidis, M. Coiled-coils in type III secretion systems: Structural flexibility, disorder and biological implications. Cell. Microbiol. 2009, 11, 719–729. [Google Scholar] [CrossRef]
- Charova, S.N.; Gazi, A.D.; Kotzabasaki, M.; Sarris, P.F.; Fadouloglou, V.E.; Panopoulos, N.J.; Kokkinidis, M. Protein Flexibility and Coiled-Coil Propensity: New Insights Into Type III and Other Bacterial Secretion Systems. In Biochemistry; InTech: London, UK, 2012. [Google Scholar]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Schubot, F.D.; Jackson, M.W.; Penrose, K.J.; Cherry, S.; Tropea, J.E.; Plano, G.V.; Waugh, D.S. Three-dimensional structure of a macromolecular assembly that regulates type III secretion in Yersinia pestis. J. Mol. Biol. 2005, 346, 1147–1161. [Google Scholar] [CrossRef]
- Archuleta, T.L.; Spiller, B.W. A Gatekeeper Chaperone Complex Directs Translocator Secretion during Type Three Secretion. Plos Pathog. 2014, 10, e1004498. [Google Scholar] [CrossRef]
- Cherradi, Y.; Schiavolin, L.; Moussa, S.; Meghraoui, A.; Meksem, A.; Biskri, L.; Azarkan, M.; Allaoui, A.; Botteaux, A. Interplay between predicted inner-rod and gatekeeper in controlling substrate specificity of the type III secretion system. Mol. Microbiol. 2013, 87, 1183–1199. [Google Scholar] [CrossRef]
- Campanacci, V.; Urvoas, A.; Cantos-Fernandes, S.; Aumont-Nicaise, M.; Arteni, A.A.; Velours, C.; Valerio-Lepiniec, M.; Dreier, B.; Plückthun, A.; Pilon, A.; et al. Insight into microtubule nucleation from tubulin-capping proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 9859–9864. [Google Scholar] [CrossRef]
- Huang, J.; Lesser, C.F.; Lory, S. The essential role of the CopN protein in Chlamydia pneumoniae intracellular growth. Nature 2008, 456, 112–115. [Google Scholar] [CrossRef]
- Nawrotek, A.; Guimarães, B.G.; Velours, C.; Subtil, A.; Knossow, M.; Gigant, B. Biochemical and structural insights into microtubule perturbation by CopN from Chlamydia pneumoniae. J. Biol. Chem. 2014, 289, 25199–25210. [Google Scholar] [CrossRef] [PubMed]
- Gendrin, C.; Contreras-Martel, C.; Bouillot, S.; Elsen, S.; Lemaire, D.; Skoufias, D.A.; Huber, P.; Attree, I.; Dessen, A. Structural basis of cytotoxicity mediated by the type III secretion toxin ExoU from pseudomonas aeruginosa. PLoS Pathog. 2012, 8, e1002637. [Google Scholar] [CrossRef]
- Halavaty, A.S.; Borek, D.; Tyson, G.H.; Veesenmeyer, J.L.; Shuvalova, L.; Minasov, G.; Otwinowski, Z.; Hauser, A.R.; Anderson, W.F. Structure of the Type III Secretion Effector Protein ExoU in Complex with Its Chaperone SpcU. PLoS ONE 2012, 7, e49388. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Frank, D.W. ExoU is a potent intracellular phospholipase. Mol. Microbiol. 2004, 53, 1279–1290. [Google Scholar] [CrossRef]
- Zhang, A.; Veesenmeyer, J.L.; Hauser, A.R. Phosphatidylinositol 4,5-bisphosphatedependent oligomerization of the Pseudomonas aeruginosa cytotoxin ExoU. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Tessmer, M.H.; Anderson, D.M.; Pickrum, A.M.; Riegert, M.O.; Frank, D.W. Identification and verification of ubiquitin-activated bacterial phospholipases. J. Bacteriol. 2019, 201, 1–16. [Google Scholar] [CrossRef]
- Tessmer, M.H.; Anderson, D.M.; Pickrum, A.M.; Riegert, M.O.; Moretti, R.; Meiler, J.; Feix, J.B.; Frank, D.W. Identification of a ubiquitin-binding interface using Rosetta and DEER. Proc. Natl. Acad. Sci. USA 2018, 115, 525–530. [Google Scholar] [CrossRef]
- Feix, J.B.; Kohn, S.; Tessmer, M.H.; Anderson, D.M.; Frank, D.W. Conformational Changes and Membrane Interaction of the Bacterial Phospholipase, ExoU: Characterization by Site-Directed Spin Labeling. Cell Biochem. Biophys. 2019, 77, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.I.; Reid, T.E.; Gies, S.L.; Feix, J.B. Interactions of the effector ExoU from pseudomonas aeruginosa with short-chain phosphatidylinositides provide insights into ExoU targeting to host membranes. J. Biol. Chem. 2019, 294, 19012–19021. [Google Scholar] [CrossRef]
- Tessmer, M.H.; Anderson, D.M.; Buchaklian, A.; Frank, D.W.; Feix, J.B. Cooperative substrate-cofactor interactions and membrane localization of the bacterial phospholipase A2 (PLA2) enzyme, ExoU. J. Biol. Chem. 2017, 292, 3411–3419. [Google Scholar] [CrossRef]
- Kohn, W.D.; Mant, C.T.; Hodges, R.S. A-Helical Protein Assembly Motifs. J. Biol. Chem. 1997, 272, 2583–2586. [Google Scholar] [CrossRef]
- Glykos, N.M.; Cesareni, G.; Kokkinidis, M. Protein plasticity to the extreme: Changing the topology of a 4-α- helical bundle with a single amino acid substitution. Structure 1999, 7, 597–603. [Google Scholar] [CrossRef]
- Fadouloglou, V.E.; Glykos, N.M.; Kokkinidis, M. Side-chain conformations in 4-α-helical bundles. Protein Eng. 2001, 14, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Amprazi, M.; Kotsifaki, D.; Providaki, M.; Kapetaniou, E.G.; Fellas, G.; Kyriazidis, I.; Pérez, J.; Kokkinidis, M. Structural plasticity of 4-α-helical bundles exemplified by the puzzle-like molecular assembly of the Rop protein. Proc. Natl. Acad. Sci. USA 2014, 111, 11049–11054. [Google Scholar] [CrossRef] [PubMed]
- Kefala, A.; Amprazi, M.; Mylonas, E.; Kotsifaki, D.; Providaki, M.; Pozidis, C.; Fotiadou, M.; Kokkinidis, M. Probing protein folding with sequence-reversed α-helical bundles. Int. J. Mol. Sci. 2021, 22, 1955. [Google Scholar] [CrossRef] [PubMed]
- Hisao, G.S.; Brothers, M.C.; Ho, M.; Wilson, B.A.; Rienstra, C.M. The membrane localization domains of two distinct bacterial toxins form a 4-helix-bundle in solution. Protein Sci. 2017, 26, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Tessmer, M.H.; DeCero, S.A.; del Alamo, D.; Riegert, M.O.; Meiler, J.; Frank, D.W.; Feix, J.B. Characterization of the ExoU activation mechanism using EPR and integrative modeling. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Müller, M.P.; Goody, R.S. Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases 2018, 9, 5–21. [Google Scholar] [CrossRef]
- Evdokimov, A.G.; Tropea, J.E.; Routzahn, K.M.; Waugh, D.S. Crystal structure of the Yersinia pestis GTPase activator YopE. Protein Sci. 2009, 11, 401–408. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Galán, J.E. Modulation of host signaling by a bacterial mimic: Structure of the Salmonella effector SptP bound to Rac1. Mol. Cell 2000, 6, 1449–1460. [Google Scholar] [CrossRef]
- Würtele, M.; Wolf, E.; Pederson, K.J.; Buchwald, G.; Ahmadian, M.R.; Barbieri, J.T.; Wittinghofer, A. How the Pseudomonas aeruginosa ExoS toxin downregulates Rac. Nat. Struct. Biol. 2001, 8, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Würtele, M.; Renault, L.; Barbieri, J.T.; Wittinghofer, A.; Wolf, E. Structure of the ExoS GTPase activating domain. Febs Lett. 2001, 491, 26–29. [Google Scholar] [CrossRef]
- Rocha, C.L.; Coburn, J.; Rucks, E.A.; Olson, J.C. Characterization of Pseudomonas aeruginosa exoenzyme S as a bifunctional enzyme in J774a.1 macrophages. Infect. Immun. 2003, 71, 5296–5305. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krall, R.; Zhang, Y.; Barbieri, J.T. Intracellular Membrane Localization of Pseudomonas ExoS and Yersinia YopE in Mammalian Cells. J. Biol. Chem. 2004, 279, 2747–2753. [Google Scholar] [CrossRef]
- Davis, J.; Wang, J.; Tropea, J.E.; Zhang, D.; Dauter, Z.; Waugh, D.S.; Wlodawer, A. Novel fold of VirA, a type III secretion system effector protein from Shigella flexneri. Protein Sci. 2008, 17, 2167–2173. [Google Scholar] [CrossRef] [PubMed]
- Germane, K.L.; Ohi, R.; Goldberg, M.B.; Spiller, B.W. Structural and functional studies indicate that Shigella VirA is not a protease and does not directly destabilize microtubules. Biochemistry 2008, 47, 10241–10243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Germane, K.L.; Spiller, B.W. Structural and functional studies indicate that the EPEC effector, EspG, directly binds p21-activated kinase. Biochemistry 2011, 50, 917–919. [Google Scholar] [CrossRef]
- Dong, N.; Zhu, Y.; Lu, Q.; Hu, L.; Zheng, Y.; Shao, F. Structurally Distinct Bacterial TBC-like GAPs Link Arf GTPase to Rab1 Inactivation to Counteract Host Defenses. Cell 2012, 150, 1029–1041. [Google Scholar] [CrossRef]
- Orchard, R.C.; Alto, N.M. Mimicking GEFs: A common theme for bacterial pathogens. Cell. Microbiol. 2012, 14, 10–18. [Google Scholar] [CrossRef]
- Buchwald, G.; Friebel, A.; Galán, J.E.; Hardt, W.D.; Wittinghofer, A.; Scheffzek, K. Structural basis for the reversible activation of a Rho protein by the bacterial toxin SopE. Embo J. 2002, 21, 3286–3295. [Google Scholar] [CrossRef]
- Williams, C.; Galyov, E.E.; Bagby, S. Solution structure, backbone dynamics, and interaction with Cdc42 of Salmonella guanine nucleotide exchange factor SopE2. Biochemistry 2004, 43, 11998–12008. [Google Scholar] [CrossRef] [PubMed]
- Ohlson, M.B.; Huang, Z.; Alto, N.M.; Blanc, M.P.; Dixon, J.E.; Chai, J.; Miller, S.I. Structure and Function of Salmonella SifA Indicate that Its Interactions with SKIP, SseJ, and RhoA Family GTPases Induce Endosomal Tubulation. Cell Host Microbe 2008, 4, 434–446. [Google Scholar] [CrossRef]
- Huang, Z.; Sutton, S.E.; Wallenfang, A.J.; Orchard, R.C.; Wu, X.; Feng, Y.; Chai, J.; Alto, N.M. Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat. Struct. Mol. Biol. 2009, 16, 853–860. [Google Scholar] [CrossRef]
- Klink, B.U.; Barden, S.; Heidler, T.V.; Borchers, C.; Ladwein, M.; Stradal, T.E.B.; Rottner, K.; Heinz, D.W. Structure of Shigella IpgB2 in complex with human RhoA: Implications for the mechanism of bacterial guanine nucleotide exchange factor mimicry. J. Biol. Chem. 2010, 285, 17197–17208. [Google Scholar] [CrossRef] [PubMed]
- Prehna, G.; Ivanov, M.I.; Bliska, J.B.; Stebbins, C.E. Yersinia Virulence Depends on Mimicry of Host Rho-Family Nucleotide Dissociation Inhibitors. Cell 2006, 126, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Kokkinidis, M.; Glykos, N.M.; Fadouloglou, V.E. Catalytic activity regulation through post-translational modification: The expanding universe of protein diversity. Adv. Protein Chem. Struct. Biol. 2020, 122, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Kinch, L.N.; Yarbrough, M.L.; Orth, K.; Grishin, N.V. Fido, a Novel AMPylation Domain Common to Fic, Doc, and AvrB. PLoS ONE 2009, 4, e5818. [Google Scholar] [CrossRef]
- Yarbrough, M.L.; Li, Y.; Kinch, L.N.; Grishin, N.V.; Ball, H.L.; Orth, K. AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 2009, 323, 269–272. [Google Scholar] [CrossRef]
- Ding, J.; Pan, X.; Du, L.; Yao, Q.; Xue, J.; Yao, H.; Wang, D.C.; Li, S.; Shao, F. Structural and Functional Insights into Host Death Domains Inactivation by the Bacterial Arginine GlcNAcyltransferase Effector. Mol. Cell 2019, 74, 922–935.e6. [Google Scholar] [CrossRef]
- Xue, J.; Hu, S.; Huang, Y.; Zhang, Q.; Yi, X.; Pan, X.; Li, S. Arg-GlcNAcylation on TRADD by NleB and SseK1 Is Crucial for Bacterial Pathogenesis. Front. Cell Dev. Biol. 2020, 8, 641. [Google Scholar] [CrossRef]
- Gazi, A.D.; Bastaki, M.; Charova, S.N.; Gkougkoulia, E.A.; Kapellios, E.A.; Panopoulos, N.J.; Kokkinidis, M. Evidence for a coiled-coil interaction mode of disordered proteins from bacterial type III secretion systems. J. Biol. Chem. 2008, 283, 34062–34068. [Google Scholar] [CrossRef] [PubMed]
- Gazi, A.D.; Charova, S.; Aivaliotis, M.; Panopoulos, N.J.; Kokkinidis, M. HrpG and HrpV proteins from the Type III secretion system of Erwinia amylovora form a stable heterodimer. Fems Microbiol. Lett. 2015, 362. [Google Scholar] [CrossRef] [PubMed]
- Tampakaki, A.P.; Fadouloglou, V.E.; Gazi, A.D.; Panopoulos, N.J.; Kokkinidis, M. Conserved features of type III secretion. Cell. Microbiol. 2004, 6, 805–816. [Google Scholar] [CrossRef]
- Mahfouz, M.M.; Li, L.; Shamimuzzaman, M.; Wibowo, A.; Fang, X.; Zhu, J.K. De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. Proc. Natl. Acad. Sci. USA 2011, 108, 2623–2628. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Yan, C.; Pan, X.; Mahfouz, M.; Wang, J.; Zhu, J.; Shi, Y. Structural Basis for Sequence-Specific Recognition of DNA by TAL effectors. Science 2012, 335, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.N.S.; Bradley, P.; Cernadas, R.A.; Bogdanove, A.J.; Stoddard, B.L. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 2012, 335, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Stella, S.; Molina, R.; Yefimenko, I.; Prieto, J.; Silva, G.; Bertonati, C.; Juillerat, A.; Duchateau, P.; Montoya, G. Structure of the AvrBs3-DNA complex provides new insights into the initial thymine-recognition mechanism. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Stella, S.; Molina, R.; López-Méndez, B.; Juillerat, A.; Bertonati, C.; Daboussi, F.; Campos-Olivas, R.; Duchateau, P.; Montoya, G. BuD, a helix-loop-helix DNA-binding domain for genome modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 2042–2052. [Google Scholar] [CrossRef]
- Sohn, K.H.; Hughes, R.K.; Piquerez, S.J.; Jones, J.D.G.; Banfield, M.J. Distinct regions of the Pseudomonas syringae coiled-coil effector AvrRps4 are required for activation of immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 16371–16376. [Google Scholar] [CrossRef]
- Xing, W.; Zou, Y.; Liu, Q.; Liu, J.; Luo, X.; Huang, Q.; Chen, S.; Zhu, L.; Bi, R.; Hao, Q.; et al. The structural basis for activation of plant immunity by bacterial effector protein AvrPto. Nature 2007, 449, 243–247. [Google Scholar] [CrossRef]
- Wulf, J.; Pascuzzi, P.E.; Fahmy, A.; Martin, G.B.; Nicholson, L.K. The solution structure of type III effector protein AvrPto reveals conformational and dynamic features important for plant pathogenesis. Structure 2004, 12, 1257–1268. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dong, J.; Xiao, F.; Fan, F.; Gu, L.; Cang, H.; Martin, G.B.; Chai, J. Crystal structure of the complex between pseudomonas effector avrptob and the tomato pto kinase reveals both a shared and a unique interface compared with avrpto-pto. Plant Cell 2009, 21, 1846–1859. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Luo, X.; Li, W.; Wang, Z.; Liu, J. The bacterial effector AvrB-induced RIN4 hyperphosphorylation is mediated by a receptor-like cytoplasmic kinase complex in arabidopsis. Mol. Plant-Microbe Interact. 2017, 30, 502–512. [Google Scholar] [CrossRef]
- Lee, C.C.; Wood, M.D.; Ng, K.; Andersen, C.B.; Liu, Y.; Luginbühl, P.; Spraggon, G.; Katagiri, F. Crystal structure of the type III effector AvrB from Pseudomonas syringae. Structure 2004, 12, 487–494. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chung, E.H.; Da Cunha, L.; Wu, A.J.; Gao, Z.; Cherkis, K.; Afzal, A.J.; MacKey, D.; Dangl, J.L. Specific threonine phosphorylation of a host target by two unrelated type III effectors activates a host innate immune receptor in plants. Cell Host Microbe 2011, 9, 125–136. [Google Scholar] [CrossRef]
- Roy, C.R.; Cherfils, J. Structure and function of Fic proteins. Nat. Rev. Microbiol. 2015, 13, 631–640. [Google Scholar] [CrossRef]
- Desveaux, D.; Singer, A.U.; Wu, A.J.; McNulty, B.C.; Musselwhite, L.; Nimchuk, Z.; Sondek, J.; Dangl, J.L. Type III effector activation via nucleotide binding, phosphorylation, and host target interaction. PLoS Pathog. 2007, 3, e90. [Google Scholar] [CrossRef]
- Han, Q.; Zhou, C.; Wu, S.; Liu, Y.; Triplett, L.; Miao, J.; Tokuhisa, J.; Deblais, L.; Robinson, H.; Leach, J.E.; et al. Crystal structure of Xanthomonas AvrRxo1-ORF1, a type III effector with a polynucleotide kinase domain, and its interactor AvrRxo1-ORF2. Structure 2015, 23, 1900–1909. [Google Scholar] [CrossRef]
- Yu, S.; Hwang, I.; Rhee, S. Crystal structure of the effector protein XOO4466 from Xanthomonas oryzae. J. Struct. Biol. 2013, 184, 361–366. [Google Scholar] [CrossRef]
- Yu, S.; Hwang, I.; Rhee, S. The crystal structure of type III effector protein XopQ from Xanthomonas oryzae complexed with adenosine diphosphate ribose. Proteins: Struct. Funct. Bioinform. 2014, 82, 2910–2914. [Google Scholar] [CrossRef]
- Schultink, A.; Qi, T.; Lee, A.; Steinbrenner, A.D.; Staskawicz, B. Roq1 mediates recognition of the Xanthomonas and Pseudomonas effector proteins XopQ and HopQ1. Plant J. 2017, 92, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Qi, T.; Zhang, H.; Liu, F.; King, M.; Toth, C.; Nogales, E.; Staskawicz, B.J. Structure of the activated ROQ1 resistosome directly recognizing the pathogen effector XopQ. Science 2020, 370. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Sheng, X.; Yin, P.; Hu, F.; Liu, Y.; Chen, C.; Li, Q.; Yan, C.; Wang, J. Structure and mechanism of a type III secretion protease, NleC. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 40–47. [Google Scholar] [CrossRef]
- Turco, M.M.; Sousa, M.C. The structure and specificity of the type III secretion system effector NleC suggest a DNA mimicry mechanism of substrate recognition. Biochemistry 2014, 53, 5131–5139. [Google Scholar] [CrossRef] [PubMed]
- Jennings, E.; Esposito, D.; Rittinger, K.; Thurston, T.L.M. Structure-function analyses of the bacterial zinc metalloprotease effector protein GtgA uncover key residues required for deactivating NF-κB. J. Biol. Chem. 2018, 293, 15316–15329. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.A.; Cadby, I.T.; Chong, Z.S.; Boelter, G.; Sevastsyanovich, Y.R.; Morris, F.C.; Cunningham, A.F.; Kritikos, G.; Meek, R.W.; Banzhaf, M.; et al. Structure-function characterization of the conserved regulatory mechanism of the Escherichia coli M48 metalloprotease BepA. J. Bacteriol. 2021, 203. [Google Scholar] [CrossRef]
- Schaeffer, R.D.; Kinch, L.N.; Liao, Y.; Grishin, N.V. Classification of proteins with shared motifs and internal repeats in the ECOD database. Protein Sci. 2016, 25, 1188–1203. [Google Scholar] [CrossRef]
- Grishin, A.M.; Cherney, M.; Anderson, D.H.; Phanse, S.; Babu, M.; Cygler, M. NleH Defines a new family of bacterial effector kinases. Structure 2014, 22, 250–259. [Google Scholar] [CrossRef]
- Halavaty, A.S.; Anderson, S.M.; Wawrzak, Z.; Kudritska, M.; Skarina, T.; Anderson, W.F.; Savchenko, A. Type III effector NleH2 from Escherichia coli O157:H7 str. Sakai features an atypical protein kinase domain. Biochemistry 2014, 53, 2433–2435. [Google Scholar] [CrossRef]
- Pruneda, J.N.; Smith, F.D.; Daurie, A.; Swaney, D.L.; Villén, J.; Scott, J.D.; Stadnyk, A.W.; Le Trong, I.; Stenkamp, R.E.; Klevit, R.E.; et al. E2∼Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. Embo J. 2014, 33, 437–449. [Google Scholar] [CrossRef]
- Takagi, K.; Kim, M.; Sasakawa, C.; Mizushima, T. Crystal structure of the substrate-recognition domain of the Shigella E3 ligase IpaH9.8. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Keszei, A.F.A.; Tang, X.; McCormick, C.; Zeqiraj, E.; Rohde, J.R.; Tyers, M.; Sicheri, F. Structure of an SspH1-PKN1 Complex Reveals the Basis for Host Substrate Recognition and Mechanism of Activation for a Bacterial E3 Ubiquitin Ligase. Mol. Cell. Biol. 2014, 34, 362–373. [Google Scholar] [CrossRef]
- Ji, C.; Du, S.; Li, P.; Zhu, Q.; Yang, X.; Long, C.; Yu, J.; Shao, F.; Xiao, J. Structural mechanism for guanylate-binding proteins (GBPs) targeting by the Shigella E3 ligase IpaH9.8. PLoS Pathog. 2019, 15, e1007876. [Google Scholar] [CrossRef] [PubMed]
- Evdokimov, A.G.; Anderson, D.E.; Routzahn, K.M.; Waugh, D.S. Unusual molecular architecture of the Yersinia pestis cytotoxin YopM: A leucine-rich repeat protein with the shortest repeating unit. J. Mol. Biol. 2001, 312, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Berneking, L.; Schnapp, M.; Rumm, A.; Trasak, C.; Ruckdeschel, K.; Alawi, M.; Grundhoff, A.; Kikhney, A.G.; Koch-Nolte, F.; Buck, F.; et al. Immunosuppressive Yersinia Effector YopM Binds DEAD Box Helicase DDX3 to Control Ribosomal S6 Kinase in the Nucleus of Host Cells. PLoS Pathog. 2016, 12, e1005660. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, H.; Hu, L.; Wang, J.; Zhou, Y.; Pang, Z.; Liu, L.; Shao, F. Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat. Struct. Mol. Biol. 2008, 15, 1302–1308. [Google Scholar] [CrossRef]
- Quezada, C.M.; Hicks, S.W.; Galán, J.E.; Erec Stebbins, C. A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2009, 106, 4864–4869. [Google Scholar] [CrossRef]
- Taylor, J.W.; Greenfield, N.J.; Wu, B.; Privalov, P.L. A calorimetric study of the folding-unfolding of an α-helix with covalently closed N and C-terminal loops. J. Mol. Biol. 1999, 291, 965–976. [Google Scholar] [CrossRef]
- Seyedarabi, A.; Sullivan, J.A.; Sasakawa, C.; Pickersgill, R.W. A disulfide driven domain swap switches off the activity of Shigella IpaH9.8 E3 ligase. Febs Lett. 2010, 584, 4163–4168. [Google Scholar] [CrossRef]
- Ihalainen, J.A.; Paoli, B.; Muff, S.; Backus, E.H.G.; Bredenbeck, J.; Woolley, G.A.; Caflisch, A.; Hamm, P. α-Helix folding in the presence of structural constraints. Proc. Natl. Acad. Sci. USA 2008, 105, 9588–9593. [Google Scholar] [CrossRef]
- Berkemeier, F.; Bertz, M.; Xiao, S.; Pinotsis, N.; Wilmanns, M.; Gräter, F.; Rief, M. Fast-folding α-helices as reversible strain absorbers in the muscle protein myomesin. Proc. Natl. Acad. Sci. USA 2011, 108, 14139–14144. [Google Scholar] [CrossRef] [PubMed]
- Schennach, M.; Schneeberger, E.M.; Breuker, K. Unfolding and Folding of the Three-Helix Bundle Protein KIX in the Absence of Solvent. J. Am. Soc. Mass Spectrom. 2016, 27, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Jesus, C.S.H.; Cruz, P.F.; Arnaut, L.G.; Brito, R.M.M.; Serpa, C. One Peptide Reveals the Two Faces of α-Helix Unfolding-Folding Dynamics. J. Phys. Chem. B 2018, 122, 3790–3800. [Google Scholar] [CrossRef]
- Zheng, W.; Peña, A.; Ilangovan, A.; Clark, J.N.B.; Frankel, G.; Egelman, E.H.; Costa, T.R.D. Cryoelectron-microscopy structure of the enteropathogenic Escherichia coli type III secretion system EspA filament. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Hou, Y.; Dai, W.; Xie, R.; Marquez-Lago, T.T.; Leier, A.; Zhou, T.; Torres, V.; Hay, I.; et al. BastionHub: A universal platform for integrating and analyzing substrates secreted by Gram-negative bacteria. Nucleic Acids Res. 2021, 49, D651–D659. [Google Scholar] [CrossRef]
- Klose, D.P.; Wallace, B.A.; Janes, R.W. 2Struc: The secondary structure server. Bioinformatics 2010, 26, 2624–2625. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Voss, N.R.; Gerstein, M. 3V: Cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010, 38, W555. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Function | T3SEs | Specific Host Target |
|---|---|---|
| Gatekeepers | Chlamydia pneumoniae CopN Shigella MxiC, Yersinia YopN/TyeA (heterodimer) | Scc3, tubulin |
| Membrane docking | Pseudomonas aeruginosa ExoU | membrane |
| GAP activity | Salmonella SptP, P. aeruginosa ExoS/T E. coli EspG, EspG2, Shigella flexneri VirA, Citrobacter rodentium EspI | Rac1 Rab |
| GEF activity | Salmonella SPI-1 SopE, SopE2, E. coli MAP Burkholderia BopE, Salmonella SPI-2 SifA, Shigella IpgB1, IpgB2 | Cdc42 |
| GDI activity | Yersinia YopO/YpkA | Rac1 |
| PTM activity | Vibrio parahaemolyticus VopS (AMPylation) P. syringae AvrB E. coli NleB, Salmonella Ssek3, Ssek1 (Arg-GlcNAcylation) | Rho, Rac, Cdc42 RIN4 DD proteins |
| Transcription activation | Xanthomonas AvrBs3, PthXo1, Hax3, Burkholderia rhizoxinica Bud | DNA |
| Host immunity activation in resistant plants | Pseudomonas syringae AvrRps4 (suppress immunity in susceptible plants, chloroplast localization is required) Pseudomonas AvrPto, AvrPtoB Xanthomonas AvrRxo1-ORF1 (T4-polynucleotide kinase) Xanthomonas campestris XopQ (suppress immunity in susceptible plants) | Pto * (Api) Rxo1 * Roq1 * (14–3-3) |
| Zinc metalloproteases | EPEC NleC, EHEC NleD, Salmonella enterica GtgA, GogA, PipA Ralstonia solanacearum RipAX2 | NF-κB |
| Kinases | EPEC NleH1, EHEC NleH2, Shigella OspG | NF-κB |
| Novel E3 Ligases | Shigella IpaH, Salmonella SspH | Ub network |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazi, A.D.; Kokkinidis, M.; Fadouloglou, V.E. α-Helices in the Type III Secretion Effectors: A Prevalent Feature with Versatile Roles. Int. J. Mol. Sci. 2021, 22, 5412. https://doi.org/10.3390/ijms22115412
Gazi AD, Kokkinidis M, Fadouloglou VE. α-Helices in the Type III Secretion Effectors: A Prevalent Feature with Versatile Roles. International Journal of Molecular Sciences. 2021; 22(11):5412. https://doi.org/10.3390/ijms22115412
Chicago/Turabian StyleGazi, Anastasia D., Michael Kokkinidis, and Vasiliki E. Fadouloglou. 2021. "α-Helices in the Type III Secretion Effectors: A Prevalent Feature with Versatile Roles" International Journal of Molecular Sciences 22, no. 11: 5412. https://doi.org/10.3390/ijms22115412
APA StyleGazi, A. D., Kokkinidis, M., & Fadouloglou, V. E. (2021). α-Helices in the Type III Secretion Effectors: A Prevalent Feature with Versatile Roles. International Journal of Molecular Sciences, 22(11), 5412. https://doi.org/10.3390/ijms22115412