
The Structure–Properties–Cytotoxicity Interplay: A Crucial Pathway to Determining Graphene Oxide Biocompatibility

 , , , ,
, , , , 
Abstract
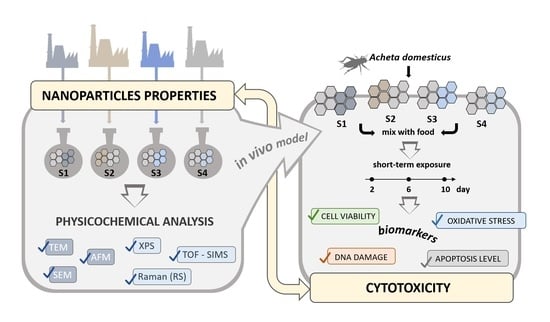
1. Introduction
- Graphene oxide from various sources does not cause significant differences in cell viability, oxidative stress level, apoptosis level, and DNA damage in A. domesticus during the 10-day exposure. The effect of all types of GO is similar, and the materials do not differ significantly in terms of physicochemical properties and toxicity.
- The selected physicochemical properties of GO are significant, and the materials significantly differentiate cell viability, the level of oxidative stress, the degree of apoptosis, and DNA damage at various time points during 10 days of exposure in A. domesticus. This may be due to the GO flakes’ size, aggregation potential, suspension stability, degree of oxidation, or the number of surface defects.
2. Results
2.1. Properties of Graphene Oxide Nanoparticles
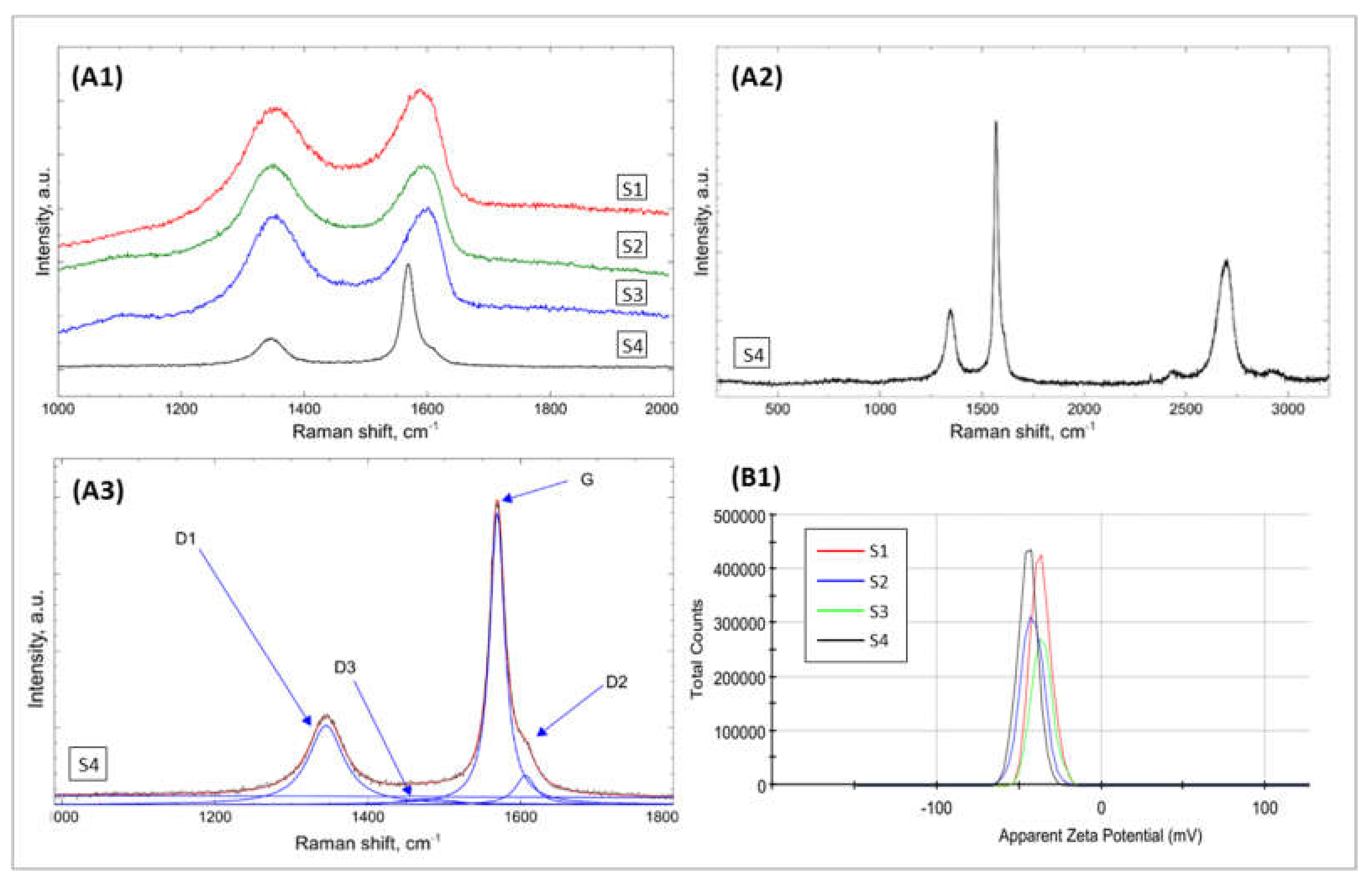
2.1.1. Morphological and Structural Analysis of GO Samples
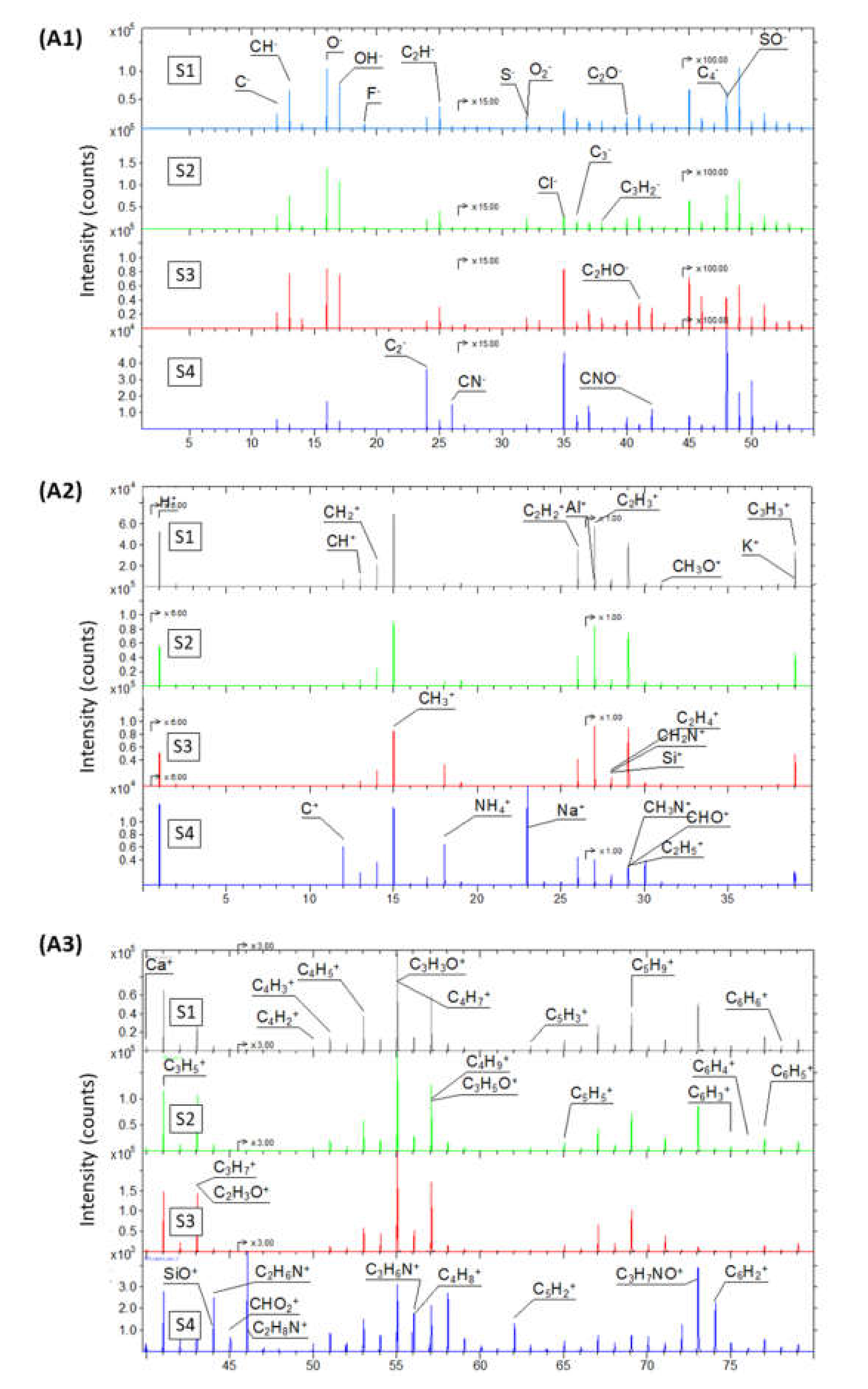
2.1.2. Chemical Composition Analysis of GO Samples
2.2. Cytotoxicity Analysis
2.2.1. Cell Viability and Oxidative Stress
2.2.2. Apoptosis Level in the Cells
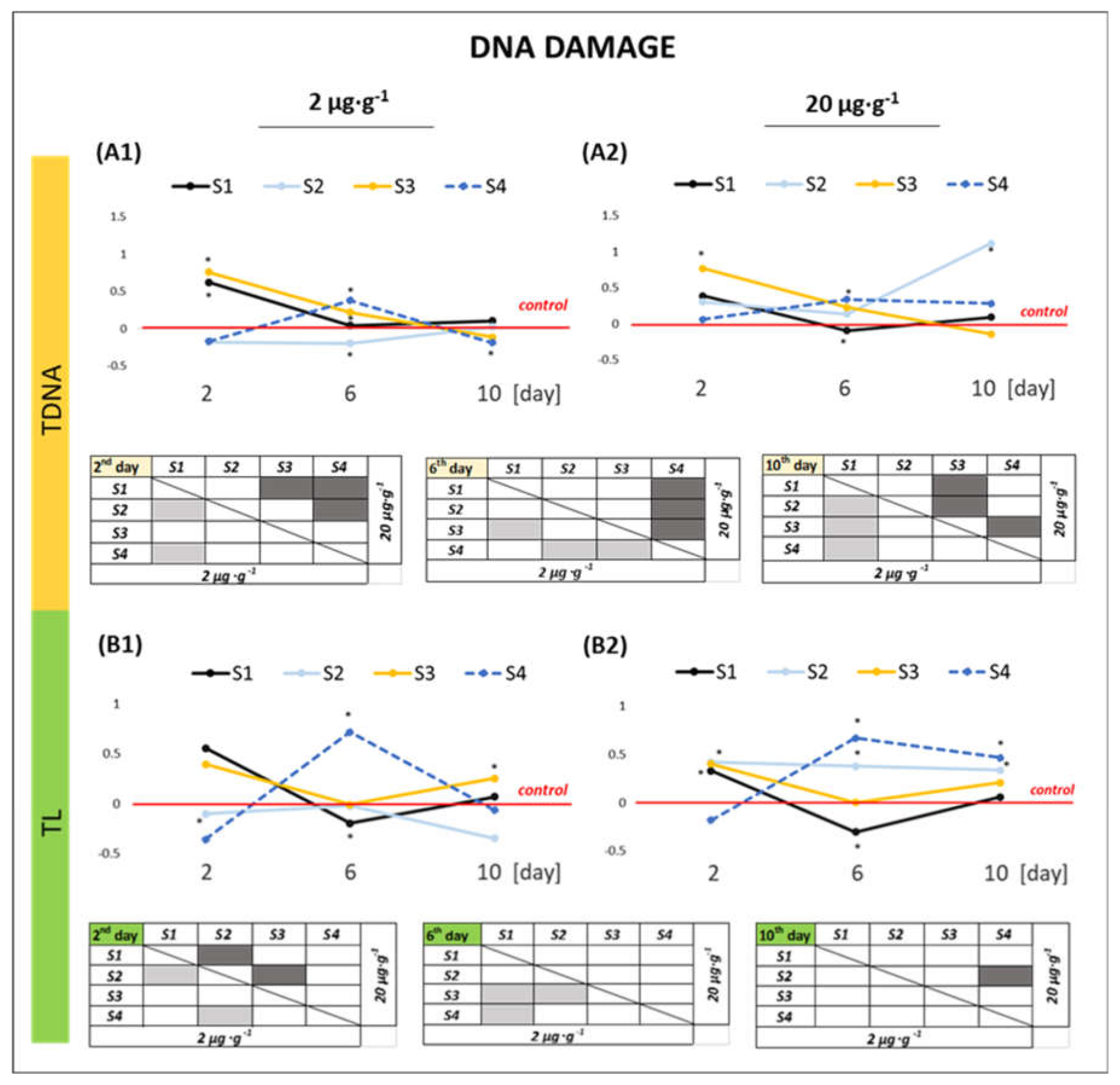
2.2.3. DNA Damage
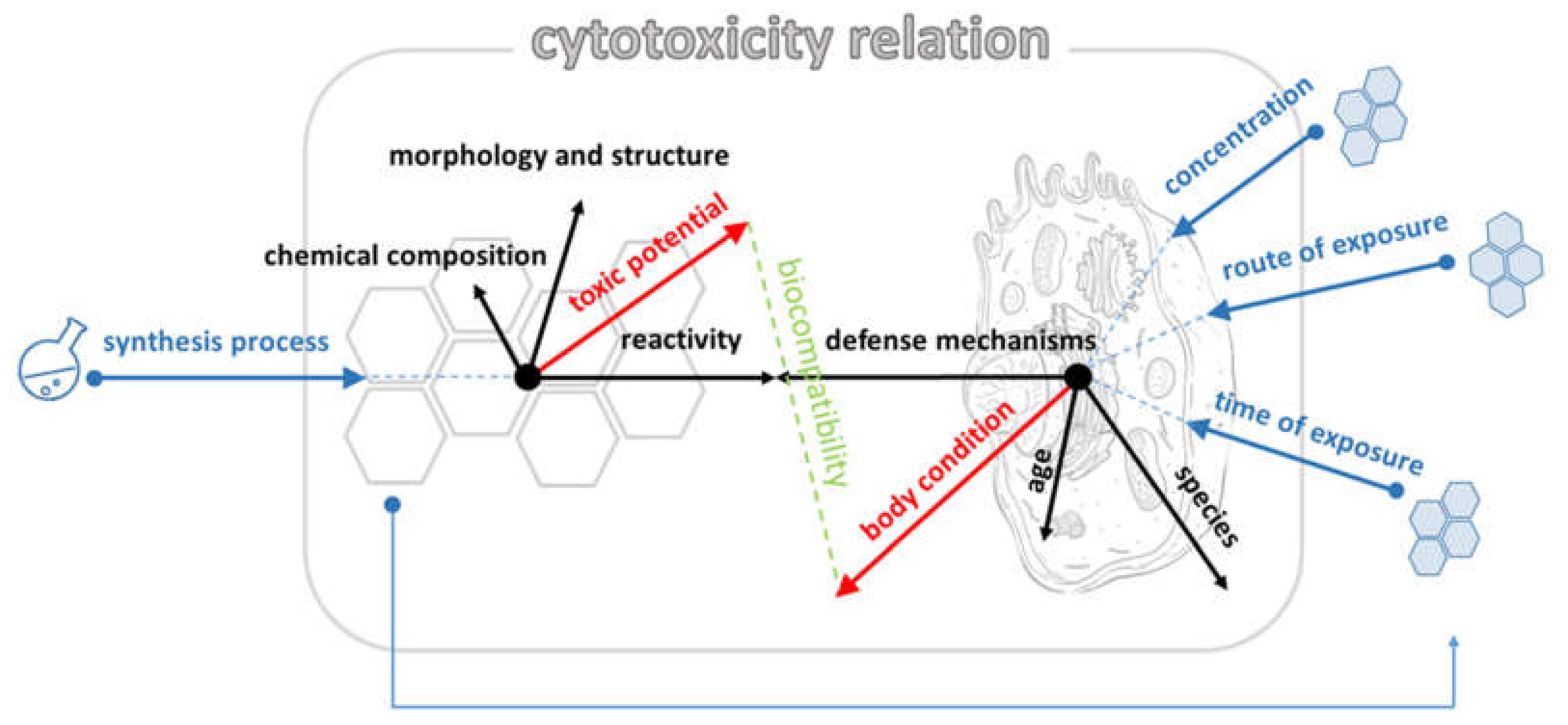
3. Discussion
3.1. Effect of GO Samples on Cell Viability and Parameters of Apoptosis
3.2. Effect of GO Samples on the Level of Oxidative Stress
3.3. Effect of GO Samples on the Genetic Material of the Cell
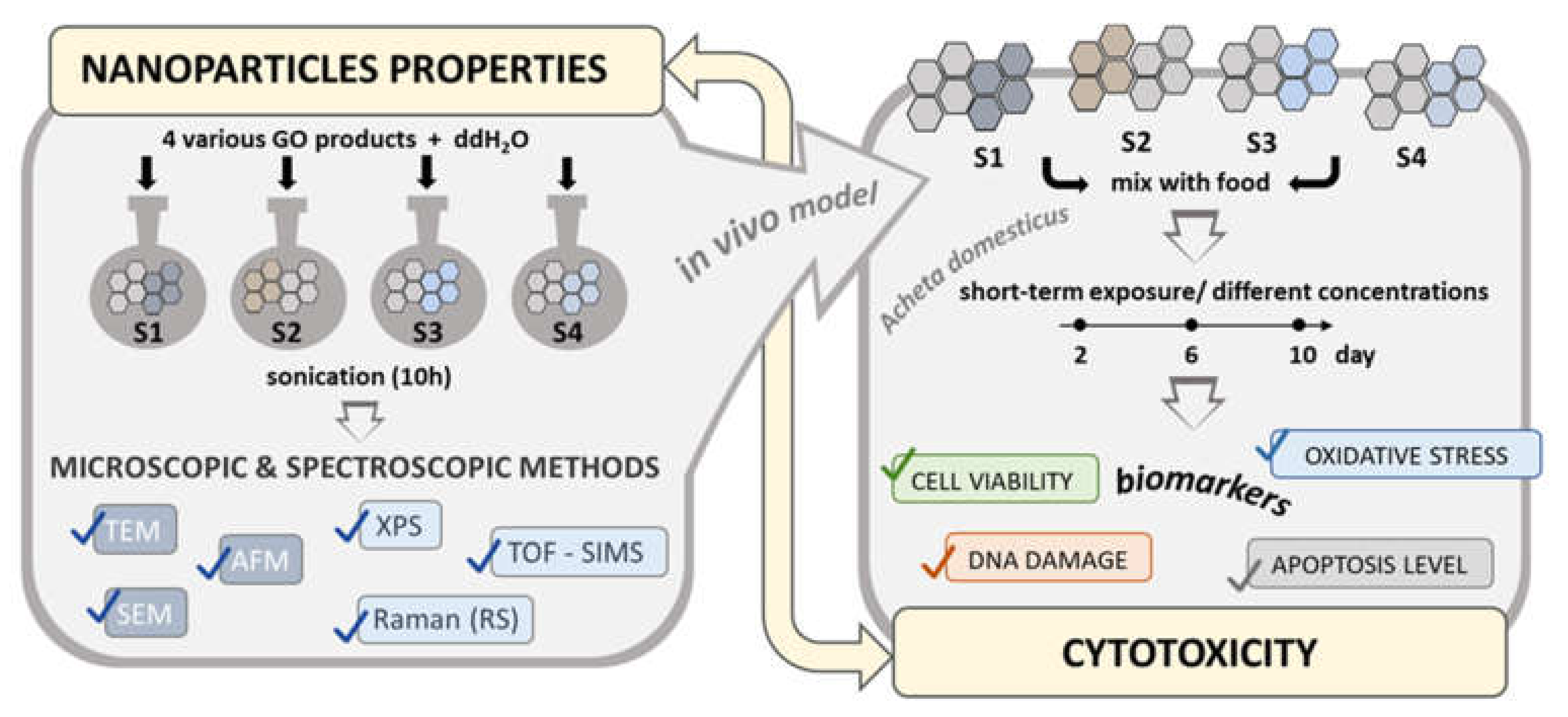
4. Materials and Methods
4.1. Experimental Model
4.2. GO Suspensions Preparation
4.3. Physicochemical Characterization of GO
4.4. Cytotoxicity Evaluation
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bands | Raman Shift/cm−1 | Structure Characteristic for a Vibration Mode |
|---|---|---|
| G | ≈1580 | ideal graphitic lattice |
| D1 (D) | ≈1350 | disordered graphitic lattice—graphene layer edges |
| D2 | ≈1620 | disordered graphitic lattice—surface graphene layers |
| D3 | ≈1500 | amorphous carbon |
| D4 | ≈1200 | disordered graphitic lattice polyenes, ionic impurities |
| Fitting Results | S1 | S2 | S3 | S4 | |
|---|---|---|---|---|---|
| D1 | Location | 1351 | 1350 | 1352 | 1345 |
| FWHM | 140 | 120 | 114 | 56 | |
| I | 62 | 60 | 62 | 35 | |
| G | Location | 1573 | 1578 | 1581 | 1569 |
| FWHM | 68 | 60 | 54 | 23 | |
| I | 22 | 20 | 19 | 55 | |
| D3 | Location | 1496 | 1513 | 1524 | 1478 |
| FWHM | 123 | 140 | 128 | 82 | |
| I | 7 | 9 | 8 | 2 | |
| D2 | Location | 1610 | 1610 | 1610 | 1606 |
| FWHM | 38 | 35 | 33 | 31 | |
| I | 9 | 10 | 11 | 7 | |
| Sample | Line |
Peak Position [eV] | Percentage Contribution | Atomic Concentration | Assignment |
|---|---|---|---|---|---|
| S1 | N1s | 399.73 | 53.53 | 0.36 | C-NH-C |
| N1s | 401.74 | 46.47 | 0.31 | pyrrolic N-H | |
| S2p3/2 | 168.26 | 100 | 0.50 | sulfate or sulfoamide S-O4 | |
| Si2p3/2 | 101.93 | 55.95 | 0.33 | SiOx | |
| Si2p3/2 | 103.79 | 44.05 | 0.26 | SiO2 | |
| O1s | 531.11 | 7.08 | 2.00 | O-C=O | |
| O1s | 532.52 | 86.32 | 24.41 | C=O | |
| O1s | 533.96 | 6.06 | 1.71 | C-O-C | |
| S2 | N1s | 399.57 | 36.35 | 0.13 | C-N |
| N1s | 401.84 | 63.65 | 0.24 | graphitic nitrogen | |
| S2p3/2 | 168.23 | 89.34 | 0.24 | sulfate or sulfoamide S-O4 | |
| S2p3/2 | 170.52 | 10.36 | 0.03 | bisulfate group (HSO4−) | |
| Si2p3/2 | 101.92 | 100 | 0.32 | SiOx | |
| O1s | 531.44 | 12.12 | 3.43 | O-C=O | |
| O1s | 532.62 | 65.92 | 18.67 | C-OH | |
| O1s | 533.91 | 21.96 | 6.22 | C-O-C | |
| S3 | N1s | 400.25 | 40.75 | 0.55 | pyrrolic nitrogen |
| N1s | 401.56 | 59.25 | 0.81 | graphitic nitrogen | |
| S2p3/2 | 168.76 | 81.82 | 2.93 | sulfate | |
| S2p3/2 | 169.54 | 18.18 | 0.65 | C-S-C | |
| Si2p3/2 | 102.62 | 100 | 0.12 | silicate | |
| O1s | 531.44 | 10.75 | 2.86 | O-C=O | |
| O1s | 532.46 | 67.20 | 17.90 | C=O | |
| O1s | 533.76 | 22.04 | 5.87 | C-O-C | |
| S4 | N1s | 400.31 | 90.09 | 0.63 | pyrrolic nitrogen |
| N1s | 402.45 | 9.91 | 0.07 | N-O | |
| S2p3/2 | 163.77 | 7.69 | 0.01 | reduced sulfide -S- | |
| S2p3/2 | 166.43 | 37.79 | 0.03 | -SOn- | |
| S2p3/2 | 168.86 | 54.52 | 0.04 | SOx | |
| Si2p3/2 | 99.41 | 43.55 | 0.19 | bulk Si | |
| Si2p3/2 | 101.76 | 30.29 | 0.13 | SiOx | |
| Si2p3/2 | 103.89 | 26.07 | 0.11 | SiO2 | |
| O1s | 531.44 | 17.21 | 0.90 | O-C=O | |
| O1s | 532.64 | 52.69 | 2.76 | C-OH | |
| O1s | 533.68 | 14.35 | 0.75 | O-H or C-O-C | |
| O1s | 534.46 | 15.76 | 0.82 | C(=O)(O–)2 |
References
- Kuhlbusch, T.A.J. Black Carbon and the Carbon Cycle. Science 1998, 280, 1903–1904. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and Graphene Oxide: Synthesis, Properties, and Applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Morales-Narváez, E.; Merkoçi, A. Graphene oxide as an optical biosensing platform. Adv. Mater. 2012, 24, 3298–3308. [Google Scholar] [CrossRef] [PubMed]
- Dai, L. Functionalization of graphene for efficient energy conversion and storage. Acc. Chem. Res. 2013, 46, 31–42. [Google Scholar] [CrossRef]
- Behbudi, G. Mini review of Graphene Oxide for medical detection and applications. Adv. Appl. NanoBio-Technol. 2020, 1, 63–66. [Google Scholar]
- Nan, Z.; Hao, C.; Ye, X.; Feng, Y.; Sun, R. Interaction of graphene oxide with bovine serum albumin: A fluorescence quenching study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 210, 348–354. [Google Scholar] [CrossRef]
- Kong, Z.; Daab, M.; Yano, H.; Huang, H.; Breu, J.; Sasaki, T.; Nguyen, S.T.; Huang, J. Visualizing Transparent 2D Sheets by Fluorescence Quenching Microscopy. Small Methods 2020, 4, 2000036. [Google Scholar] [CrossRef]
- Georgakilas, V.; Otyepka, M.; Bourlinos, A.B.; Chandra, V.; Kim, N.; Kemp, K.C.; Hobza, P.; Zboril, R.; Kim, K.S. Functionalization of Graphene: Covalent and Non-Covalent Approaches, Derivatives and Applications. Chem. Rev. 2012, 112, 6156–6214. [Google Scholar] [CrossRef]
- Tissera, N.D.; Wijesena, R.N.; Perera, J.R.; de Silva, K.M.N.; Amaratunge, G.A.J. Hydrophobic cotton textile surfaces using an amphiphilic graphene oxide (GO) coating. Appl. Surf. Sci. 2015, 324, 455–463. [Google Scholar] [CrossRef]
- Lopez, A.; Liu, J. Covalent and Noncovalent Functionalization of Graphene Oxide with DNA for Smart Sensing. Adv. Intell. Syst. 2020, 2, 2000123. [Google Scholar] [CrossRef]
- Kuila, T.; Bose, S.; Mishra, A.K.; Khanra, P.; Kim, N.H.; Lee, J.H. Chemical functionalization of graphene and its applications. Prog. Mater. Sci. 2012, 57, 1061–1105. [Google Scholar] [CrossRef]
- Nebol’sin, V.A.; Galstyan, V.; Silina, Y.E. Graphene oxide and its chemical nature: Multi-stage interactions between the oxygen and graphene. Surf. Interfaces 2020, 21, 100763. [Google Scholar] [CrossRef]
- Nishina, Y.; Eigler, S. Chemical and electrochemical synthesis of graphene oxide—A generalized view. Nanoscale 2020, 12, 12731–12740. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Tour, J.M. Mechanism of Graphene Oxide Formation. ACS Nano 2014, 8, 3060–3068. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, B.; Bulin, C.; Li, R.; Xing, R. High-efficient Synthesis of Graphene Oxide Based on Improved Hummers Method. Sci. Rep. 2016, 6, 36143. [Google Scholar] [CrossRef]
- Kozhemyakina, N.V.; Eigler, S.; Dinnebier, R.E.; Inayat, A.; Schwieger, W.; Hirsch, A. Effect of the Structure and Morphology of Natural, Synthetic and Post-processed Graphites on Their Dispersibility and Electronic Properties. Fullerenes, Nanotub. Carbon Nanostructures 2013, 21, 804–823. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Walter, J.; Nacken, T.J.; Damm, C.; Thajudeen, T.; Eigler, S.; Peukert, W. Determination of the lateral dimension of graphene oxide nanosheets using analytical ultracentrifugation. Small 2015, 11, 814–825. [Google Scholar] [CrossRef]
- Dimiev, A.; Kosynkin, D.V.; Alemany, L.B.; Chaguine, P.; Tour, J.M. Pristine Graphite Oxide. J. Am. Chem. Soc. 2012, 134, 2815–2822. [Google Scholar] [CrossRef]
- Morimoto, N.; Suzuki, H.; Takeuchi, Y.; Kawaguchi, S.; Kunisu, M.; Bielawski, C.W.; Nishina, Y. Real-Time, In Situ Monitoring of the Oxidation of Graphite: Lessons Learned. Chem. Mater. 2017, 29, 2150–2156. [Google Scholar] [CrossRef]
- Qi, Y.; Chen, W.; Liu, F.; Liu, J.; Zhang, T.; Chen, W. Aggregation morphology is a key factor determining protein adsorption on graphene oxide and reduced graphene oxide nanomaterials. Environ. Sci. Nano 2019, 6, 1303–1309. [Google Scholar] [CrossRef]
- Park, S.; Ruoff, R.S. Chemical methods for the production of graphenes. Nat. Nanotechnol. 2009, 4, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Whitby, R.L.D. Chemical Control of Graphene Architecture: Tailoring Shape and Properties. ACS Nano 2014, 8, 9733–9754. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Liu, X.; Zhang, W.; Zeng, Z.; Liu, Z.; Zhang, C.; Liu, Y.; Shao, B.; Liang, Q.; Tang, W.; et al. Advances in the application, toxicity and degradation of carbon nanomaterials in environment: A review. Environ. Int. 2020, 134, 105298. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhang, S.; Huang, T.; Cui, F.; Xing, B. Effects of pH and electrolytes on the sheet-to-sheet aggregation mode of graphene oxide in aqueous solutions. Environ. Sci. Nano 2020, 7, 984–995. [Google Scholar] [CrossRef]
- Dziewięcka, M.; Flasz, B.; Rost-Roszkowska, M.; Kędziorski, A.; Kochanowicz, A.; Augustyniak, M. Graphene oxide as a new anthropogenic stress factor—Multigenerational study at the molecular, cellular, individual and population level of Acheta domesticus. J. Hazard. Mater. 2020, 396. [Google Scholar] [CrossRef] [PubMed]
- Dziewięcka, M.; Witas, P.; Karpeta-Kaczmarek, J.; Kwaśniewska, J.; Flasz, B.; Balin, K.; Augustyniak, M. Reduced fecundity and cellular changes in Acheta domesticus after multigenerational exposure to graphene oxide nanoparticles in food. Sci. Total Environ. 2018, 635, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Dziewięcka, M.; Karpeta-Kaczmarek, J.; Augustyniak, M.; Majchrzycki, Ł.; Augustyniak-Jabłokow, M.A. Evaluation of In Vivo graphene oxide toxicity for Acheta domesticus in relation to nanomaterial purity and time passed from the exposure. J. Hazard. Mater. 2015, 305, 30–40. [Google Scholar] [CrossRef]
- Dziewięcka, M.; Karpeta-Kaczmarek, J.; Augustyniak, M.; Rost-Roszkowska, M. Short-term In Vivo exposure to graphene oxide can cause damage to the gut and testis. J. Hazard. Mater. 2017, 328, 80–89. [Google Scholar] [CrossRef]
- Karpeta-Kaczmarek, J.; Dziewięcka, M.; Augustyniak, M.; Rost-Roszkowska, M. Effects of short-term exposure of Acheta domesticus to nanodiamonds in food: DNA damage but no histological alteration in tissues. Carbon 2016, 110, 458–468. [Google Scholar] [CrossRef]
- Karpeta-Kaczmarek, J.; Andrzej, K.; Augustyniak-jab, M.A.; Dziewi, M.; Augustyniak, M. Chronic toxicity of nanodiamonds can disturb development and reproduction of Acheta domesticus L. Environ. Res. 2018, 166, 602–609. [Google Scholar] [CrossRef]
- Flasz, B.; Dziewięcka, M.; Kędziorski, A.; Tarnawska, M.; Augustyniak, M. Multigenerational graphene oxide intoxication results in reproduction disorders at the molecular level of vitellogenin protein expression in Acheta domesticus. Chemosphere. 2021, 280, 130772. [Google Scholar] [CrossRef]
- Karpeta-Kaczmarek, J.; Augustyniak, M.; Rost-Roszkowska, M. Ultrastructure of the gut epithelium in Acheta domesticus after long-term exposure to nanodiamonds supplied with food. Arthropod Struct. Dev. 2016, 45, 253–264. [Google Scholar] [CrossRef]
- Flasz, B.; Dziewięcka, M.; Kędziorski, A.; Tarnawska, M.; Augustyniak, M. Vitellogenin expression, DNA damage, health status of cells and catalase activity in Acheta domesticus selected according to their longevity after graphene oxide treatment. Sci. Total Environ. 2020, 737. [Google Scholar] [CrossRef]
- Arif, T.; Colas, G.; Filleter, T. Effect of Humidity and Water Intercalation on the Tribological Behavior of Graphene and Graphene Oxide. ACS Appl. Mater. Interfaces 2018, 10, 22537–22544. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology. X-ray Photoelectron Spectroscopy Database. Version 4.1. Available online: http://srdata.nist.gov/xps/ (accessed on 17 March 2021).
- Wang, K.; Ruan, J.; Song, H.; Zhang, J.; Wo, Y.; Guo, S.; Cui, D. Biocompatibility of Graphene Oxide. Nanoscale Res. Lett. 2011, 6, 8. [Google Scholar] [CrossRef]
- Jastrzebska, A.M.; Olszyna, A.R. The ecotoxicity of graphene family materials: Current status, knowledge gaps and future needs. J. Nanoparticle Res. 2015, 17. [Google Scholar] [CrossRef]
- Akhavan, O.; Ghaderi, E.; Akhavan, A. Size-dependent genotoxicity of graphene nanoplatelets in human stem cells. Biomaterials 2012, 33, 8017–8025. [Google Scholar] [CrossRef]
- Nirmal, N.K.; Awasthi, K.K.; John, P.J. Effects of Nano-Graphene Oxide on Testis, Epididymis and Fertility of Wistar Rats. Basic Clin. Pharmacol. Toxicol. 2017, 121, 202–210. [Google Scholar] [CrossRef]
- Lv, X.; Yang, Y.; Tao, Y.; Jiang, Y.; Chen, B.; Zhu, X.; Cai, Z.; Li, B. A mechanism study on toxicity of graphene oxide to Daphnia magna: Direct link between bioaccumulation and oxidative stress. Environ. Pollut. 2018, 234, 953–959. [Google Scholar] [CrossRef]
- Albert, E.L.; Che Abdullah, C.A.; Shiroshaki, Y. Synthesis and characterization of graphene oxide functionalized with magnetic nanoparticle via simple emulsion method. Results Phys. 2018, 11, 944–950. [Google Scholar] [CrossRef]
- Muniyalakshmi, M.; Sethuraman, K.; Silambarasan, D. Synthesis and characterization of graphene oxide nanosheets. Mater. Today Proc. 2020, 21, 408–410. [Google Scholar] [CrossRef]
- Subrahmanyam, K.S.; Vivekchand, S.R.C.; Govindaraj, A.; Rao, C.N.R. A study of graphenes prepared by different methods: Characterization, properties and solubilization. J. Mater. Chem. 2008, 18, 1517–1523. [Google Scholar] [CrossRef]
- Tabish, T.A.; Pranjol, M.Z.I.; Hayat, H.; Rahat, A.A.M.; Abdullah, T.M.; Whatmore, J.L.; Zhang, S. In Vitro toxic effects of reduced graphene oxide nanosheets on lung cancer cells. Nanotechnology 2017, 28, 504001. [Google Scholar] [CrossRef]
- Yang, K.; Gong, H.; Shi, X.; Wan, J.; Zhang, Y.; Liu, Z. In Vivo biodistribution and toxicology of functionalized nano-graphene oxide in mice after oral and intraperitoneal administration. Biomaterials 2013, 34, 2787–2795. [Google Scholar] [CrossRef]
- Tu, Z.; Achazi, K.; Schulz, A.; Mülhaupt, R.; Thierbach, S.; Rühl, E.; Adeli, M.; Haag, R. Combination of Surface Charge and Size Controls the Cellular Uptake of Functionalized Graphene Sheets. Adv. Funct. Mater. 2017, 27. [Google Scholar] [CrossRef]
- Iversen, T.-G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Canton, I.; Battaglia, G. Endocytosis at the nanoscale. Chem. Soc. Rev. 2012, 41, 2718–2739. [Google Scholar] [CrossRef]
- Shang, L.; Nienhaus, K.; Jiang, X.; Yang, L.; Landfester, K.; Mailänder, V.; Simmet, T.; Nienhaus, G.U. Nanoparticle interactions with live cells: Quantitative fluorescence microscopy of nanoparticle size effects. Beilstein J. Nanotechnol. 2014, 5, 2388–2397. [Google Scholar] [CrossRef]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef]
- Wang, A.; Pu, K.; Dong, B.; Liu, Y.; Zhang, L.; Zhang, Z.; Duan, W.; Zhu, Y. Role of surface charge and oxidative stress in cytotoxicity and genotoxicity of graphene oxide towards human lung fibroblast cells. J. Appl. Toxicol. 2013, 33, 1156–1164. [Google Scholar] [CrossRef]
- Manke, A.; Wang, L.; Rojanasakul, Y. Mechanisms of nanoparticle-induced oxidative stress and toxicity. Biomed. Res. Int. 2013, 2013, 942916. [Google Scholar] [CrossRef]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, P.; Zhou, Z.; Wei, T. Interactions of graphene with mammalian cells: Molecular mechanisms and biomedical insights. Adv. Drug Deliv. Rev. 2016, 105, 145–162. [Google Scholar] [CrossRef]
- Jia, P.; Sun, T.; Junaid, M.; Yang, L.; Ma, Y.; Cui, Z.; Wei, D.; Shi, H.; Pei, D. Nanotoxicity of different sizes of graphene (G) and graphene oxide (GO) In Vitro and In Vivo. Environ. Pollut. 2019, 247, 595–606. [Google Scholar] [CrossRef]
- Xia, T.; Kovochich, M.; Brant, J.; Hotze, M.; Sempf, J.; Oberley, T.; Sioutas, C.; Yeh, J.I.; Wiesner, M.R.; Nel, A.E. Comparison of the abilities of ambient and manufactured nanoparticles to induce cellular toxicity according to an oxidative stress paradigm. Nano Lett. 2006, 6, 1794–1807. [Google Scholar] [CrossRef]
- Knaapen, A.M.; Borm, P.J.A.; Albrecht, C.; Schins, R.P.F. Inhaled particles and lung cancer. Part A Mech. Int. J. Cancer 2004, 109, 799–809. [Google Scholar] [CrossRef]
- Hu, X.; Yu, Y.; Hou, W.; Zhou, J.; Song, L. Effects of particle size and pH value on the hydrophilicity of graphene oxide. Appl. Surf. Sci. 2013, 273, 118–121. [Google Scholar] [CrossRef]
- Magdolenova, Z.; Collins, A.; Kumar, A.; Dhawan, A.; Stone, V.; Dusinska, M. Mechanisms of genotoxicity. A review of In Vitro and In Vivo studies with engineered nanoparticles. Nanotoxicology 2014, 8, 233–278. [Google Scholar] [CrossRef]
- Hackenberg, S.; Friehs, G.; Froelich, K.; Ginzkey, C.; Koehler, C.; Scherzed, A.; Burghartz, M.; Hagen, R.; Kleinsasser, N. Intracellular distribution, geno- and cytotoxic effects of nanosized titanium dioxide particles in the anatase crystal phase on human nasal mucosa cells. Toxicol. Lett. 2010, 195, 9–14. [Google Scholar] [CrossRef]
- Wilson, N.R.; Pandey, P.A.; Beanland, R.; Rourke, J.P.; Lupo, U.; Rowlands, G.; Römer, R.A. On the structure and topography of free-standing chemically modified graphene. New J. Phys. 2010, 12, 125010. [Google Scholar] [CrossRef]
- Jung, I.; Field, D.A.; Clark, N.J.; Zhu, Y.; Yang, D.; Piner, R.D.; Stankovich, S.; Dikin, D.A.; Geisler, H.; Ventrice, C.A.; et al. Reduction Kinetics of Graphene Oxide Determined by Electrical Transport Measurements and Temperature Programmed Desorption. J. Phys. Chem. C 2009, 113, 18480–18486. [Google Scholar] [CrossRef]
- Den Boer, D.; Weis, J.G.; Zuniga, C.A.; Sydlik, S.A.; Swager, T.M. Apparent Roughness as Indicator of (Local) Deoxygenation of Graphene Oxide. Chem. Mater. 2014, 26, 4849–4855. [Google Scholar] [CrossRef]
- Tuinstra, F.; Koenig, J.L. Raman Spectrum of Graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef]
- Thomsen, C.; Reich, S. Double resonant raman scattering in graphite. Phys. Rev. Lett. 2000, 85, 5214–5217. [Google Scholar] [CrossRef]
- Knight, D.S.; White, W.B. Characterization of diamond films by Raman spectroscopy. J. Mater. Res. 1989, 4, 385–393. [Google Scholar] [CrossRef]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Qureshi, U.A.; Khatri, Z.; Ahmed, F.; Ibupoto, A.S.; Khatri, M.; Mahar, F.K.; Brohi, R.Z.; Kim, I.S. Highly efficient and robust electrospun nanofibers for selective removal of acid dye. J. Mol. Liq. 2017, 244, 478–488. [Google Scholar] [CrossRef]
- Benninghoven, A. Chemical Analysis of Inorganic and Organic Surfaces and Thin Films by Static Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS). Angew. Chem. Int. Ed. Engl. 1994, 33, 1023–1043. [Google Scholar] [CrossRef]
- Xu, Z.; Gao, H.; Guoxin, H. Solution-based synthesis and characterization of a silver nanoparticle-graphene hybrid film. Carbon 2011, 49, 4731–4738. [Google Scholar] [CrossRef]
- Daniyal, W.M.E.M.M.; Fen, Y.W.; Saleviter, S.; Chanlek, N.; Nakajima, H.; Abdullah, J.; Yusof, N.A. X-ray Photoelectron Spectroscopy Analysis of Chitosan-Graphene Oxide-Based Composite Thin Films for Potential Optical Sensing Applications. Polymers 2021, 13, 478. [Google Scholar] [CrossRef]
- Tarnawska, D.; Balin, K.; Jastrzebska, M.; Talik, A.; Wrzalik, R. Physicochemical analysis of sediments formed on the surface of hydrophilic intraocular lens after Descemet’s stripping endothelial keratoplasty. Materials 2020, 13, 4145. [Google Scholar] [CrossRef]
- Karpeta-Kaczmarek, J.; Dziewięcka, M.; Augustyniak, M.; Rost-Roszkowska, M.; Pawlyta, M. Oxidative stress and genotoxic effects of diamond nanoparticles. Environ. Res. 2016, 148, 264–272. [Google Scholar] [CrossRef]
- Augustyniak, M.; Gladysz, M.; Dziewięcka, M. Mutation Research/Reviews in Mutation Research. The Comet assay in insects—Status. Prospect. Benefits Sci. 2016, 767, 67–76. [Google Scholar] [CrossRef]











| Element | Atomic Concentration [at.%] | |||
|---|---|---|---|---|
| S1 | S2 | S3 | S4 | |
| C | 69.88 | 70.72 | 68.32 | 93.57 |
| N | 0.67 | 0.37 | 1.36 | 0.70 |
| O | 28.28 | 28.32 | 26.63 | 5.23 |
| Al | 0.08 | - | - | - |
| Si | 0.59 | 0.32 | 0.12 | 0.43 |
| S | 0.50 | 0.27 | 3.58 | 0.07 |
| Ratio of atomic concentrations C/O | ||||
| C/O | 2.47 | 2.5 | 2.57 | 17.89 |
| Sample | Peak Position | Percentage Contribution | Atomic Concentration | Assignment |
|---|---|---|---|---|
| S1 | 284.76 | 51.61 | 35.55 | C-C |
| 286.74 | 38.39 | 26.44 | C-O | |
| 288.48 | 10.00 | 6.89 | C=O | |
| S2 | 284.79 | 36.05 | 25.49 | C-C |
| 286.75 | 52.26 | 36.96 | C-O | |
| 288.40 | 10.69 | 7.56 | C=O | |
| 290.30 | 0.99 | 0.70 | C=O (OH) | |
| S3 | 284.79 | 59.37 | 40.56 | C-C |
| 286.76 | 31.00 | 21.18 | C-O | |
| 288.74 | 9.62 | 6.57 | C=O | |
| S4 | 284.03 | 3.87 | 3.59 | C=N |
| 284.81 | 54.82 | 51.30 | C-C | |
| 285.44 | 13.99 | 13.09 | C-OH | |
| 286.11 | 8.66 | 8.10 | C-N | |
| 287.06 | 6.51 | 6.08 | carbonyl>C=O | |
| 289.03 | 8.23 | 7.70 | O-C=O | |
| 291.56 | 3.92 | 3.67 | HO-C=O or π–π* satellite |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziewięcka, M.; Pawlyta, M.; Majchrzycki, Ł.; Balin, K.; Barteczko, S.; Czerkawska, M.; Augustyniak, M. The Structure–Properties–Cytotoxicity Interplay: A Crucial Pathway to Determining Graphene Oxide Biocompatibility. Int. J. Mol. Sci. 2021, 22, 5401. https://doi.org/10.3390/ijms22105401
Dziewięcka M, Pawlyta M, Majchrzycki Ł, Balin K, Barteczko S, Czerkawska M, Augustyniak M. The Structure–Properties–Cytotoxicity Interplay: A Crucial Pathway to Determining Graphene Oxide Biocompatibility. International Journal of Molecular Sciences. 2021; 22(10):5401. https://doi.org/10.3390/ijms22105401
Chicago/Turabian StyleDziewięcka, Marta, Mirosława Pawlyta, Łukasz Majchrzycki, Katarzyna Balin, Sylwia Barteczko, Martyna Czerkawska, and Maria Augustyniak. 2021. "The Structure–Properties–Cytotoxicity Interplay: A Crucial Pathway to Determining Graphene Oxide Biocompatibility" International Journal of Molecular Sciences 22, no. 10: 5401. https://doi.org/10.3390/ijms22105401
APA StyleDziewięcka, M., Pawlyta, M., Majchrzycki, Ł., Balin, K., Barteczko, S., Czerkawska, M., & Augustyniak, M. (2021). The Structure–Properties–Cytotoxicity Interplay: A Crucial Pathway to Determining Graphene Oxide Biocompatibility. International Journal of Molecular Sciences, 22(10), 5401. https://doi.org/10.3390/ijms22105401