
Viruses and Skin Cancer

 , ,
, , 
Abstract
1. Introduction
2. Human Papillomavirus
2.1. Historical Perspective
2.2. Structure and Pathogenicity Mechanisms
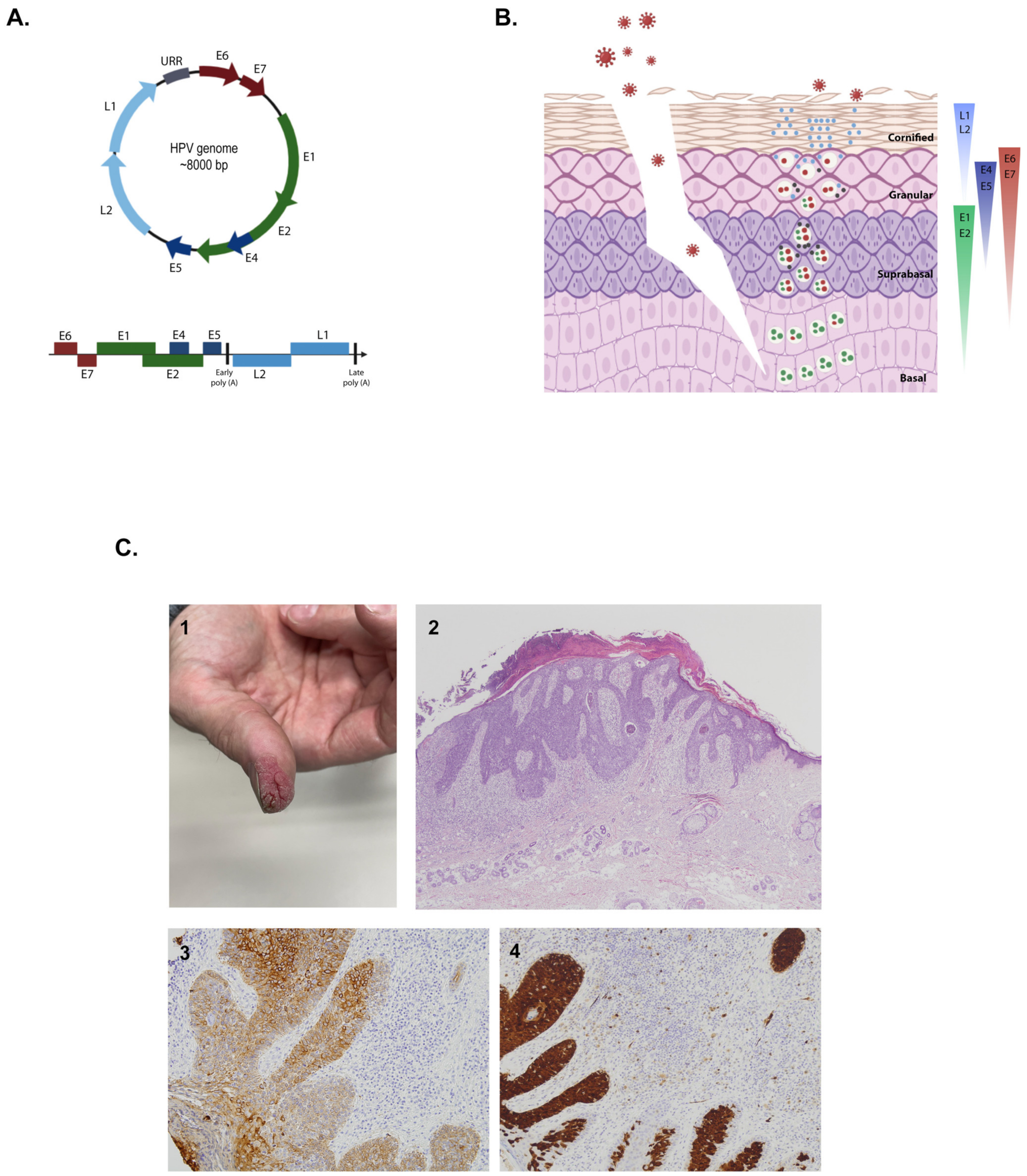
2.2.1. Structure and Genome
2.2.2. Infective Cycle
2.3. Clinical Symptoms, Prognosis, and Treatment
2.3.1. Infection of the Anogenital Region
Benign Lesions: Genital Warts
Malignant Lesions
Cervical Cancer
Penile and Vulvar Carcinoma
Verrucous cSCC of the Anogenital Region: Buschke–Löwenstein Tumor
2.3.2. Epidermodysplasia Verruciformis (Lewandowsky–Lutz Dysplasia)
2.3.3. Cutaneous Squamous Cell Carcinoma
3. Merkel Cell Polyomavirus and Merkel Cell Carcinoma
3.1. Historical Perspective
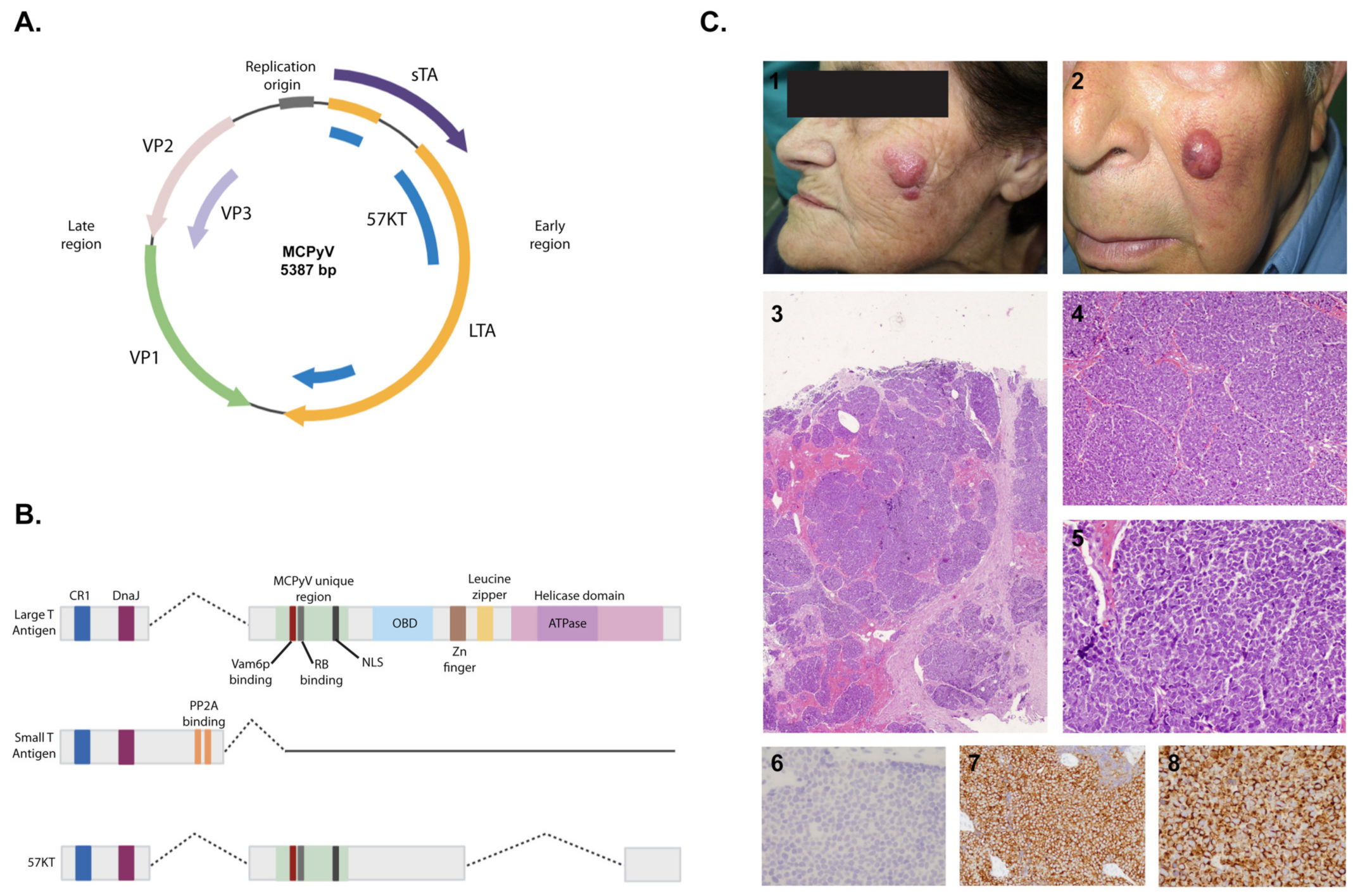
3.2. Structure and Pathogenicity Mechanisms
3.3. Merkel Cell Carcinoma
3.3.1. Epidemiology
3.3.2. Clinical Features
3.3.3. Prognosis
3.3.4. Treatment
4. Human Herpesvirus 8 and Kaposi’s Sarcoma
4.1. Historical Perspective
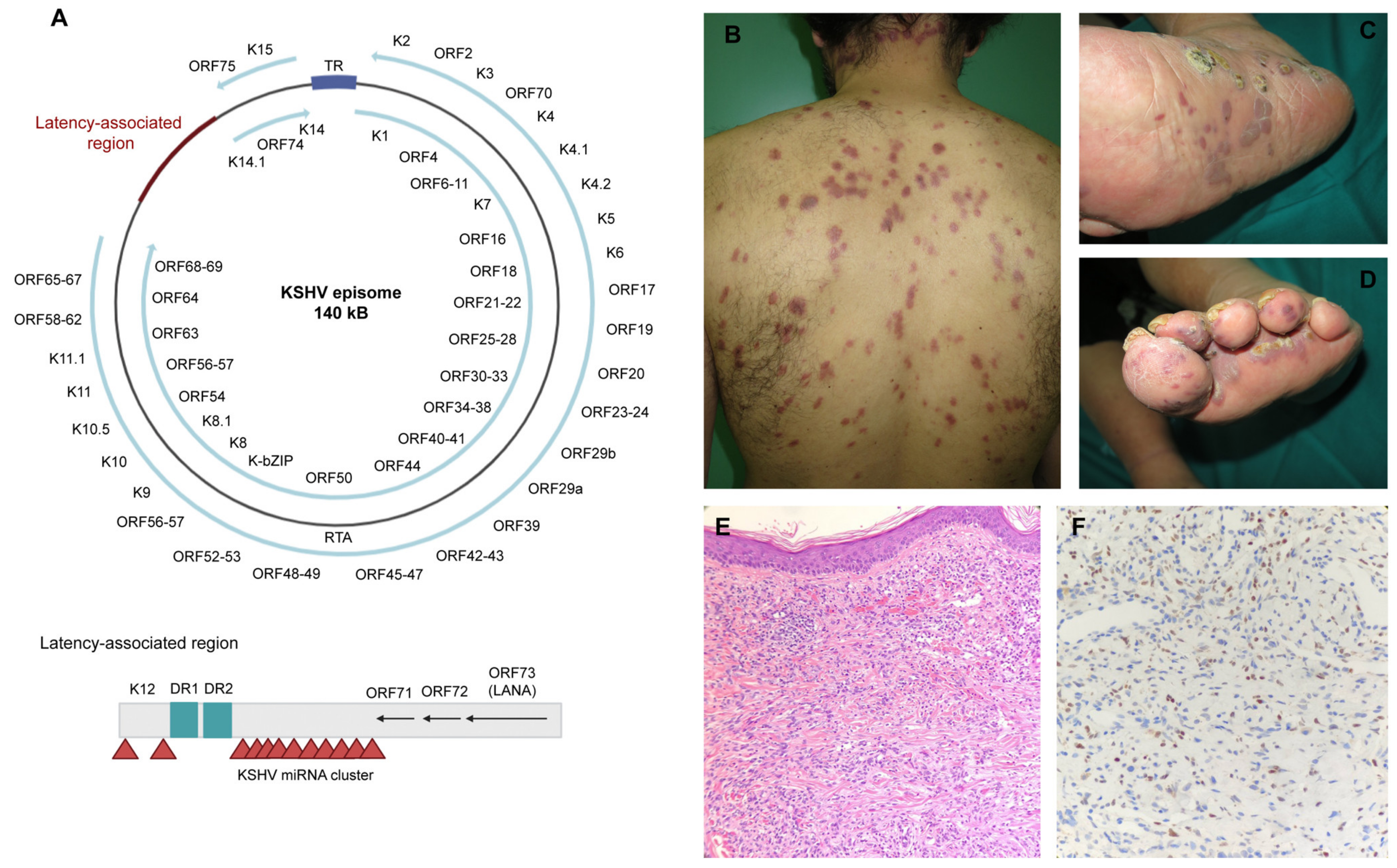
4.2. Structure and Pathogenicity Mechanisms
4.2.1. Structure
4.2.2. Infective Cycle
4.3. Kaposi’s Sarcoma
Epidemiology of HHV8 Infection
5. Conclusions
Funding
Conflicts of Interest
References
- Rous, P. A Transmissible Avian Neoplasm. (Sarcoma of the Common Fowl). J. Exp. Med. 1910, 12, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Javier, R.T.; Butel, J.S. The history of tumor virology. Cancer Res. 2008, 68, 7693–7706. [Google Scholar] [CrossRef] [PubMed]
- Bravo, I.G.; Felez-Sanchez, M. Papillomaviruses: Viral evolution, cancer and evolutionary medicine. Evol. Med. Public Health 2015, 2015, 32–51. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Condylomata acuminata and human genital cancer. Cancer Res. 1976, 36, 794. [Google Scholar]
- zur Hausen, H. Human papillomaviruses and their possible role in squamous cell carcinomas. Curr. Top. Microbiol. Immunol. 1977, 78, 1–30. [Google Scholar]
- zur Hausen, H.; Meinhof, W.; Scheiber, W.; Bornkamm, G.W. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int. J. Cancer 1974, 13, 650–656. [Google Scholar] [CrossRef]
- Meisels, A.; Fortin, R. Condylomatous lesions of the cervix and vagina. I. Cytologic patterns. Acta Cytol. 1976, 20, 505–509. [Google Scholar] [CrossRef]
- Meisels, A.; Roy, M.; Fortier, M.; Morin, C.; Casas-Cordero, M.; Shah, K.V.; Turgeon, H. Human papillo-mavirus infection of the cervix: The atypical condyloma. Acta Cytol. 1981, 25, 7–16. [Google Scholar]
- Della Torre, G.; Pilotti, S.; de Palo, G.; Rilke, F. Viral particles in cervical condylomatous lesions. Tumori 1978, 64, 549–553. [Google Scholar] [CrossRef]
- Durst, M.; Gissmann, L.; Ikenberg, H.; zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef]
- Boshart, M.; Gissmann, L.; Ikenberg, H.; Kleinheinz, A.; Scheurlen, W.; zur Hausen, H. A new type of pap-illomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984, 3, 1151–1157. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Orth, G.; Jablonska, S.; Favre, M.; Croissant, O.; Jarzabek-Chorzelska, M.; Rzesa, G. Characterization of two types of human papillomaviruses in lesions of epidermodysplasia verruciformis. Proc. Natl. Acad. Sci. USA 1978, 75, 1537–1541. [Google Scholar] [CrossRef]
- Orth, G.; Jablonska, S.; Jarzabek-Chorzelska, M.; Obalek, S.; Rzesa, G.; Favre, M.; Croissant, O. Character-istics of the lesions and risk of malignant conversion associated with the type of human papillomavirus in-volved in epidermodysplasia verruciformis. Cancer Res. 1979, 39, 1074–1082. [Google Scholar]
- Gissmann, L.; zur Hausen, H. Partial characterization of viral DNA from human genital warts (Condylomata acuminata). Int. J. Cancer 1980, 25, 605–609. [Google Scholar] [CrossRef]
- Gissmann, L.; Diehl, V.; Schultz-Coulon, H.J.; zur Hausen, H. Molecular cloning and characterization of human papilloma virus DNA derived from a laryngeal papilloma. J. Virol. 1982, 44, 393–400. [Google Scholar] [CrossRef]
- Gissmann, L.; deVilliers, E.M.; zur Hausen, H. Analysis of human genital warts (Condylomata acuminata) and other genital tumors for human papillomavirus type 6 DNA. Int. J. Cancer 1982, 29, 143–146. [Google Scholar] [CrossRef]
- Ikenberg, H.; Gissmann, L.; Gross, G.; Grussendorf-Conen, E.I.; zur Hausen, H. Human papillomavirus type-16-related DNA in genital Bowen’s disease and in Bowenoid papulosis. Int. J. Cancer 1983, 32, 563–565. [Google Scholar] [CrossRef]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef]
- Garcia-Vallve, S.; Alonso, A.; Bravo, I.G. Papillomaviruses: Different genes have different histories. Trends Microbiol. 2005, 13, 514–521. [Google Scholar] [CrossRef]
- Munger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Rochat, A.; Zeltner, R.; Borenstein, L.; Barrandon, Y.; Wettstein, F.O.; Iftner, T. The primary target cells of the high-risk cottontail rabbit papillomavirus colocalize with hair follicle stem cells. J. Virol. 1996, 70, 1912–1922. [Google Scholar] [CrossRef]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32 (Suppl. 1), S7–S15. [Google Scholar] [CrossRef]
- Cheng, S.; Schmidt-Grimminger, D.C.; Murant, T.; Broker, T.R.; Chow, L.T. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal dif-ferentiated keratinocytes. Genes Dev. 1995, 9, 2335–2349. [Google Scholar] [CrossRef]
- Sherman, L.; Jackman, A.; Itzhaki, H.; Stoppler, M.C.; Koval, D.; Schlegel, R. Inhibition of serum- and cal-cium-induced differentiation of human keratinocytes by HPV16 E6 oncoprotein: Role of p53 inactivation. Virology 1997, 237, 296–306. [Google Scholar] [CrossRef]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef]
- Bedell, M.A.; Hudson, J.B.; Golub, T.R.; Turyk, M.E.; Hosken, M.; Wilbanks, G.D.; Laimins, L.A. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 1991, 65, 2254–2260. [Google Scholar] [CrossRef]
- Florin, L.; Sapp, C.; Streeck, R.E.; Sapp, M. Assembly and translocation of papillomavirus capsid proteins. J. Virol. 2002, 76, 10009–10014. [Google Scholar] [CrossRef]
- Kohler, A.; Forschner, T.; Meyer, T.; Ulrich, C.; Gottschling, M.; Stockfleth, E.; Nindl, I. Multifocal distribution of cutaneous human papillomavirus types in hairs from different skin areas. Br. J. Dermatol. 2007, 156, 1078–1080. [Google Scholar] [CrossRef]
- Plasmeijer, E.I.; Neale, R.E.; Buettner, P.G.; de Koning, M.N.; Ter Schegget, J.; Quint, W.G.; Green, A.C.; Feltkamp, M.C. Betapapillomavirus infection profiles in tissue sets from cutaneous squamous cell-carcinoma patients. Int. J. Cancer 2010, 126, 2614–2621. [Google Scholar] [CrossRef]
- Workowski, K.A.; Bolan, G.A. Sexually transmitted diseases treatment guidelines. MMWR Recomm. Rep. 2015, 64, 1–137. [Google Scholar] [CrossRef]
- Saslow, D.; Solomon, D.; Lawson, H.W.; Killackey, M.; Kulasingam, S.L.; Cain, J.; Garcia, F.A.R.; Moriarty, A.T.; Waxman, A.G.; Wilbur, D.C.; et al. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer, J. Clin. 2012, 62, 147–172. [Google Scholar] [CrossRef]
- Boshart, M.; zur Hausen, H. Human papillomaviruses in Buschke-Lowenstein tumors: Physical state of the DNA and identification of a tandem duplication in the noncoding region of a human papillomavirus 6 subtype. J. Virol. 1986, 58, 963–966. [Google Scholar] [CrossRef]
- Burger, B.; Itin, P.H. Epidermodysplasia verruciformis. Curr. Probl. Dermatol. 2014, 45, 123–131. [Google Scholar]
- Yabe, Y.; Sadakane, H. The virus of epidermodysplasia verruciformis: Electron microscopic and fluorescent antibody studies. J. Investig. Dermatol. 1975, 65, 324–330. [Google Scholar] [CrossRef]
- Yabe, Y.; Yasui, M.; Yoshino, N.; Fujiwara, T.; Ohkuma, N.; Nohara, N. Epidermodysplasia verruciformis: Viral particles in early malignant lesions. J. Investig. Dermatol. 1978, 71, 225–228. [Google Scholar] [CrossRef]
- Ostrow, R.S.; Bender, M.; Niimura, M.; Seki, T.; Kawashima, M.; Pass, F.; Faras, A.J. Human papillomavirus DNA in cutaneous primary and metastasized squamous cell carcinomas from patients with epidermodys-plasia verruciformis. Proc. Natl. Acad. Sci. USA 1982, 79, 1634–1638. [Google Scholar] [CrossRef]
- Sawaya, G.F.; Smith-McCune, K. Cervical Cancer Screening. Obstet. Gynecol. 2016, 127, 459–467. [Google Scholar] [CrossRef]
- Patel, D.A.; Saraiya, M.; Copeland, G.; Cote, M.L.; Datta, S.D.; Sawaya, G.F. Treatment patterns for cervical carcinoma in situ in Michigan, 1998–2003. J. Regist. Manag. 2013, 40, 84–92. [Google Scholar]
- Paraskevaidis, E.; Kyrgiou, M.; Martin-Hirsch, P. Commentary: Have we dismissed ablative treatment too soon in colposcopy practice? BJOG Int. J. Obstet. Gynaecol. 2006, 114, 3–4. [Google Scholar] [CrossRef]
- Tainio, K.; Athanasiou, A.; Tikkinen, K.A.O.; Aaltonen, R.; Cárdenas, J.; Glazer-Livson, S.; Jakobsson, M.; Joronen, K.; Kiviharju, M.; Louvanto, K.; et al. Clinical course of untreated cervical intraepithelial neoplasia grade 2 under active surveillance: Systematic review and meta-analysis. BMJ 2018, 360, k499. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Bieber, A.K.; Stein, J.A.; Pomeranz, M.K. Diagnosis and management of vulvar cancer: A review. J. Am. Acad. Dermatol. 2019, 81, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.; Gomez-Martinez, R.A. Vulvar Cancer. Obstet. Gynecol Clin. N. Am. 2019, 46, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.A.; Anderson, L.; Eva, L.; Scurry, J. Clinical and molecular classification of vulvar squamous pre-cancers. Int. J. Gynecol. Cancer 2019, 29, 821–828. [Google Scholar] [CrossRef]
- Calmon, M.F.; Tasso Mota, M.; Vassallo, J.; Rahal, P. Penile carcinoma: Risk factors and molecular alterations. Sci. World J. 2011, 11, 269–282. [Google Scholar] [CrossRef]
- Cubilla, A.L.; Velazquez, E.F.; Amin, M.B.; Epstein, J.; Berney, D.M.; Corbishley, C.M.; Members of the ISUP Penile Tumor Panel. The World Health Organisation 2016 classification of penile carcinomas: A review and update from the International Society of Urological Pathology expert-driven recommendations. Histopathology 2018, 72. [Google Scholar] [CrossRef]
- Shimizu, A.; Tamura, A.; Abe, M.; Motegi, S.; Nagai, Y.; Ishikawa, O.; Nakatani, Y.; Yamamoto, Y.; Uezato, H.; Hoshino, H. Detection of human papillomavirus type 56 in Bowen’s disease involving the nail matrix. Br. J. Dermatol. 2008, 158, 1273–1279. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tanese, K.; Kubo, A.; Matsumoto-Mochimaru, N.; Sasaki, A.; Kameyama, K.; Amagai, M.; Umegaki-Arao, N. Identification of a human papillomavirus type 58 lineage in multiple Bowen’s disease on the fingers: Case report and published work review. J. Dermatol. 2018, 45, 1195–1198. [Google Scholar] [CrossRef]
- Perruchoud, D.L.; Varonier, C.; Haneke, E.; Hunger, R.E.; Beltraminelli, H.; Borradori, L.; Ehnis Pérez, A. Bowen disease of the nail unit: A retrospective study of 12 cases and their association with human papil-lomaviruses. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1503–1506. [Google Scholar] [CrossRef]
- Baek, Y.S.; Jeon, J.; Kim, A.; Song, H.J.; Kim, C. Human papillomavirus is more frequently detected in the pelvic than non-pelvic area in patients with squamous cell carcinoma in situ (Bowen’s disease). Eur. J. Der-matol. 2020, 30, 111–118. [Google Scholar] [CrossRef]
- Dika, E.; Venturoli, S.; Patrizi, A.; Piraccini, B.M.; Fanti, P.A.; Barbieri, D.; Nocera, M.; Chessa, M.A.; Landini, M.P.; La Placa, M. The detection of human papillomavirus–16 in squamous cell carcinoma of the nail unit: A case series. J. Am. Acad. Dermatol. 2017, 76, 354–356. [Google Scholar] [CrossRef]
- Dika, E.; Starace, M.; Patrizi, A.; Fanti, P.A.; Piraccini, B.M. Squamous Cell Carcinoma of the Nail Unit: A Clinical Histopathologic Study and a Proposal for Classification. Dermatol. Surg. 2019, 45, 365–370. [Google Scholar] [CrossRef]
- Lutzner, M.A.; Blanchet-Bardon, C.; Orth, G. Clinical observations, virologic studies, and treatment trials in patients with epidermodysplasia verruciformis, a disease induced by specific human papillomaviruses. J. Investig. Dermatol. 1984, 83, 18s–25s. [Google Scholar] [CrossRef]
- Ramoz, N.; Rueda, L.A.; Bouadjar, B.; Favre, M.; Orth, G. A susceptibility locus for epidermodysplasia verruciformis, an abnormal predisposition to infection with the oncogenic human papillomavirus type 5, maps to chromosome 17qter in a region containing a psoriasis locus. J. Investig. Dermatol. 1999, 112, 259–263. [Google Scholar] [CrossRef]
- Ramoz, N.; Favre, M.; Orth, G.; Taieb, A.; Rueda, L.A.; Montoya, L.S.; Bouadjar, B. Evidence for a nonallelic heterogeneity of epidermodysplasia verruciformis with two susceptibility loci mapped to chromosome regions 2p21-p24 and 17q25. J. Investig. Dermatol. 2000, 114, 1148–1153. [Google Scholar] [CrossRef]
- Orth, G. Genetics of epidermodysplasia verruciformis: Insights into host defense against papillomaviruses. Semin. Immunol. 2006, 18, 362–374. [Google Scholar] [CrossRef]
- Lazarczyk, M.; Cassonnet, P.; Pons, C.; Jacob, Y.; Favre, M. The EVER proteins as a natural barrier against papillomaviruses: A new insight into the pathogenesis of human papillomavirus infections. Microbiol. Mol. Biol. Rev. 2009, 73, 348–370. [Google Scholar] [CrossRef]
- Reuschenbach, M.; Tran, T.; Faulstich, F.; Hartschuh, W.; Vinokurova, S.; Kloor, M.; Krautkramer, E.; Zeier, M.; Doeberitz, M.V.K.; Sommerer, C. High-risk human papillomavirus in non-melanoma skin lesions from renal allograft recipients and immunocompetent patients. Br. J. Cancer 2011, 104, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, C.; Kanitakis, J.; Stockfleth, E.; Euvrard, S. Skin cancer in organ transplant recipients—Where do we stand today? Am. J. Transplant. 2008, 8, 2192–2198. [Google Scholar] [CrossRef] [PubMed]
- Veness, M.J.; Quinn, D.I.; Ong, C.S.; Keogh, A.M.; Macdonald, P.S.; Cooper, S.G.; Morgan, G.W. Aggressive cutaneous malignancies following cardiothoracic transplantation: The Australian experience. Cancer 1999, 85, 1758–1764. [Google Scholar] [CrossRef]
- Euvrard, S.; Kanitakis, J.; Pouteil-Noble, C.; Claudy, A.; Touraine, J.L. Skin cancers in organ transplant re-cipients. Ann. Transplant. 1997, 2, 28–32. [Google Scholar]
- Euvrard, S.; Kanitakis, J.; Decullier, E.; Butnaru, A.C.; Lefrancois, N.; Boissonnat, P.; Sebbag, L.; Garnier, J.-L.; Pouteil-Noble, C.; Cahen, R.; et al. Subsequent skin cancers in kidney and heart transplant recipients after the first squamous cell carcinoma. Transplantation 2006, 81, 1093–1100. [Google Scholar] [CrossRef]
- Martinez, J.C.; Otley, C.C.; Stasko, T.; Euvrard, S.; Brown, C.; Schanbacher, C.F.; Weaver, A.L. Defining the clinical course of metastatic skin cancer in organ transplant recipients: A multicenter collaborative study. Arch. Dermatol. 2003, 139, 301–306. [Google Scholar] [CrossRef]
- Veness, M.J.; Palme, C.E.; Morgan, G.J. High-risk cutaneous squamous cell carcinoma of the head and neck: Results from 266 treated patients with metastatic lymph node disease. Cancer 2006, 106, 2389–2396. [Google Scholar] [CrossRef]
- Wieland, U.; Kreuter, A.; Pfister, H. Human papillomavirus and immunosuppression. Curr. Probl. Dermatol. 2014, 45, 154–165. [Google Scholar]
- Euvrard, S.; Chardonnet, Y.; Pouteil-Noble, C.; Kanitakis, J.; Chignol, M.C.; Thivolet, J.; Touraine, J.L. As-sociation of skin malignancies with various and multiple carcinogenic and noncarcinogenic human papil-lomaviruses in renal transplant recipients. Cancer 1993, 72, 2198–2206. [Google Scholar] [CrossRef]
- Shamanin, V.; zur Hausen, H.; Lavergne, D.; Proby, C.M.; Leigh, I.M.; Neumann, C.; Hamm, H.; Goos, M.; Haustein, U.-F.; Jung, E.G.; et al. Human papillomavirus infections in nonmelanoma skin cancers from renal transplant recipients and nonimmunosuppressed patients. J. Natl. Cancer Inst. 1996, 88, 802–811. [Google Scholar] [CrossRef]
- Shamanin, V.; Glover, M.; Rausch, C.; Proby, C.; Leigh, I.M.; zur Hausen, H.; De Villiers, E.M. Specific types of human papillomavirus found in benign proliferations and carcinomas of the skin in immunosuppressed patients. Cancer Res. 1994, 54, 4610–4613. [Google Scholar]
- Ganzenmueller, T.; Yakushko, Y.; Kluba, J.; Henke-Gendo, C.; Gutzmer, R.; Schulz, T.F. Next-generation sequencing fails to identify human virus sequences in cutaneous squamous cell carcinoma. Int. J. Cancer 2012, 131, E1173–E1179. [Google Scholar] [CrossRef]
- Arron, S.T.; Ruby, J.G.; Dybbro, E.; Ganem, D.; Derisi, J.L. Transcriptome sequencing demonstrates that human papillomavirus is not active in cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2011, 131, 1745–1753. [Google Scholar] [CrossRef]
- Harwood, C.A.; Surentheran, T.; McGregor, J.M.; Spink, P.J.; Leigh, I.M.; Breuer, J.; Proby, C.M. Human papillomavirus infection and non-melanoma skin cancer in immunosuppressed and immunocompetent in-dividuals. J. Med. Virol. 2000, 61, 289–297. [Google Scholar] [CrossRef]
- La Placa, M.; Venturoli, S.; Barbieri, D.; Nocera, M.; Patrizi, A.; Landini, M.; Fanti, P.; Dika, E. Presence of cutaneous human papillomavirus DNA in squamous cell carcinoma of the scalp: A case series. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 369–371. [Google Scholar] [CrossRef]
- Schaper, I.D.; Marcuzzi, G.P.; Weissenborn, S.J.; Kasper, H.U.; Dries, V.; Smyth, N.; Fuchs, P.; Pfister, H. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 2005, 65, 1394–1400. [Google Scholar] [CrossRef]
- De Andrea, M.; Ritta, M.; Landini, M.M.; Borgogna, C.; Mondini, M.; Kern, F.; Ehrenreiter, K.; Baccarini, M.; Marcuzzi, G.P.; Smola, S.; et al. Keratinocyte-specific stat3 heterozygosity impairs development of skin tu-mors in human papillomavirus 8 transgenic mice. Cancer Res. 2010, 70, 7938–7948. [Google Scholar] [CrossRef]
- Marcuzzi, G.P.; Hufbauer, M.; Kasper, H.U.; Weissenborn, S.J.; Smola, S.; Pfister, H. Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J. Gen. Virol. 2009, 90, 2855–2864. [Google Scholar] [CrossRef]
- Wang, J.; Aldabagh, B.; Yu, J.; Arron, S.T. Role of human papillomavirus in cutaneous squamous cell car-cinoma: A meta-analysis. J. Am. Acad. Dermatol. 2014, 70, 621–629. [Google Scholar] [CrossRef]
- Toker, C. Trabecular carcinoma of the skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Tang, C.K.; Toker, C. Trabecular carcinoma of the skin: An ultrastructural study. Cancer 1978, 42, 2311–2321. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Penas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Rigel, D.S.; Merrick, R.; Friedman, R.; Cockerell, C.; Lim, H.; Kirkwood, J. Cancer of the Skin; Rigel, D.S., Merrick, R., Friedman, R., Cockerell, C., Lim, H., Kirkwood, J., Eds.; Saunders Elsevier: Philadelphia, PA, USA, 2011. [Google Scholar]
- Agelli, M.; Clegg, L.X. Epidemiology of primary Merkel cell carcinoma in the United States. J. Am. Acad. Dermatol. 2003, 49, 832–841. [Google Scholar] [CrossRef]
- Llombart, B.; Requena, C.; Cruz, J. Update on Merkel Cell Carcinoma: Epidemiology, Etiopathogenesis, Clinical Features, Diagnosis, and Staging. Actas Dermosifiliogr. 2017, 108, 108–119. [Google Scholar] [CrossRef]
- Guler-Nizam, E.; Leiter, U.; Metzler, G.; Breuninger, H.; Garbe, C.; Eigentler, T.K. Clinical course and prognostic factors of Merkel cell carcinoma of the skin. Br. J. Dermatol. 2009, 161, 90–94. [Google Scholar] [CrossRef]
- Kempf, W.; Mertz, K.D.; Hofbauer, G.F.; Tinguely, M. Skin cancer in organ transplant recipients. Pathobiology 2013, 80, 302–309. [Google Scholar] [CrossRef]
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef]
- Lanoy, E.; Costagliola, D.; Engels, E.A. Skin cancers associated with HIV infection and solid-organ trans-plantation among elderly adults. Int. J. Cancer 2010, 126, 1724–1731. [Google Scholar]
- Kaae, J.; Hansen, A.V.; Biggar, R.J.; Boyd, H.A.; Moore, P.S.; Wohlfahrt, J.; Melbye, M. Merkel cell carcinoma: Incidence, mortality, and risk of other cancers. J. Natl. Cancer Inst. 2010, 102, 793–801. [Google Scholar] [CrossRef]
- Brenner, B.; Sulkes, A.; Rakowsky, E.; Feinmesser, M.; Yukelson, A.; Bar-Haim, E.; Katz, A.; Idelevich, E.; Neuman, A.; Barhana, M.; et al. Second neoplasms in patients with Merkel cell carcinoma. Cancer 2001, 91, 1358–1362. [Google Scholar] [CrossRef]
- Eng, T.Y.; Boersma, M.G.; Fuller, C.D.; Goytia, V.; Jones, W.E., 3rd; Joyner, M.; Nguyen, D.D. A compre-hensive review of the treatment of Merkel cell carcinoma. Am. J. Clin. Oncol. 2007, 30, 624–636. [Google Scholar] [CrossRef]
- Medina-Franco, H.; Urist, M.M.; Fiveash, J.; Heslin, M.J.; Bland, K.I.; Beenken, S.W. Multimodality treatment of Merkel cell carcinoma: Case series and literature review of 1024 cases. Ann. Surg. Oncol. 2001, 8, 204–208. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef]
- Fitzgerald, T.L.; Dennis, S.; Kachare, S.D.; Vohra, N.A.; Wong, J.H.; Zervos, E.E. Dramatic Increase in the Incidence and Mortality from Merkel Cell Carcinoma in the United States. Am. Surg. 2015, 81, 802–806. [Google Scholar] [CrossRef]
- Vlad, R.; Woodlock, T.J. Merkel cell carcinoma after chronic lymphocytic leukemia: Case report and literature review. Am. J. Clin. Oncol. 2003, 26, 531–534. [Google Scholar] [CrossRef]
- Buell, J.F.; Trofe, J.; Hanaway, M.J.; Beebe, T.M.; Gross, T.G.; Alloway, R.R.; First, M.; Woodle, E. Immuno-suppression and Merkel cell cancer. Transplant. Proc. 2002, 34, 1780–1781. [Google Scholar] [CrossRef]
- Moll, I.; Bladt, U.; Jung, E.G. Presence of Merkel cells in sun-exposed and not sun-exposed skin: A quantitative study. Arch. Dermatol. Res. 1990, 282, 213–216. [Google Scholar] [CrossRef]
- Popp, S.; Waltering, S.; Herbst, C.; Moll, I.; Boukamp, P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int. J. Cancer 2002, 99, 352–360. [Google Scholar] [CrossRef]
- Allen, P.J.; Bowne, W.B.; Jaques, D.P.; Brennan, M.F.; Busam, K.; Coit, D.G. Merkel cell carcinoma: Prognosis and treatment of patients from a single institution. J. Clin. Oncol. 2005, 23, 2300–2309. [Google Scholar] [CrossRef]
- Gupta, S.G.; Wang, L.C.; Penas, P.F.; Gellenthin, M.; Lee, S.J.; Nghiem, P. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: The Dana-Farber experience and me-ta-analysis of the literature. Arch. Dermatol. 2006, 142, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Lebbe, C.; Zur Hausen, A.; Avril, M.F.; Hariharan, S.; Bharmal, M.; Becker, J.C. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. Eur. J. Cancer 2017, 71, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Storer, B.E.; Iyer, J.G.; Moshiri, A.; Parvathaneni, U.; Byrd, D.; Sober, A.J.; Sondak, V.K.; Gershenwald, J.E.; Nghiem, P. Adjuvant Radiation Therapy and Chemotherapy in Merkel Cell Carcinoma: Survival Analyses of 6908 Cases From the National Cancer Data Base. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Veness, M.J.; Perera, L.; McCourt, J.; Shannon, J.; Hughes, T.M.; Morgan, G.J.; Gebski, V. Merkel cell carci-noma: Improved outcome with adjuvant radiotherapy. ANZ J. Surg. 2005, 75, 275–281. [Google Scholar] [CrossRef]
- Lewis, K.G.; Weinstock, M.A.; Weaver, A.L.; Otley, C.C. Adjuvant local irradiation for Merkel cell carcinoma. Arch. Dermatol. 2006, 142, 693–700. [Google Scholar] [CrossRef]
- Finnigan, R.; Hruby, G.; Wratten, C.; Keller, J.; Tripcony, L.; Dickie, G.; Rischin, D.; Poulsen, M. The impact of preradiation residual disease volume on time to locoregional failure in cutaneous Merkel cell carcinoma—A TROG substudy. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 91–95. [Google Scholar] [CrossRef]
- Sims, J.R.; Grotz, T.E.; Pockaj, B.A.; Joseph, R.W.; Foote, R.L.; Otley, C.C.; Weaver, A.L.; Jakub, J.W.; Price, D.L. Sentinel lymph node biopsy in Merkel cell carcinoma: The Mayo Clinic experience of 150 patients. Surg. Oncol. 2018, 27, 11–17. [Google Scholar] [CrossRef]
- Lebbe, C.; Becker, J.C.; Grob, J.J.; Malvehy, J.; Del Marmol, V.; Pehamberger, H.; Peris, K.; Saiag, P.; Mid-dleton, M.R.; Bastholt, L. Diagnosis and treatment of Merkel Cell Carcinoma. European consensus-based interdisciplinary guideline. Eur. J. Cancer 2015, 51, 2396–2403. [Google Scholar] [CrossRef]
- Kachare, S.D.; Wong, J.H.; Vohra, N.A.; Zervos, E.E.; Fitzgerald, T.L. Sentinel lymph node biopsy is associ-ated with improved survival in Merkel cell carcinoma. Ann. Surg. Oncol. 2014, 21, 1624–1630. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- EMD Serono Inc. and Pfizer Inc. Bavencio® (Avelumab) Injection: US Prescribing Information. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761049s000lbl.pdf (accessed on 23 March 2017).
- US FDA. FDA Approves First Treatment for Rare Form of Skin Cancer [Media Release]. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-rare-form-skin-cancer (accessed on 23 March 2017).
- Kaposi, M. Idiopathisches multiples Pigmentsarkom der Haut. Arch. Dermat. Syph. 1872, 4, 265–273. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P. Twenty years of KSHV. Viruses 2014, 6, 4258–4264. [Google Scholar] [CrossRef]
- Durack, D.T. Opportunistic infections and Kaposi’s sarcoma in homosexual men. N. Engl. J. Med. 1981, 305, 1465–1467. [Google Scholar] [CrossRef]
- Centers for Disease, C. Kaposi’s sarcoma and Pneumocystis pneumonia among homosexual men-New York City and California. MMWR Morb. Mortal Wkly Rep. 1981, 30, 305–308. [Google Scholar]
- Jaffe, H.W.; Choi, K.; Thomas, P.A.; Haverkos, H.W.; Auerbach, D.M.; Guinan, M.E.; Rogers, M.F.; Spira, T.J.; Darrow, W.W.; Kramer, M.A.; et al. National case-control study of Kaposi’s sarcoma and Pneumocystis ca-rinii pneumonia in homosexual men: Part Epidemiologic results. Ann. Intern. Med. 1983, 99, 145–151. [Google Scholar] [CrossRef]
- Rogers, M.F.; Morens, D.M.; Stewart, J.A.; Kaminski, R.M.; Spira, T.J.; Feorino, P.M.; Larsen, S.A.; Francis, D.P.; Wilson, M.; Kaufman, L. National case-control study of Kaposi’s sarcoma and Pneumocystis carinii pneumonia in homosexual men: Part Laboratory results. Ann. Intern. Med. 1983, 99, 151–158. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and those without HIV infection. N. Engl. J. Med. 1995, 332, 1181–1185. [Google Scholar] [CrossRef]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef]
- Renne, R.; Lagunoff, M.; Zhong, W.; Ganem, D. The size and conformation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J. Virol. 1996, 70, 8151–8154. [Google Scholar] [CrossRef]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef]
- Neipel, F.; Albrecht, J.C.; Fleckenstein, B. Human herpesvirus 8–the first human Rhadinovirus. J. Natl. Cancer Inst. 1998, 1998, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Hensler, H.; Tomaszewski, M.; Rappocciolo, G.; Rinaldo, C.; Jenkins, F. Human herpesvirus 8 glycoprotein B binds the entry receptor DC-SIGN. Virus Res. 2014, 190, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, E.; López, L.; Navarrete, G. Sarcoma de Kaposi. Revisión de la literatura, un enfoque en la eti-opatogenia. Dermatol. Cosmét. Méd. Quir. 2018, 16, 128–133. [Google Scholar]
- Ye, F.C.; Lei, X.; Gao, S.J. Mechanisms of Kaposi’s Sarcoma-Associated Herpesvirus Latency and Reactiva-tion. Adv. Virol. 2011, 2011, 193860. [Google Scholar] [CrossRef] [PubMed]
- Dupin, N.; Fisher, C.; Kellam, P.; Ariad, S.; Tulliez, M.; Franck, N.; Van Marck, E.; Salmon, D.; Gorin, I.; Escande, J.P.; et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multi-centric Castleman’s disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 1999, 96, 4546–4551. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.; Lu, J.; Robertson, E. Molecular Biology of Kaposi’s Sarcoma-associated Herpesvirus and Related Oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Akula, S.M.; Pramod, N.P.; Wang, F.-Z.; Chandran, B. Human herpesvirus 8 envelope-associated glyco-protein B interacts with heparan sulfate-like moieties. Virology 2001, 284, 235–249. [Google Scholar] [CrossRef]
- Akula, S.M.; Pramod, N.P.; Wang, F.-Z.; Chandran, B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 2002, 108, 407–419. [Google Scholar] [CrossRef]
- Kaleeba, J.A.; Berger, E.A. Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: Cystine trans-porter xCT. Science 2006, 311, 1921–1924. [Google Scholar] [CrossRef]
- Rappocciolo, G.; Jenkins, F.J.; Hensler, H.R.; Piazza, P.; Jais, M.; Borowski, L.; Watkins, S.C.; Rinaldo, C.R. DC-SIGN Is a Receptor for Human Herpesvirus 8 on Dendritic Cells and Macrophages. J. Immunol. 2006, 176, 1741–1749. [Google Scholar] [CrossRef]
- Chandran, B. Early Events in Kaposi’s Sarcoma-Associated Herpesvirus Infection of Target Cells. J. Virol. 2010, 84, 2188–2199. [Google Scholar] [CrossRef]
- Giffin, L.; Damania, B. Advances in Virus Research; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Chapter Two-KSHV: Pathways to Tumorigenesis and Persistent Infection; pp. 111–139. [Google Scholar]
- Barbera, A.J.; Ballestas, M.E.; Kaye, K.M. The Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen 1 N Terminus Is Essential for Chromosome Association, DNA Replication, and Episome Persistence. J. Virol. 2004, 78, 294–301. [Google Scholar] [CrossRef]
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Joukov, V.; Walter, J.C.; Luger, K.; Kaye, K.M. The Nucleosomal Surface as a Docking Station for Kaposi’s Sarcoma Herpesvirus LANA. Science 2006, 311, 856–861. [Google Scholar] [CrossRef]
- Si, H.; Robertson, E.S. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Latency-Associated Nuclear An-tigen Induces Chromosomal Instability through Inhibition of p53 Function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma–E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef]
- Fujimuro, M.; Wu, F.Y.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mecha-nism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef]
- Verma, S.C.; Borah, S.; Robertson, E.S. Latency-Associated Nuclear Antigen of Kaposi’s Sarcoma-Associated Herpesvirus Up-Regulates Transcription of Human Telomerase Reverse Transcriptase Promoter through Interaction with Transcription Factor Sp1. J. Virol. 2004, 78, 10348–10359. [Google Scholar] [CrossRef]
- Di Bartolo, D.L.; Cannon, M.; Liu, Y.-F.; Renne, R.; Chadburn, A.; Boshoff, C.; Cesarman, E. KSHV LANA inhibits TGF-B signaling through epigenetic silencing of the TGF-B type II receptor. Blood 2008, 111, 4731–4740. [Google Scholar] [CrossRef]
- Cai, Q.-L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. EC 5 S Ubiquitin Complex Is Recruited by KSHV Latent Antigen LANA for Degradation of the VHL and p53 Tumor Suppressors. PLoS Pathog. 2006, 2, 1002–1012. [Google Scholar] [CrossRef]
- Aneja, K.K.; Yuan, Y. Reactivation and Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus: An Update. Front. Microbiol. 2017, 8, 1–23. [Google Scholar] [CrossRef]
- Sun, R.; Lin, S.-F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of Kaposi’s Sar-coma-Associated Herpesvirus Gene Expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Bais, C.; Santomasso, B.; Coso, O.; Arvanitakis, L.; Geras Raaka, E.; Gutkind, J.S.; Asch, A.S.; Cesarman, E.; Gerhengorn, M.C.; Mesri, E.A. G-protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 1998, 391, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Brodie, C.; Sarid, R. An essential role of ERK signalling in TPA-induced reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2006, 87, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Harada, J.N.; Brown, H.J.; Deng, H.; Jung Song, M.; Wu, T.-T.; Kato-Stankiewicz, J.; Nelson, C.G.; Vieira, J.; Tamanoi, F.; et al. Systematic Identification of Cellular Signals Reactivating Kaposi Sarcoma–Associated Herpesvirus. PLoS Pathog. 2007, 3. [Google Scholar] [CrossRef]
- Sun, R.; Lin, S.-F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef]
- West, J.T.; Wood, C. The role of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 2003, 22, 5150–5163. [Google Scholar] [CrossRef]
- Song, M.J.; Deng, H.; Sun, R. Comparative Study of Regulation of RTA-Responsive Genes in Kaposi’s Sar-coma-Associated Herpesvirus/Human Herpesvirus 8. J. Virol. 2003, 77, 9451–9462. [Google Scholar] [CrossRef]
- Guito, J.; Lukac, D.M. KSHV Reactivation and Novel Implications of Protein Isomerization on Lytic Switch Control. Viruses 2015, 7, 72–109. [Google Scholar] [CrossRef]
- Guito, J.; Gavina, A.; Palmeri, D.; Lukac, D.M. The Cellular Peptidyl-Prolyl cis/trans Isomerase Pin1 Regulates Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus from Latency. J. Virol. 2014, 88, 547–558. [Google Scholar] [CrossRef]
- Lan, K.; kuppers, D.A.; Verma, S.C.; Robertson, E.S. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Latency-Associated Nuclear Antigen Inhibits Lytic Replication by Targeting Rta: A Potential Mechanism for Virus-Mediated Control of Latency. J. Virol. 2004, 78, 6585–6594. [Google Scholar] [CrossRef]
- Tornesello, M.L.; Biryahwaho, B.; Downing, R.; Hatzakis, A.; Alessi, E.; Cusini, M.; Ruocco, V.; Katongo-le-Mbidde, E.; Loquercio, G.; Buonaguro, L.; et al. Human herpesvirus type 8 variants circulating in Europe, Africa and North America in classic, endemic and epidemic Kaposi’s sarcoma lesions during pre-AIDS and AIDS era. Virology 2010, 398, 280–289. [Google Scholar] [CrossRef]
- Manns, A.; Strickler, H.D.; Hanchard, B.; Manassaram, D.M.; Waters, D.; Ablashi, D.V. Age-and sex-specific seroprevalence of human herpesvirus 8 in Jamaica. J. Natl. Cancer Inst. 1998, 90, 1102–1104. [Google Scholar] [CrossRef][Green Version]
- Kedes, D.H.; Operskalski, E.; Busch, M.; Kohn, R.; Flood, J.; Ganem, D. The seroepidemiology of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus): Distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 1996, 2, 918–924. [Google Scholar] [CrossRef]
- Lennette, E.T.; Blackbourn, D.J.; Levy, J.A. Antibodies to human herpesvirus type 8 in the general population and in Kaposi’s sarcoma patients. Lancet 1996, 348, 858–861. [Google Scholar] [CrossRef]
- Martin, J.N.; Ganem, D.E.; Osmond, D.H.; Page-Shafer, K.A.; Macrae, D.; Kedes, D.H. Sexual transmission and the natural history of human herpesvirus 8 infection. N. Engl. J. Med. 1998, 338, 948–954. [Google Scholar] [CrossRef]
- Cattani, P.; Capuano, M.; Cerimele, F.; La Parola, I.L.; Santangelo, R.; Masini, C.; Cerimele, D.; Fadda, G. Human herpesvirus 8 seroprevalence and evaluation of nonsexual transmission routes by detection of DNA in clinical specimens from human immunodeficiency virus-seronegative patients from central and southern Italy, with and without Kaposi’s sarcoma. J. Clin. Microbiol. 1999, 37, 1150–1153. [Google Scholar] [CrossRef]
- Pauk, J.; Huang, M.L.; Brodie, S.J.; Wald, A.; Koelle, D.M.; Schacker, T.; Celum, C.; Selke, S.; Corey, L. Mu-cosal shedding of human herpesvirus 8 in men. N. Engl. J. Med. 2000, 343, 1369–1377. [Google Scholar] [CrossRef]
- Koelle, D.M.; Huang, M.L.; Chandran, B.; Vieira, J.; Piepkorn, M.; Corey, L. Frequent detection of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in saliva of human immunodeficiency vi-rus-infected men: Clinical and immunologic correlates. J. Infect. Dis. 1997, 176, 94–102. [Google Scholar] [CrossRef]
- Casper, C.; Krantz, E.; Selke, S.; Kuntz, S.R.; Wang, J.; Huang, M.L.; Pauk, J.S.; Corey, L.; Wald, A. Frequent and asymptomatic oropharyngeal shedding of human herpesvirus 8 among immunocompetent men. J. Infect. Dis. 2007, 195, 30–36. [Google Scholar] [CrossRef]
- Bender Ignacio, R.A.; Goldman, J.D.; Magaret, A.S.; Selke, S.; Huang, M.L.; Gantt, S.; Johnston, C.; Phipps, W.T.; Schiffer, J.T.; Zuckerman, R.A.; et al. Patterns of human herpesvirus-8 oral shedding among diverse cohorts of human herpesvirus-8 seropositive persons. Infect. Agent. Cancer 2016, 11, 7. [Google Scholar] [CrossRef]
- Hladik, W.; Dollard, S.C.; Mermin, J.; Fowlkes, A.L.; Downing, R.; Amin, M.M.; Banage, F.; Nzaro, E.; Kataaha, P.; Dondero, T.J.; et al. Transmission of human herpesvirus 8 by blood transfusion. N. Engl. J. Med. 2006, 355, 1331–1338. [Google Scholar] [CrossRef]
- Antman, K.; Chang, Y. Kaposi’s sarcoma. N. Engl. J. Med. 2000, 342, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, P.; Carbone, A. Kaposi’s Sarcoma Herpesvirus: Twenty years after its discovery. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1288–1294. [Google Scholar]
- Pinzone, M.R.; Berretta, M.; Cacopardo, B.; Nunnari, G. Epstein-barr virus- and Kaposi sarcoma-associated herpesvirus-related malignancies in the setting of human immunodeficiency virus infection. Semin. Oncol. 2015, 42, 258–271. [Google Scholar] [CrossRef]
- Radu, O.; Pantanowitz, L. Kaposi sarcoma. Arch. Pathol. Lab. Med. 2013, 137, 289–294. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Prim. 2019, 5, 9. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Alpha | Skin and mucosa | High-risk types | Pre- and malignant lesions (immortalize human keratinocytes) | HPV16 | Cervical squamous cell carcinoma (~50) Cervical adenocarcinoma (~35) Oropharyngeal cancer (~25) |
| HPV18 | Cervical squamous cell carcinoma (~20) Cervical adenocarcinoma (~35) | ||||
| HPV31, 33, 35, 39, 45, 51, 52, 56, 58, 59 | Cervical squamous cell carcinoma (~30) | ||||
| Low-risk types | Benign lesions (do not immortalize human keratinocytes) | HPV6, 11 | Benign genital lesions Respiratory papillomatosis | ||
| HPV13, 32 | Oral focal epithelial hyperplasia | ||||
| HPV2, 3, 27, 57 | Skin warts | ||||
| Beta | Skin | Latent infections in the general population activated under conditions of immune suppression. Strangely, these viruses can cause epidermodysplasia verruciformis (EV), an aggressive growth of benign and malignant neoplasias of the skin, in genetically predisposed individuals | HPV5, 8 | First beta HPV types isolated from SCC of EV individuals | |
| HPV9, 12, 14, 15, 17, 19–25, 36–38, 47, 49, 75, 76, 80, 92, 93, 96, 98–100, 104, 105, 107, 110, 111, 113, 115, 118, 120, 122, 124, 143, 145, 150–152, 159 | Likely associated with SCC in EV patients as well as in immunocompromised and immunocompetent individuals | ||||
| Gamma | Skin | Benign lesions. Histologically distinguishable by intracytoplasmic inclusion bodies specific to type species | HPV4, 48, 50, 60, 65, 88, 95, 101, 103, 108, 109, 112, 115, 116, 119, 121, 123, 126–142, 144, 146–149, 153–158, 161–170 | Skin warts and papilomas | |
| Mu | Skin and mucosa | Mostly cause clinically latent infections. Histologically distinguishable by intracytoplasmic inclusion bodies specific to type species | HPV1, 63, 204 | Palmoplantar warts | |
| Nu | Skin | Benign and malignant lesions | HPV41 | The first and the only member of a new subgroup of HPVs. It has been detected in skin warts, but also in skin carcinomas and premalignant keratoses | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becerril, S.; Corchado-Cobos, R.; García-Sancha, N.; Revelles, L.; Revilla, D.; Ugalde, T.; Román-Curto, C.; Pérez-Losada, J.; Cañueto, J. Viruses and Skin Cancer. Int. J. Mol. Sci. 2021, 22, 5399. https://doi.org/10.3390/ijms22105399
Becerril S, Corchado-Cobos R, García-Sancha N, Revelles L, Revilla D, Ugalde T, Román-Curto C, Pérez-Losada J, Cañueto J. Viruses and Skin Cancer. International Journal of Molecular Sciences. 2021; 22(10):5399. https://doi.org/10.3390/ijms22105399
Chicago/Turabian StyleBecerril, Sara, Roberto Corchado-Cobos, Natalia García-Sancha, Leonor Revelles, David Revilla, Tatiana Ugalde, Concepción Román-Curto, Jesús Pérez-Losada, and Javier Cañueto. 2021. "Viruses and Skin Cancer" International Journal of Molecular Sciences 22, no. 10: 5399. https://doi.org/10.3390/ijms22105399
APA StyleBecerril, S., Corchado-Cobos, R., García-Sancha, N., Revelles, L., Revilla, D., Ugalde, T., Román-Curto, C., Pérez-Losada, J., & Cañueto, J. (2021). Viruses and Skin Cancer. International Journal of Molecular Sciences, 22(10), 5399. https://doi.org/10.3390/ijms22105399