
Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression




Abstract
1. Introduction
2. Histone Acetylation
3. Histone Deacetylase (HDAC) Families and Classes
4. HDAC and Depression
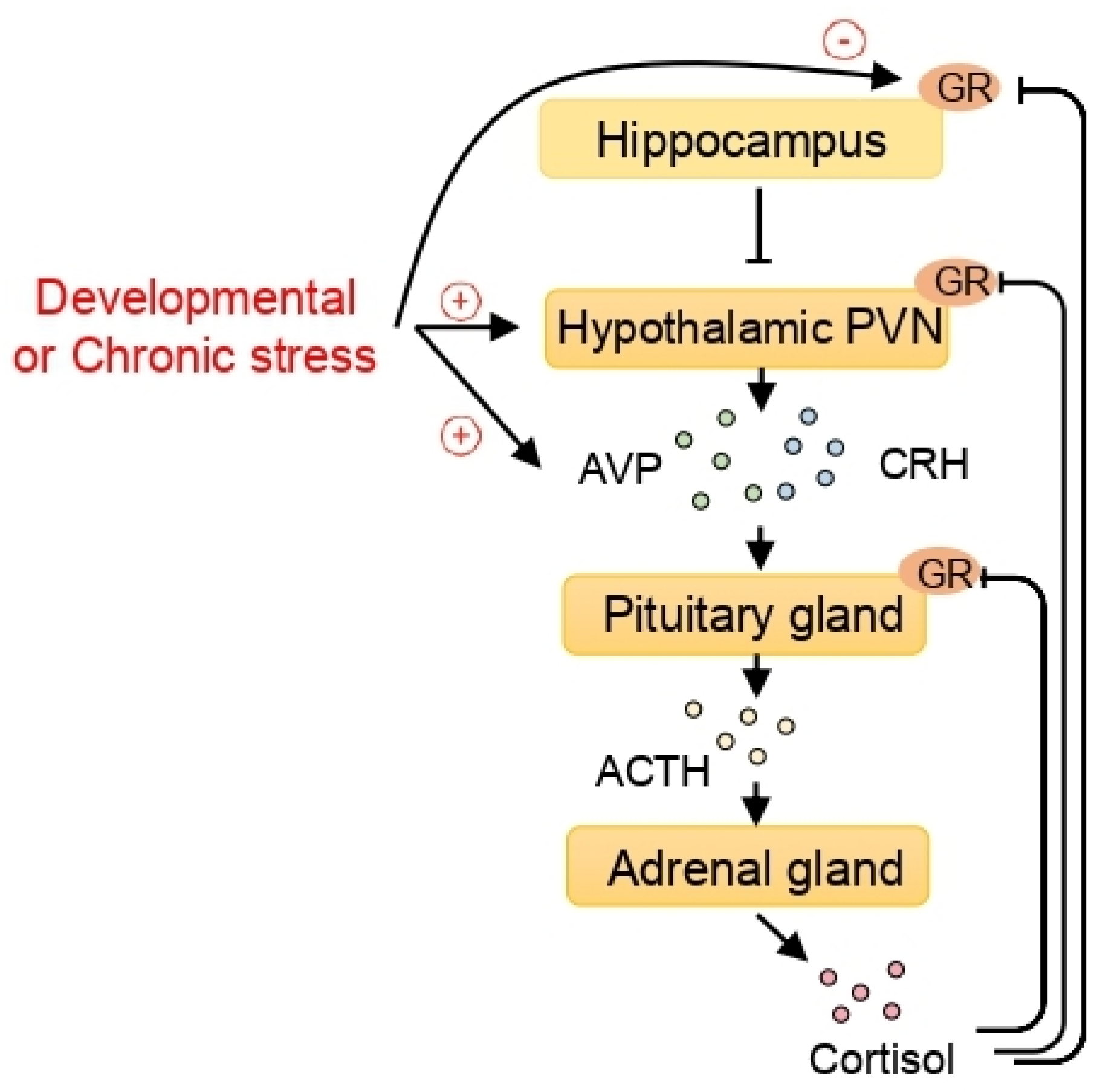
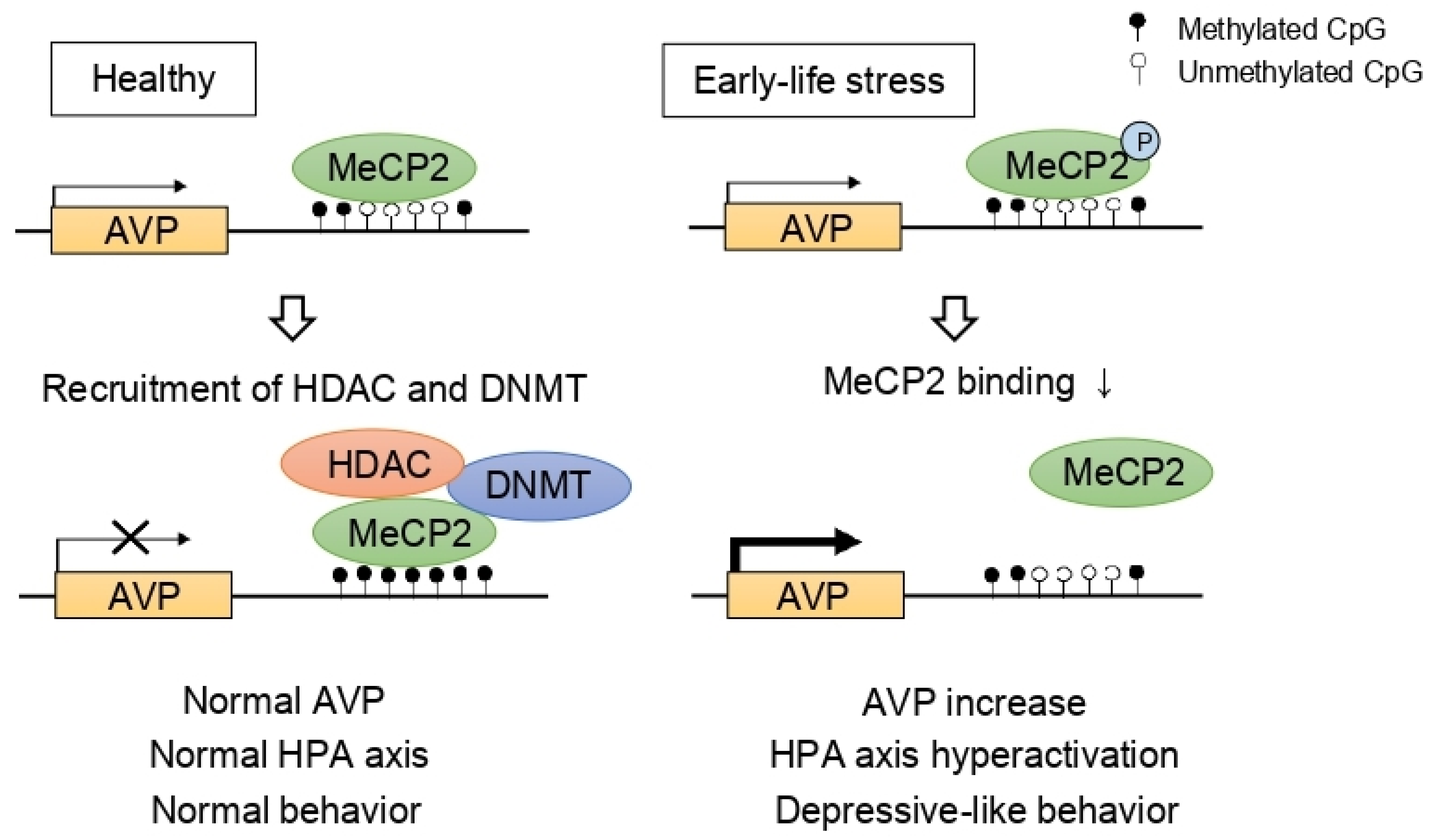
5. HDAC and the Hypothalamic-Pituitary-Adrenal (HPA) Axis
6. HDAC and Brain-Derived Neurotrophic Factor
7. HDAC and Neuronal Plasticity
8. Molecular Diagnosis of Depression: An Epigenetic Perspective
9. Molecular Therapeutics of Depression: An Epigenetic Perspective
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Burcusa, S.L.; Iacono, W.G. Risk for recurrence in depression. Clin. Psychol. Rev. 2007, 27, 959–985. [Google Scholar] [CrossRef]
- Liu, Q.; He, H.; Yang, J.; Feng, X.; Zhao, F.; Lyu, J. Changes in the global burden of depression from 1990 to 2017: Findings from the Global Burden of Disease study. J. Psychiatry Res. 2020, 126, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Levcht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef]
- Calabro, M.; Fabbri, C.; Kasper, S.; Zohar, J.; Souery, D.; Montgomery, S.; Albani, D.; Forloni, G.; Ferentinos, P.; Rujescu, D.; et al. Research Domain Criteria (RDoC): A Perspective to Probe the Biological Background behind Treatment Efficacy in Depression. Curr. Med. Chem. 2021, 28, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Insel, T.; Cuthbert, B.; Garvey, M.; Heinssen, R.; Pine, D.S.; Quinn, K.; Sanislow, C.; Wang, P. Research Domain Criteria (RDoC): Toward a New Classification Framework for Research on Mental Disorders. Am. J. Psychiatry 2010, 167, 748–751. [Google Scholar] [CrossRef]
- Bruckl, T.M.; Spoormaker, V.I.; Samann, P.G.; Brem, A.-K.; Henco, L.; Czamara, D.; Elbau, I.; Grandi, N.C.; Jollans, L.; Kuhnel, A.; et al. The biological classification of mental disorders (BeCOME) study: A protocol for an observational deep-phenotyping study for the identification of biological subtypes. BMC Psychiatry 2020, 20, 213. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef]
- Hasler, G. Pathophysiology of depression: Do we have any solid evidence of interest to clinicians? World Psychiatry 2010, 9, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Nestler, E.J. Linking Molecules to Mood: New Insight Into the Biology of Depression. Am. J. Psychiatry 2010, 167, 1305–1320. [Google Scholar] [CrossRef]
- Schlaepfer, T.E.; Cohen, M.X.; Frick, C.; Kosel, M.M.; Brodesser, D.; Axmacher, N.; Joe, A.Y.; Kreft, M.; Lenartz, D.; Sturm, V. Deep Brain Stimulation to Reward Circuitry Alleviates Anhedonia in Refractory Major Depression. Neuropsychopharmacology 2007, 33, 368–377. [Google Scholar] [CrossRef]
- Price, J.L.; Drevets, W.C. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn. Sci. 2012, 16, 61–71. [Google Scholar] [CrossRef]
- Clapp, M.; Aurora, N.; Herrera, L.; Bhatia, M.; Wilen, E.; Wakefield, S. Gut Microbiota’s Effect on Mental Health: The Gut-Brain Axis. Clin. Pract. 2017, 7, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.J.K.; Milev, R. The effects of probiotics on depressive symptoms in humans: A systematic review. Ann. Gen. Psychiatry 2017, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Penninx, B.W.J.H. Oxidative stress in major depressive and anxiety disorders, and the association with antidepressant use; results from a large adult cohort. Psychol. Med. 2017, 47, 936–948. [Google Scholar] [CrossRef]
- Navarrete, F.; Garcia-Gutierrez, M.S.; Jurado-Barba, R.; Rubio, G.; Gasparyan, A.; Austrich-Olivares, A.; Manzanares, J. Endocannabinoid System Components as Potential Biomarkers in Psychiatry. Front. Psychiatry 2020, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Kennis, M.; Gerritsen, L.; Van Dalen, M.; Williams, A.; Cuijpers, P.; Bockting, C. Prospective biomarkers of major depressive disorder: A systematic review and meta-analysis. Mol. Psychiatry 2020, 25, 321–338. [Google Scholar] [CrossRef]
- Opel, N.; Cearns, M.; Clark, S.; Toben, C.; Grotegerd, D.; Heindel, W.; Kugel, H.; Teuber, A.; Minnerup, H.; Berger, K.; et al. Large-scale evidence for an association between low-grade peripheral inflammation and brain structural alterations in major depression in the BiDirect study. J. Psychiatry Neurosci. 2019, 44, 423–431. [Google Scholar] [CrossRef]
- Green, C.; Shen, X.; Stevenson, A.J.; Conole, E.L.; Harris, M.A.; Barbu, M.C.; Hawkins, E.L.; Adams, M.J.; Hillary, R.F.; Lawrie, S.M.; et al. Structural brain correlates of serum and epigenetic markers of inflammation in major depressive disorder. Brain Behav. Immun. 2021, 92, 39–48. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic Epidemiology of Major Depression: Review and Meta-Analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef]
- Bosker, F.J.; Hartman, C.A.; Nolte, I.M.; Prins, B.P.; Terpstra, P.; Posthuma, D.; van Veen, T.; Willemsen, G.; DeRijk, R.H.; de Geus, E.J.; et al. Poor replication of candidate genes for major depressive disorder using genome-wide association data. Mol. Psychiatry 2010, 16, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Pergadia, M.L.; Blackwood, D.H.R.; Penninx, B.W.J.H.; Gordon, S.D.; Nyholt, D.R.; Ripke, S.; MacIntyre, D.J.; McGhee, K.A.; Maclean, A.W.; et al. Genome-wide association study of major depressive disorder: New results, meta-analysis, and lessons learned. Mol. Psychiatry 2010, 17, 36–48. [Google Scholar] [CrossRef]
- Uher, R.; Investigators, G.; Investigators, M.; Investigators, S.D. Common Genetic Variation and Antidepressant Efficacy in Major Depressive Disorder: A Meta-Analysis of Three Genome-Wide Pharmacogenetic Studies. Am. J. Psychiatry 2013, 170, 207–217. [Google Scholar]
- Cai, N.; Bigdeli, T.B.; Kretzschmar, W.; Li, Y.; Liang, J.; Song, L.; Hu, J.; Li, Q.; Jin, W.; Hu, Z.; et al. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015, 523, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Okbay, A.; Baselmans, B.M.L.; De Neve, J.E.; Turley, P.; Nivard, M.G.; Fontana, M.A.; Meddens, S.F.W.; Linner, R.K.; Rietveld, C.A.; Derringer, J.; et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 2016, 48, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Hyde, C.L.; Nagle, M.W.; Tian, C.; Chen, X.; Paciga, S.A.; Wendland, J.R.; Tung, J.Y.; Hinds, D.A.; Perlis, R.H.; Winslow, A.R. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C. Mega-Analysis of Genome-Wide Association Studies in Major Depressive Disorder: Mdd Working Group of the Psychiatric Genomics Consortium. Eur. Neuropsychopharm. 2017, 27, S119. [Google Scholar]
- Howard, D.M.; Adams, M.J.; Shirali, M.; Clarke, T.K.; Marioni, R.E.; Davies, G.; Coleman, J.R.I.; Alloza, C.; Shen, X.Y.; Barbu, M.C.; et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 2018, 9, 1470. [Google Scholar] [CrossRef]
- Saveanu, R.V.; Nemeroff, C.B. Etiology of Depression: Genetic and Environmental Factors. Psychiatry Clin. N. Am. 2012, 35, 51–71. [Google Scholar] [CrossRef]
- Bruce, M.L. Psychosocial risk factors for depressive disorders in late life. Biol. Psychiatry 2002, 52, 175–184. [Google Scholar] [CrossRef]
- Cheptou, P.O.; Donohue, K. Epigenetics as a new avenue for the role of inbreeding depression in evolutionary ecology. Heredity 2013, 110, 205–206. [Google Scholar] [CrossRef]
- Shapero, B.G.; Black, S.K.; Liu, R.T.; Klugman, J.; Bender, R.E.; Abramson, L.Y.; Alloy, L.B. Stressful Life Events and Depression Symptoms: The Effect of Childhood Emotional Abuse on Stress Reactivity. J. Clin. Psychol. 2014, 70, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kennedy, P.J.; Nestler, E.J. Epigenetics of the Depressed Brain: Role of Histone Acetylation and Methylation. Neuropsychopharmacology 2013, 38, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Tsai, S.-J. Epigenetics and Depression: An Update. Psychiatry Investig. 2019, 16, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Petronis, A. Molecular studies of major depressive disorder: The epigenetic perspective. Mol. Psychiatry 2007, 12, 799–814. [Google Scholar] [CrossRef]
- O’Donnell, K.J.; Meaney, M.J. Epigenetics, Development, and Psychopathology. Annu. Rev. Clin. Psychol. 2020, 16, 327–350. [Google Scholar] [CrossRef]
- Meaney, M.J.; Ferguson-Smith, A.C. Epigenetic regulation of the neural transcriptome: The meaning of the marks. Nat. Neurosci. 2010, 13, 1313–1318. [Google Scholar] [CrossRef]
- Ciuculete, D.M.; Voisin, S.; Kular, L.; Jonsson, J.; Rask-Andersen, M.; Mwinyi, J.; Schioth, H.B. meQTL and ncRNA functional analyses of 102 GWAS-SNPs associated with depression implicate HACE1 and SHANK2 genes. Clin. Epigenetics 2020, 12, 99. [Google Scholar] [CrossRef]
- Wade, P.A.; Pruss, D.; Wolffe, A.P. Histone acetylation: Chromatin in action. Trends Biochem. Sci. 1997, 22, 128–132. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Marks, P.A.; Miller, T.; Richon, V.M. Histone deacetylases. Curr. Opin. Pharmacol. 2003, 3, 344–351. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Bio. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A Mammalian Histone Deacetylase Related to the Yeast Transcriptional Regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Ayer, D.E. Histone deacetylases: Transcriptional repression with SINers and NuRDs. Trends Cell Biol. 1999, 9, 193–198. [Google Scholar] [CrossRef]
- Wen, Y.D.; Perissi, V.; Staszewski, L.M.; Yang, W.M.; Krones, A.; Glass, C.K.; Rosenfeld, M.G.; Seto, E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2000, 97, 7202–7207. [Google Scholar] [CrossRef]
- Hu, E.; Chen, Z.X.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.-F.; Johanson, K.; Sung, C.-M.; Liu, R.G.; Winkler, J. Cloning and Characterization of a Novel Human Class I Histone Deacetylase That Functions as a Transcription Repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef]
- Muslin, A.J.; Xing, H.M. 14-3-3 proteins: Regulation of subcellular localization by molecular interference. Cell. Signal 2000, 12, 703–709. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Schreiber, S.L. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA 2000, 97, 7835–7840. [Google Scholar] [CrossRef]
- Wang, A.H.; Kruhlak, M.J.; Wu, J.; Bertos, N.R.; Vezmar, M.; Posner, B.I.; Bazett-Jones, D.P.; Yang, X.-J. Regulation of Histone Deacetylase 4 by Binding of 14-3-3 Proteins. Mol. Cell. Biol. 2000, 20, 6904–6912. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA 2000, 97, 14400–14405. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.-Y.; Downes, M.; Ordentlich, P.; Evans, R.M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Gene Dev. 2000, 14, 55–66. [Google Scholar]
- Zhang, H.; Okada, S.; Hatano, M.; Okabe, S.; Tokuhisa, T. A new functional domain of Bcl6 family that recruits histone deacetylases. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2001, 1540, 188–200. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and Functional Characterization of HDAC11, a Novel Member of the Human Histone Deacetylase Family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, C.B.; Sherman, J.M.; Devine, S.E.; Cameron, E.E.; Pillus, L.; Boeke, J.D. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell-cycle progression, and chromosome stability. Genes Dev. 1995, 9, 2888–2902. [Google Scholar] [CrossRef]
- Frye, R.A. Characterization of Five Human cDNAs with Homology to the Yeast SIR2 Gene: Sir2-like Proteins (Sirtuins) Metabolize NAD and May Have Protein ADP-Ribosyltransferase Activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Choi, B.H.; He, B.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef]
- LaPlant, Q.; Vialou, V.; Covington, H.E., 3rd; Dumitriu, D.; Feng, J.; Warren, B.L.; Maze, I.; Dietz, D.M.; Watts, E.L.; Iniguez, S.D.; et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1137–1143. [Google Scholar] [CrossRef]
- Hodes, G.E.; Pfau, M.L.; Purushothaman, I.; Ahn, H.F.; Golden, S.A.; Christoffel, D.J.; Magida, J.; Brancato, A.; Takahashi, A.; Flanigan, M.E.; et al. Sex Differences in Nucleus Accumbens Transcriptome Profiles Associated with Susceptibility versus Resilience to Subchronic Variable Stress. J. Neurosci. 2015, 35, 16362–16376. [Google Scholar] [CrossRef]
- Han, L.K.M.; Aghajani, M.; Clark, S.L.; Chan, R.F.; Hattab, M.W.; Shabalin, A.A.; Zhao, M.; Kumar, G.; Xie, L.Y.; Jansen, R.; et al. Epigenetic Aging in Major Depressive Disorder. Am. J. Psychiatry 2018, 175, 774–782. [Google Scholar] [CrossRef]
- Covington, H.E.; Maze, I.; LaPlant, Q.C.; Vialou, V.F.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.J.; Wu, E.Y.; et al. Antidepressant Actions of Histone Deacetylase Inhibitors. J. Neurosci. 2009, 29, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Covington, H.E.; Vialou, V.F.; LaPlant, Q.; Ohnishi, Y.N.; Nestler, E.J. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci. Lett. 2011, 493, 122–126. [Google Scholar] [CrossRef]
- Covington, H.E.; Maze, I.; Vialou, V.; Nestler, E.J. Antidepressant action of HDAC inhibition in the prefrontal cortex. Neuroscience 2015, 298, 329–335. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.E.; Akbarian, S. Antidepressant-Like Effects of the Histone Deacetylase Inhibitor, Sodium Butyrate, in the Mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef]
- Hobara, T.; Uchida, S.; Otsuki, K.; Matsubara, T.; Funato, H.; Matsuo, K.; Suetsugi, M.; Watanabe, Y. Altered gene expression of histone deacetylases in mood disorder patients. J. Psychiatry Res. 2010, 44, 263–270. [Google Scholar] [CrossRef]
- Singh, H.; Wray, N.; Schappi, J.M.; Rasenick, M.M. Disruption of lipid-raft localized Galphas/tubulin complexes by antidepressants: A unique feature of HDAC6 inhibitors, SSRI and tricyclic compounds. Neuropsychopharmacology 2018, 43, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Chmura, J.; Bhaumik, R.; Pandey, G.N.; Rasenick, M.M. Membrane-Associated α-Tubulin Is Less Acetylated in Postmortem Prefrontal Cortex from Depressed Subjects Relative to Controls: Cytoskeletal Dynamics, HDAC6, and Depression. J. Neurosci. 2020, 40, 4033–4041. [Google Scholar] [CrossRef]
- Borba, L.A.; Broseghini, L.D.; Manosso, L.M.; de Moura, A.B.; Botelho, M.E.M.; Arent, C.O.; Behenck, J.P.; Hilsendeger, A.; Kammer, L.H.; Valvassori, S.S.; et al. Environmental enrichment improves lifelong persistent behavioral and epigenetic changes induced by early-life stress. J. Psychiatry Res. 2021, 138, 107–116. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in Neurodevelopment and Brain Senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef]
- Michan, S.; Li, Y.; Chou, M.M.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 Is Essential for Normal Cognitive Function and Synaptic Plasticity. J. Neurosci. 2010, 30, 9695–9707. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Yoshimura, R.; Kitajima, T.; Okochi, T.; Okumura, T.; Tsunoka, T.; Yamanouchi, Y.; Kinoshita, Y.; Kawashima, K.; Fukuo, Y.; et al. SIRT1 gene is associated with major depressive disorder in the Japanese population. J. Affect. Disord. 2010, 126, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Abe-Higuchi, N.; Uchida, S.; Yamagata, H.; Higuchi, F.; Hobara, T.; Hara, K.; Kobayashi, A.; Watanabe, Y. Hippocampal Sirtuin 1 Signaling Mediates Depression-like Behavior. Biol. Psychiatry 2016, 80, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-D.; Hesterman, J.; Call, T.; Magazu, S.; Keeley, E.; Armenta, K.; Kronman, H.; Neve, R.L.; Nestler, E.J.; Ferguson, D. SIRT1 Mediates Depression-Like Behaviors in the Nucleus Accumbens. J. Neurosci. 2016, 36, 8441–8452. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, Y.; Wang, Y.; Liu, L.; Zhang, X.; Li, B.; Cui, R. The Effects of Psychological Stress on Depression. Curr. Neuropharmacol. 2015, 13, 494–504. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Spengler, D. Epigenetics of Early Child Development. Front. Psychiatry 2011, 2, 16. [Google Scholar] [CrossRef]
- Sandi, C.; Haller, J. Stress and the social brain: Behavioural effects and neurobiological mechanisms. Nat. Rev. Neurosci. 2015, 16, 290–304. [Google Scholar] [CrossRef]
- McEwen, B.S.; Bowles, N.P.; Gray, J.D.; Hill, M.N.; Hunter, R.G.; Karatsoreos, I.N.; Nasca, C. Mechanisms of stress in the brain. Nat. Neurosci. 2015, 18, 1353–1363. [Google Scholar] [CrossRef]
- Godoy, L.D.; Rossignoli, M.T.; Delfino-Pereira, P.; Garcia-Cairasco, N.; de Lima Umeoka, E.H. A Comprehensive Overview on Stress Neurobiology: Basic Concepts and Clinical Implications. Front. Behav. Neurosci. 2018, 12, 127. [Google Scholar] [CrossRef]
- Paul, S.; Jeon, W.K.; Bizon, J.L.; Han, J.S. Interaction of basal forebrain cholinergic neurons with the glucocorticoid system in stress regulation and cognitive impairment. Front. Aging Neurosci. 2015, 7, 43. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Spengler, D. Epigenetic programming of the HPA axis: Early life decides. Stress 2011, 14, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.M.; Swiergiel, A.H.; Lisowski, P. Epigenetics of stress adaptations in the brain. Brain Res. Bull. 2013, 98, 76–92. [Google Scholar] [CrossRef]
- Klengel, T.; Binder, E.B. Epigenetics of Stress-Related Psychiatric Disorders and Gene x Environment Interactions. Neuron 2015, 86, 1343–1357. [Google Scholar] [CrossRef]
- Levine, A.; Worrell, T.R.; Zimnisky, R.; Schmauss, C. Early life stress triggers sustained changes in histone deacetylase expression and histone H4 modifications that alter responsiveness to adolescent antidepressant treatment. Neurobiol. Dis. 2012, 45, 488–498. [Google Scholar] [CrossRef]
- Tesone-Coelho, C.; Morel, L.J.; Bhatt, J.; Estevez, L.; Naudon, L.; Giros, B.; Zwiller, J.; Dauge, V. Vulnerability to opiate intake in maternally deprived rats: Implication of MeCP2 and of histone acetylation. Addict. Biol. 2013, 20, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmuhl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Neumann, I.D.; Landgraf, R. Balance of brain oxytocin and vasopressin: Implications for anxiety, depression, and social behaviors. Trends Neurosci. 2012, 35, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Y.; Labonte, B.; Wen, X.L.; Turecki, G.; Meaney, M.J. Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans. Neuropsychopharmacology 2013, 38, 111–123. [Google Scholar] [CrossRef]
- Seo, M.K.; Kim, S.G.; Seog, D.H.; Bahk, W.M.; Kim, S.H.; Park, S.W.; Lee, J.G. Effects of Early Life Stress on Epigenetic Changes of the Glucocorticoid Receptor 17 Promoter during Adulthood. Int. J. Mol. Sci. 2020, 21, 6331. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef]
- Hing, B.; Sathyaputri, L.; Potash, J.B. A comprehensive review of genetic and epigenetic mechanisms that regulate BDNF expression and function with relevance to major depressive disorder. Am. J. Med. Genet. B 2018, 177, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Misztak, P.; Panczyszyn-Trzewik, P.; Nowak, G.; Sowa-Kucma, M. Epigenetic marks and their relationship with BDNF in the brain of suicide victims. PLoS ONE 2020, 15, e0239335. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-W.; Chen, L.Y. Epigenetic Regulation of BDNF Gene during Development and Diseases. Int. J. Mol. Sci. 2017, 18, 571. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fan, W.D.; Zhang, X.Q.; Dong, E.B. Gestational stress induces depressive-like and anxiety-like phenotypes through epigenetic regulation of BDNF expression in offspring hippocampus. Epigenetics 2016, 11, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.K.; Ly, N.N.; Lee, C.H.; Cho, H.Y.; Choi, C.M.; Nhu, L.H.; Lee, J.G.; Lee, B.J.; Kim, G.M.; Yoon, B.J.; et al. Early life stress increases stress vulnerability through BDNF gene epigenetic changes in the rat hippocampus. Neuropharmacology 2016, 105, 388–397. [Google Scholar] [CrossRef]
- Su, C.L.; Su, C.W.; Hsiao, Y.H.; Gean, P.W. Epigenetic regulation of BDNF in the learned helplessness-induced animal model of depression. J. Psychiatry Res. 2016, 76, 101–110. [Google Scholar] [CrossRef]
- Gassen, N.C.; Fries, G.R.; Zannas, A.S.; Hartmann, J.; Zschocke, J.; Hafner, K.; Carrillo-Roa, T.; Steinbacher, J.; Preissinger, S.N.; Hoeijmakers, L.; et al. Chaperoning epigenetics: FKBP51 decreases the activity of DNMT1 and mediates epigenetic effects of the antidepressant paroxetine. Sci. Signal. 2015, 8, ra119. [Google Scholar] [CrossRef]
- Varela, R.B.; Resende, W.R.; Dal-Pont, G.C.; Gava, F.F.; Tye, S.J.; Quevedo, J.; Valvassori, S.S. HDAC inhibitors reverse mania-like behavior and modulate epigenetic regulatory enzymes in an animal model of mania induced by Ouabain. Pharmacol. Biochem. Behav. 2020, 193, 172917. [Google Scholar] [CrossRef]
- Zocchi, L.; Sassone-Corsi, P. SIRT1-mediated deacetylation of MeCP2 contributes to BDNF expression. Epigenetics 2012, 7, 695–700. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krysta, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef]
- Uchida, S.; Yamagata, H.; Seki, T.; Watanabe, Y. Epigenetic mechanisms of major depression: Targeting neuronal plasticity. Psychiatry Clin. Neurosci. 2018, 72, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.J.; Bagot, R.C.; Labonte, B.; Nestler, E.J. Epigenetic Signaling in Psychiatric Disorders. J. Mol. Biol. 2014, 426, 3389–3412. [Google Scholar] [CrossRef] [PubMed]
- Labonte, B.; Engmann, O.; Purushothaman, I.; Menard, C.; Wang, J.S.; Tan, C.F.; Scarpa, J.R.; Moy, G.; Loh, Y.H.E.; Cahill, M.; et al. Sex-specific transcriptional signatures in human depression. Nat. Med. 2017, 23, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Oitzl, M.S.; Joels, M. Stress and cognition: Are corticosteroids good or bad guys? Trends Neurosci. 1999, 22, 422–426. [Google Scholar] [CrossRef]
- Guan, Z.H.; Giustetto, M.; Lomvardas, S.; Kim, J.H.; Miniaci, M.C.; Schwartz, J.H.; Thanos, D.; Kandel, E.R. Integration of Long-Term-Memory-Related Synaptic Plasticity Involves Bidirectional Regulation of Gene Expression and Chromatin Structure. Cell 2002, 111, 483–493. [Google Scholar] [CrossRef]
- Levenson, J.M.; O’Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of Histone Acetylation during Memory Formation in the Hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- Bolger, T.A.; Yao, T.P. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 2005, 25, 9544–9553. [Google Scholar] [CrossRef]
- Chen, B.; Cepko, C.L. HDAC4 Regulates Neuronal Survival in Normal and Diseased Retinas. Science 2009, 323, 256–259. [Google Scholar] [CrossRef]
- Sando, R.; Gounko, N.; Pieraut, S.; Liao, L.J.; Yates, J.; Maximov, A. HDAC4 Governs a Transcriptional Program Essential for Synaptic Plasticity and Memory. Cell 2012, 151, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Akhtar, M.W.; Adachi, M.; Mahgoub, M.; Bassel-Duby, R.; Kavalali, E.T.; Olson, E.N.; Monteggia, L.M. An Essential Role for Histone Deacetylase 4 in Synaptic Plasticity and Memory Formation. J. Neurosci. 2012, 32, 10879–10886. [Google Scholar] [CrossRef]
- Campbell, R.R.; Kramar, E.A.; Pham, L.; Beardwood, J.H.; Augustynski, A.S.; Lopez, A.J.; Chitnis, O.S.; Delima, G.; Banihani, J.; Matheos, D.P.; et al. HDAC3 Activity within the Nucleus Accumbens Regulates Cocaine-Induced Plasticity and Behavior in a Cell-Type-Specific Manner. J. Neurosci. 2021, 41, 2814–2827. [Google Scholar] [CrossRef] [PubMed]
- Schmaal, L.; Pozzi, E.; Ho, T.C.; van Velzen, L.S.; Veer, I.M.; Opel, N.; Van Someren, E.J.W.; Han, L.K.M.; Aftanas, L.; Aleman, A.; et al. ENIGMA MDD: Seven years of global neuroimaging studies of major depression through worldwide data sharing. Transl. Psychiatry 2020, 10, 172. [Google Scholar] [CrossRef]
- Erjavec, G.N.; Sagud, M.; Perkovic, M.N.; Strac, D.S.; Konjevod, M.; Tudor, L.; Uzun, S.; Pivac, N. Depression: Biological markers and treatment. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2021, 105, 110139. [Google Scholar] [CrossRef]
- Gadad, B.S.; Jha, M.K.; Czysz, A.; Furman, J.L.; Mayes, T.L.; Emslie, M.P.; Trivedi, M.H. Peripheral biomarkers of major depression and antidepressant treatment response: Current knowledge and future outlooks. J. Affect. Disord. 2018, 233, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Le-Niculescu, H.; Roseberry, K.; Gill, S.S.; Levey, D.F.; Phalen, P.L.; Mullen, J.; Williams, A.; Bhairo, S.; Voegtline, T.; Davis, H.; et al. Precision medicine for mood disorders: Objective assessment, risk prediction, pharmacogenomics, and repurposed drugs. Mol. Psychiatry 2021, 1–29. [Google Scholar] [CrossRef]
- Himmerich, H.; Milenovic, S.; Fulda, S.; Plumakers, B.; Sheldrick, A.J.; Michel, T.M.; Kircher, T.; Rink, L. Regulatory T cells increased while IL-1 beta decreased during antidepressant therapy. J. Psychiatr. Res. 2010, 44, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Gennarelli, M.; Uher, R.; Breen, G.; Farmer, A.; Aitchison, K.J.; Craig, I.W.; Anacker, C.; Zunsztain, P.A.; McGuffin, P.; et al. Candidate Genes Expression Profile Associated with Antidepressants Response in the GENDEP Study: Differentiating between Baseline ‘Predictors’ and Longitudinal ‘Targets’. Neuropsychopharmacology 2013, 38, 377–385. [Google Scholar] [CrossRef]
- Cattaneo, A.; Ferrari, C.; Uher, R.; Bocchio-Chiavetto, L.; Riva, M.A.; Pariante, C.M. Absolute Measurements of Macrophage Migration Inhibitory Factor and Interleukin-1-beta mRNA Levels Accurately Predict Treatment Response in Depressed Patients. Int. J. Neuropsychoph. 2016, 19, pyw045. [Google Scholar] [CrossRef]
- Belzeaux, R.; Lin, R.X.; Ju, C.; Chay, M.A.; Fiori, L.M.; Lutz, P.E.; Turecki, G. Transcriptomic and epigenomic biomarkers of antidepressant response. J. Affect. Disord. 2018, 233, 36–44. [Google Scholar] [CrossRef]
- Le-Niculescu, H.; Kurian, S.M.; Yehyawi, N.; Dike, C.; Patel, S.D.; Edenberg, H.J.; Tsuang, M.T.; Salomon, D.R.; Nurnberger, J.I., Jr.; Niculescu, A.B. Identifying blood biomarkers for mood disorders using convergent functional genomics. Mol. Psychiatry 2009, 14, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Buch, A.M.; Liston, C. Dissecting diagnostic heterogeneity in depression by integrating neuroimaging and genetics. Neuropsychopharmacology 2021, 46, 156–175. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.K.; Cohen, J.D. An Integrative Theory of Prefrontal Cortex Function. Annu. Rev. Neurosci. 2001, 24, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.M.; Meador-Woodruff, J.H. Post-translational protein modifications in schizophrenia. NPJ Schizophr. 2020, 6, 1–16. [Google Scholar] [CrossRef]
- Jovanova, O.S.; Nedeljkovic, I.; Spieler, D.; Walker, R.M.; Liu, C.Y.; Luciano, M.; Bressler, J.; Brody, J.; Drake, A.J.; Evans, K.L.; et al. DNA Methylation Signatures of Depressive Symptoms in Middle-aged and Elderly Persons Meta-analysis of Multiethnic Epigenome-wide Studies. JAMA Psychiatry 2018, 75, 949–959. [Google Scholar] [CrossRef]
- Li, M.; D’Arcy, C.; Li, X.; Zhang, T.; Joober, R.; Meng, X. What do DNA methylation studies tell us about depression? A systematic review. Transl. Psychiatry 2019, 9, 68. [Google Scholar] [CrossRef]
- Lopez, J.P.; Mamdani, F.; Labonte, B.; Beaulieu, M.M.; Yang, J.P.; Berlim, M.T.; Ernst, C.; Turecki, G. Epigenetic regulation of BDNF expression according to antidepressant response. Mol. Psychiatry 2013, 18, 398–399. [Google Scholar] [CrossRef]
- Iga, J.; Ueno, S.; Yamauchi, K.; Numata, S.; Kinouchi, S.; Tayoshi-Shibuya, S.; Song, H.W.; Ohmori, T. Altered HDAC5 and CREB mRNA expressions in the peripheral leukocytes of major depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2007, 31, 628–632. [Google Scholar] [CrossRef]
- Nasca, C.; Bigio, B.; Lee, F.S.; Young, S.P.; Kautz, M.M.; Albright, A.; Beasley, J.; Millington, D.S.; Mathé, A.A.; Kocsis, J.H.; et al. Acetyl-l-carnitine deficiency in patients with major depressive disorder. Proc. Natl. Acad. Sci. USA 2018, 115, 8627–8632. [Google Scholar] [CrossRef]
- Wey, H.-Y.; Wang, C.; Schroeder, F.A.; Logan, J.; Price, J.C.; Hooker, J.M. Kinetic Analysis and Quantification of [11C]Martinostat for in Vivo HDAC Imaging of the Brain. ACS Chem. Neurosci. 2015, 6, 708–715. [Google Scholar] [CrossRef]
- Wey, H.Y.; Gilbert, T.M.; Zurcher, N.R.; She, A.; Bhanot, A.; Taillon, B.D.; Schroeder, F.A.; Wang, C.; Haggarty, S.J.; Hooker, J.M. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci. Transl. Med. 2016, 8, 351ra106. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, T.M.; Zürcher, N.R.; Wu, C.J.; Bhanot, A.; Hightower, B.G.; Kim, M.; Albrecht, D.S.; Wey, H.-Y.; Schroeder, F.A.; Rodriguez-Thompson, A.; et al. PET neuroimaging reveals histone deacetylase dysregulation in schizophrenia. J. Clin. Investig. 2019, 129, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-E.J.; Gilbert, T.M.; Catanese, M.C.; Hightower, B.G.; Peters, A.T.; Parmar, A.J.; Kim, M.; Wang, C.; Roffman, J.L.; Brown, H.E.; et al. In vivo human brain expression of histone deacetylases in bipolar disorder. Transl. Psychiatry 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Toth, M. Epigenetic Neuropharmacology: Drugs Affecting the Epigenome in the Brain. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Ezra-Nevo, G.; Regev, L.; Neufeld-Cohen, A.; Chen, A. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat. Neurosci. 2010, 13, 1351–1353. [Google Scholar] [CrossRef]
- Ookubo, M.; Kanai, H.; Aoki, H.; Yamada, N. Antidepressants and mood stabilizers effects on histone deacetylase expression in C57BL/6 mice: Brain region specific changes. J. Psychiatry Res. 2013, 47, 1204–1214. [Google Scholar] [CrossRef]
- Deussing, J.M.; Jakovcevski, M. Histone Modifications in Major Depressive Disorder and Related Rodent Models. Adv. Exp. Med. Biol. 2017, 978, 169–183. [Google Scholar] [CrossRef]
- Fuchikami, M.; Yamamoto, S.; Morinobu, S.; Okada, S.; Yamawaki, Y.; Yamawaki, S. The potential use of histone deacetylase inhibitors in the treatment of depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016, 64, 320–324. [Google Scholar] [CrossRef]
- Uchida, S.; Hara, K.; Kobayashi, A.; Otsuki, K.; Yamagata, H.; Hobara, T.; Suzuki, T.; Miyata, N.; Watanabe, Y. Epigenetic Status of Gdnf in the Ventral Striatum Determines Susceptibility and Adaptation to Daily Stressful Events. Neuron 2011, 69, 359–372. [Google Scholar] [CrossRef]
- Golden, S.A.; Christoffel, D.J.; Heshmati, M.; Hodes, G.E.; Magida, J.; Davis, K.; Cahill, M.E.; Dias, C.; Ribeiro, E.; Ables, J.L.; et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat. Med. 2013, 19, 337–344. [Google Scholar] [CrossRef]
- Han, A.; Sung, Y.-B.; Chung, S.-Y.; Kwon, M.-S. Possible additional antidepressant-like mechanism of sodium butyrate: Targeting the hippocampus. Neuropharmacology 2014, 81, 292–302. [Google Scholar] [CrossRef]
- Sada, N.; Fujita, Y.; Mizuta, N.; Ueno, M.; Furukawa, T.; Yamashita, T. Inhibition of HDAC increases BDNF expression and promotes neuronal rewiring and functional recovery after brain injury. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Athira, K.V.; Madhana, R.M.; Bais, A.K.; Singh, V.B.; Malik, A.; Sinha, S.; Lahkar, M.; Kumar, P.; Samudrala, P.K. Cognitive Improvement by Vorinostat through Modulation of Endoplasmic Reticulum Stress in a Corticosterone-Induced Chronic Stress Model in Mice. ACS Chem. Neurosci. 2020, 11, 2649–2657. [Google Scholar] [CrossRef]
- Ershadi, A.S.B.; Amini-Khoei, H.; Hosseini, M.-J.; Dehpour, A.R. SAHA Improves Depressive Symptoms, Cognitive Impairment and Oxidative Stress: Rise of a New Antidepressant Class. Neurochem. Res. 2021, 46, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Vinarskaya, A.K.; Balaban, P.M.; Roshchin, M.V.; Zuzina, A.B. Sodium butyrate as a selective cognitive enhancer for weak or impaired memory. Neurobiol. Learn. Mem. 2021, 180, 107414. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Bishton, M.J.; Harrison, S.J. Clinical Studies of Histone Deacetylase Inhibitors. Clin. Cancer Res. 2009, 15, 3958–3969. [Google Scholar] [CrossRef]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef]
- Majchrzak-Celinska, A.; Warych, A.; Szoszkiewicz, M. Novel Approaches to Epigenetic Therapies: From Drug Combinations to Epigenetic Editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef]
- Karnib, N.; El-Ghandour, R.; El Hayek, L.; Nasrallah, P.; Khalifeh, M.; Barmo, N.; Jabre, V.; Ibrahim, P.; Bilen, M.; Stephan, J.; et al. Lactate is an antidepressant that mediates resilience to stress by modulating the hippocampal levels and activity of histone deacetylases. Neuropsychopharmacology 2019, 44, 1152–1162. [Google Scholar] [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9, 477. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Class | Protein (S. cerevisiae) | Protein (Human) | Subcellular Localization |
|---|---|---|---|
| Class I | Rpd3 | HDAC1 | Nucleus |
| HDAC2 | Nucleus | ||
| HDAC3 | Nucleus | ||
| HDAC8 | Nucleus | ||
| Class IIa | Hda1 | HDAC4 | Nucleus/cytoplasm |
| HDAC5 | Nucleus/cytoplasm | ||
| HDAC7 | Nucleus/cytoplasm | ||
| HDAC9 | Nucleus/cytoplasm | ||
| Class IIb | Hda1 | HDAC6 | Cytoplasm |
| HDAC10 | Cytoplasm | ||
| Class IV | Hos3 | HDAC11 | Nucleus/cytoplasm |
| Class III | Sir2 | SIRT1 | Nucleus/cytoplasm |
| SIRT2 | Nucleus/cytoplasm | ||
| SIRT3 | Nucleus/mitochondria | ||
| SIRT4 | Mitochondria | ||
| SIRT5 | Mitochondria | ||
| SIRT6 | Nucleus | ||
| SIRT7 | Nucleus |
| HDAC Inhibitor | Animal Model | Measurement of Antidepressant Effect | Molecular Mechanisms of Action | Ref. |
|---|---|---|---|---|
| MS-275 | Chronic social defeat stress | Social avoidance, sucrose preference, FST | acH3 ↑ in the NAc | [62] |
| Chronic social defeat stress | Sucrose preference test, social avoidance (combined with social enrichment) | acH3 ↑ in the hippocampus | [63] | |
| Chronic social defeat stress | Social avoidance, FST | acH3 ↑ in the mPFC | [64] | |
| Chronic social defeat stress | Social avoidance | Rac1 ↑ in the NAc synapse structural plasticity normalization | [141] | |
| SAHA | Chronic social defeat stress | Social avoidance, sucrose preference, FST | acH3 ↑ in the NAc | [62] |
| Chronic unpredictable mild stress | Social interaction, sucrose preference test, novelty-suppressed test, FST | HDAC2 inhibition, Gdnf ↑ in the NAc | [140] | |
| Sodium butyrate | Behavioral despair paradigm | TST | acH3 ↑ in the hippocampus, Bdnf↑ in the frontal cortex | [65] |
| Chronic social defeat stress | Social avoidance | HDAC5 inhibition, acH3 ↑ in Bdnf gene P3, P4 promotor | [66] | |
| Chronic restraint stress | Sucrose preference test, Light/dark test, TST, FST | HDAC2 ↑, pCREB ↑, AcH3 ↑, BDNF ↑ in the hippocampus | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-S.; Kim, J.; Ahn, S.H.; Ryu, H.-Y. Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression. Int. J. Mol. Sci. 2021, 22, 5398. https://doi.org/10.3390/ijms22105398
Park H-S, Kim J, Ahn SH, Ryu H-Y. Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression. International Journal of Molecular Sciences. 2021; 22(10):5398. https://doi.org/10.3390/ijms22105398
Chicago/Turabian StylePark, Hyun-Sun, Jongmin Kim, Seong Hoon Ahn, and Hong-Yeoul Ryu. 2021. "Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression" International Journal of Molecular Sciences 22, no. 10: 5398. https://doi.org/10.3390/ijms22105398
APA StylePark, H.-S., Kim, J., Ahn, S. H., & Ryu, H.-Y. (2021). Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression. International Journal of Molecular Sciences, 22(10), 5398. https://doi.org/10.3390/ijms22105398