
A Negative Energy Balance Is Associated with Metabolic Dysfunctions in the Hypothalamus of a Humanized Preclinical Model of Alzheimer’s Disease, the 5XFAD Mouse

 ,
,  ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Results
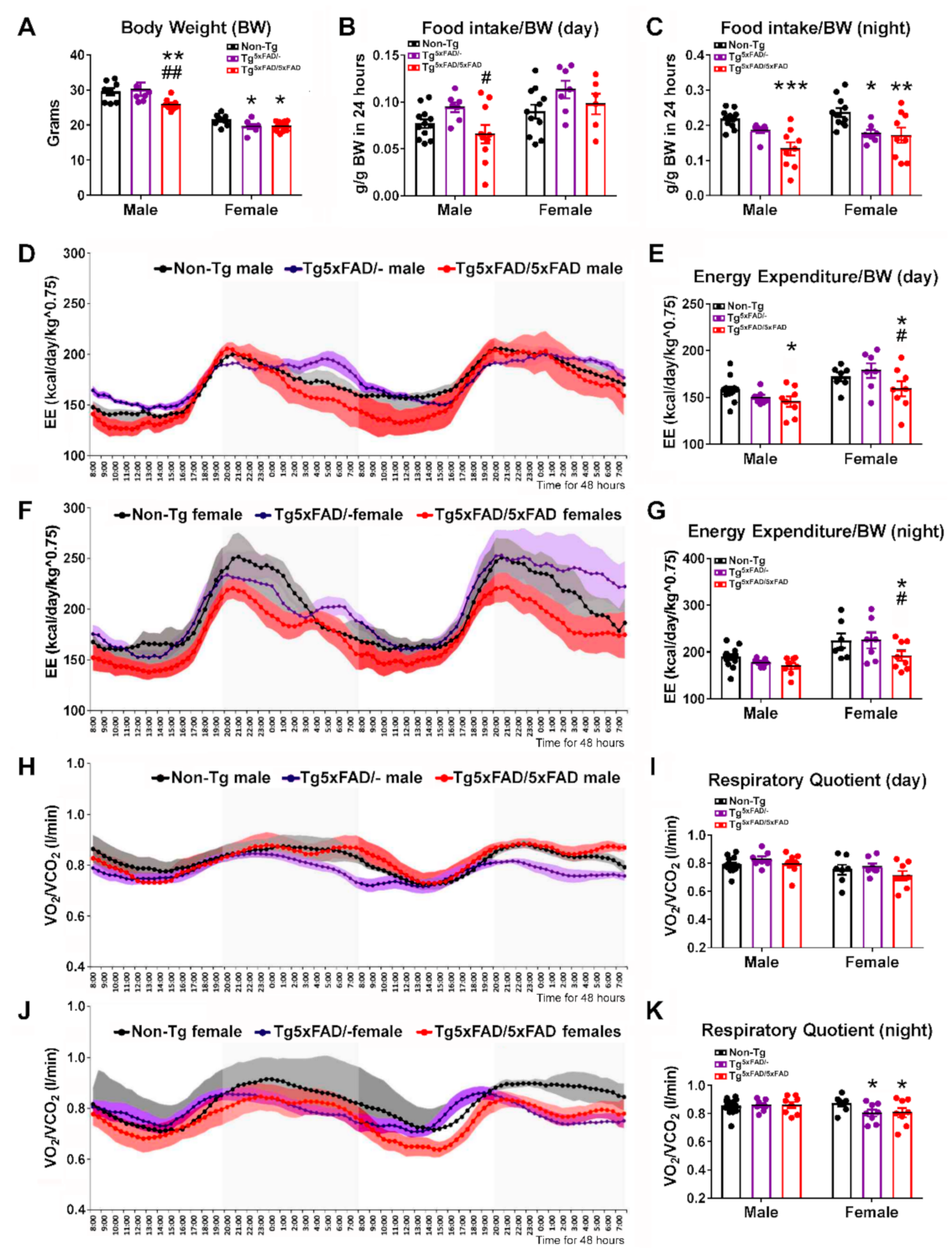
2.1. Heterozygous (Tg5xFAD/-) and Homozygous (Tg5xFAD/5xFAD) Transgenic 5xFAD Mice Showed Decreased Body Weight, Food Intake, and Energy Expenditure at 6 Months
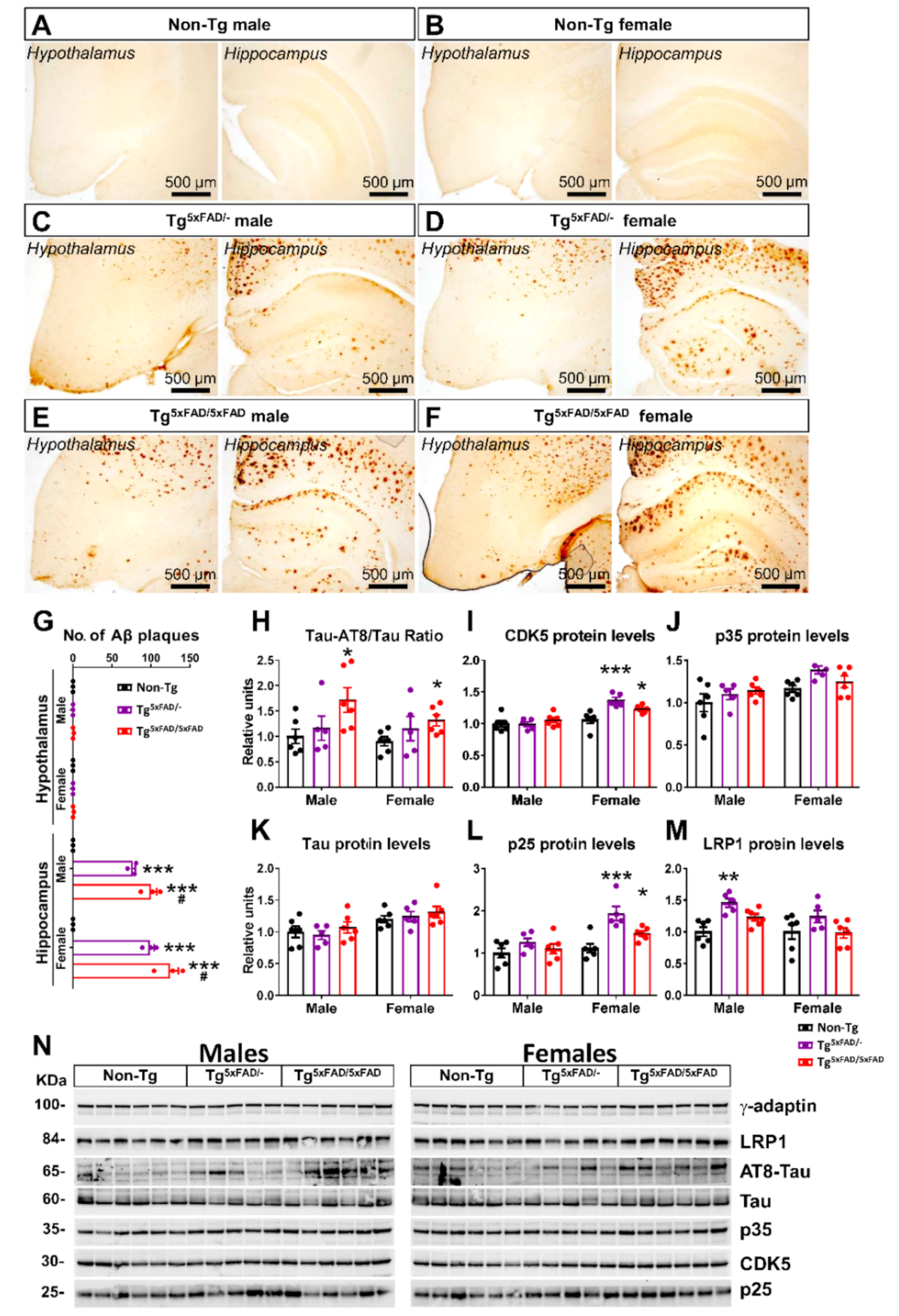
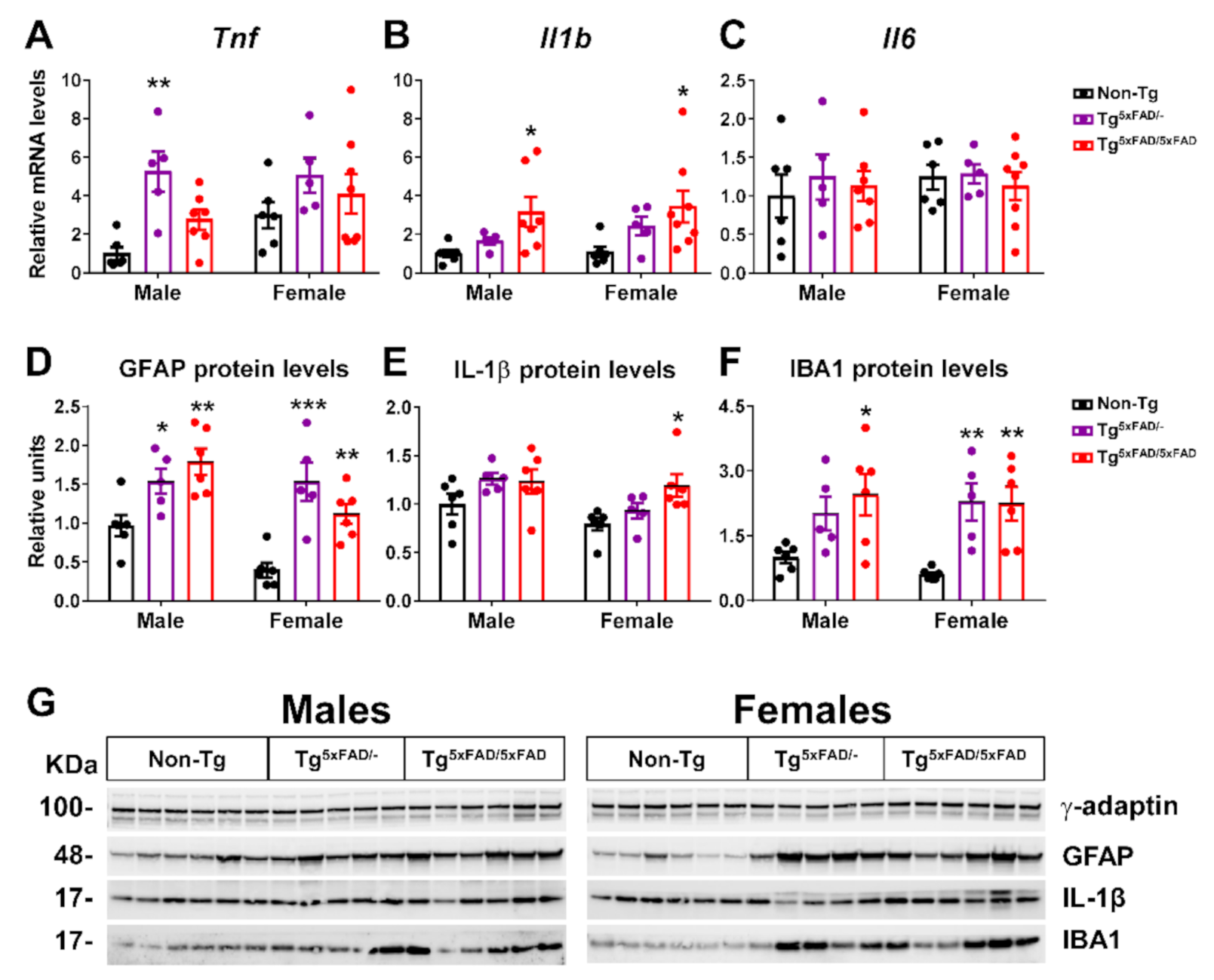
2.2. Heterozygous (Tg5xFAD/-) and Homozygous (Tg5xFAD/5xFAD) Transgenic 5xFAD Mice Showed Increased Hypothalamic Tau Phosphorylation and Inflammation Despite Absence of Aβ Plaques in the Hypothalamus
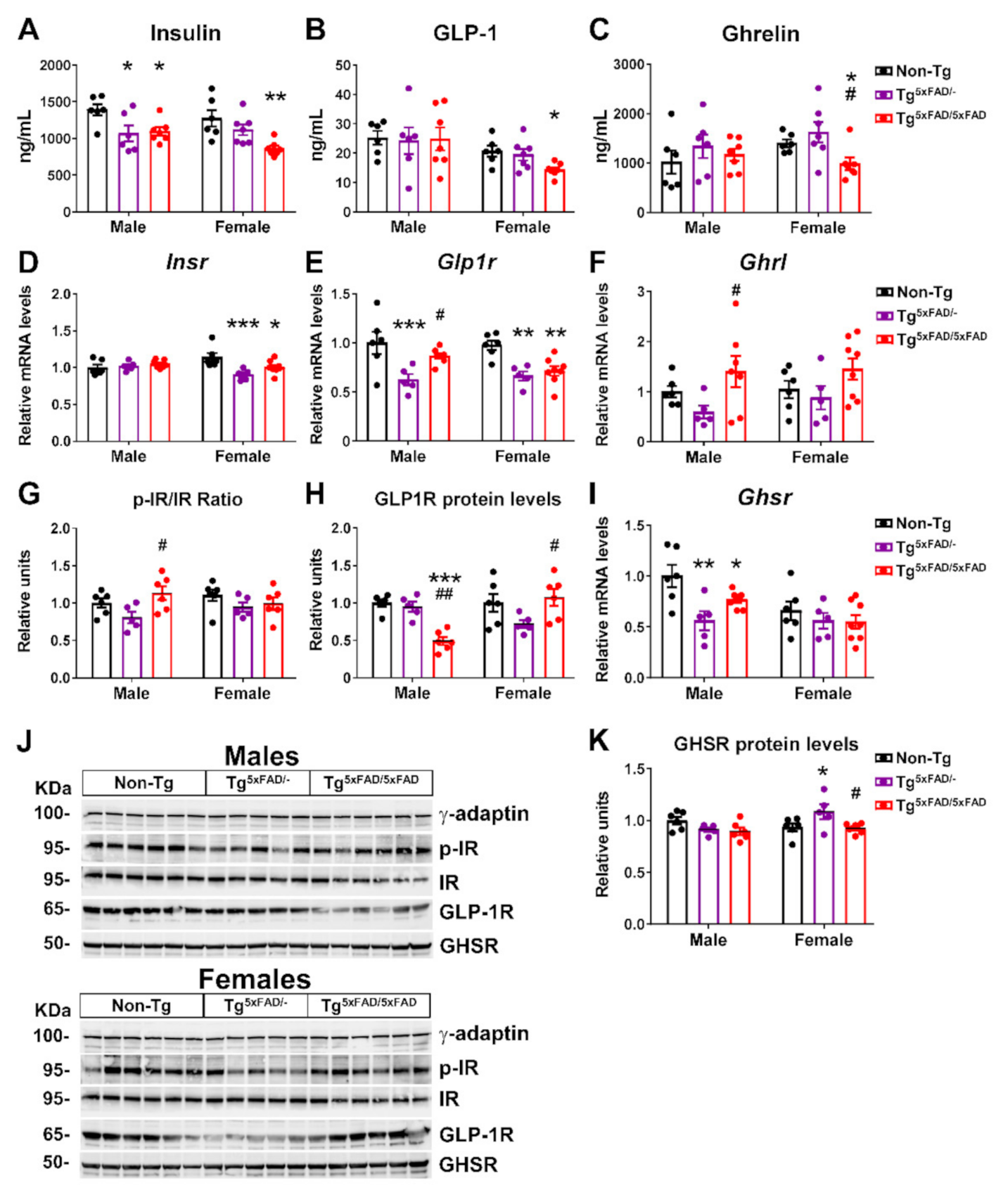
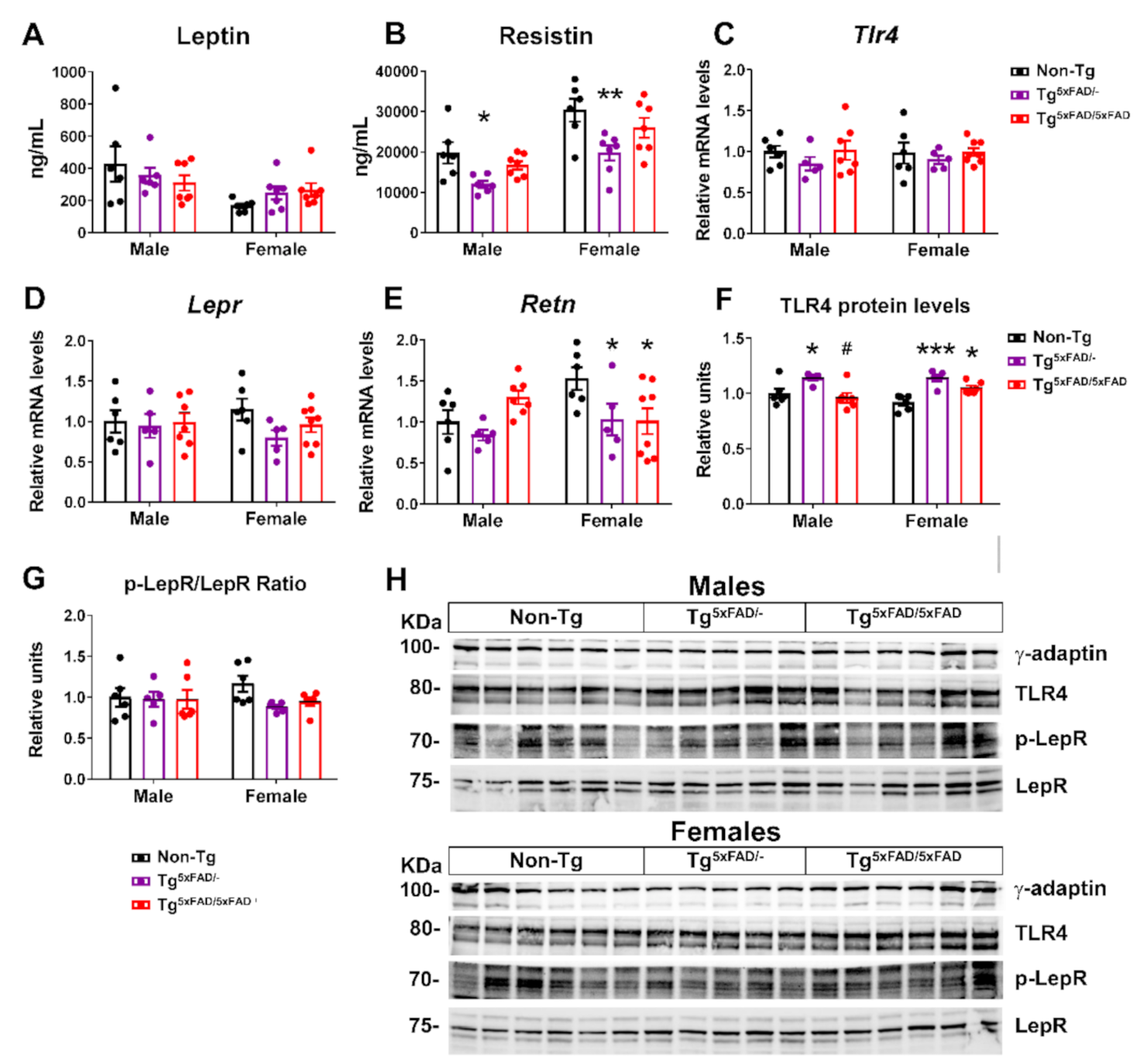
2.3. Transgenic 5xFAD Mice Had Low Circulating Levels of Insulin, GLP-1, Ghrelin, and Resistin, as Well as Altered Activity and Expression of Hormone Receptors in the Hypothalamus
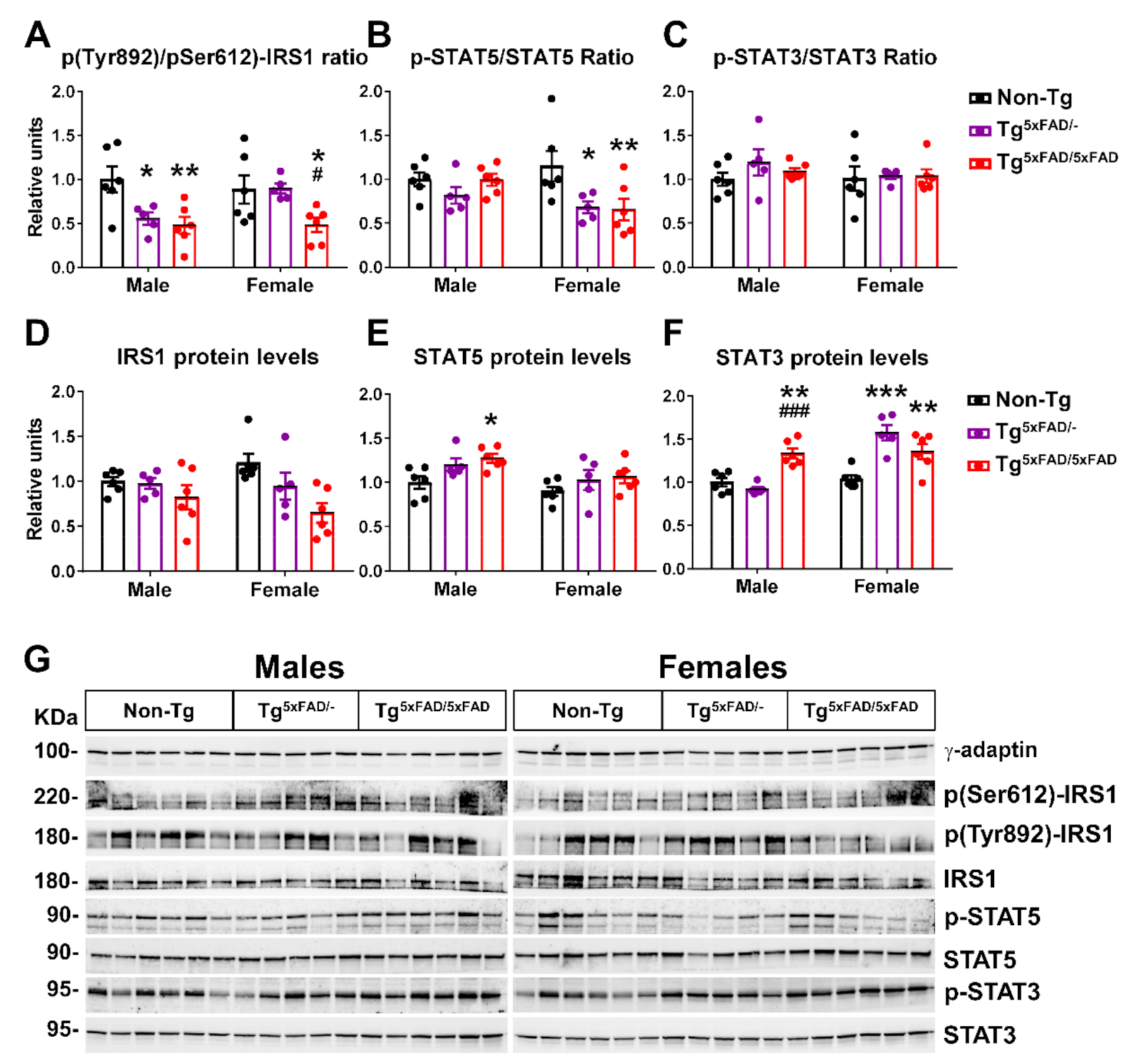
2.4. Insulin and Leptin Signaling Was Decreased in Hypothalamus of Transgenic 5xFAD Mice at 6 Months of Age
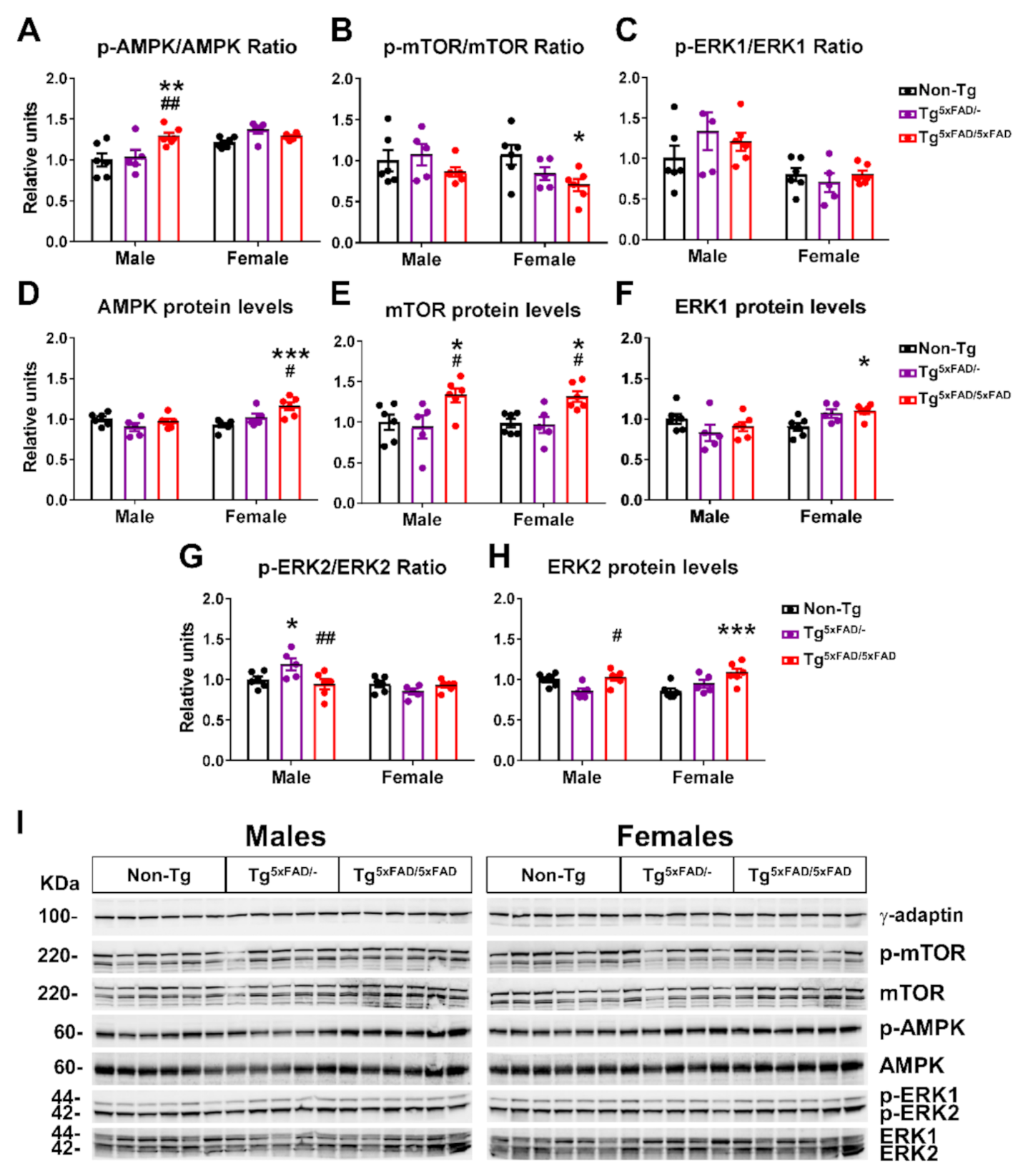
2.5. Hypothalamic Energy Sensors Were Dysregulated in Transgenic 5xFAD Mice at 6 Months of Age
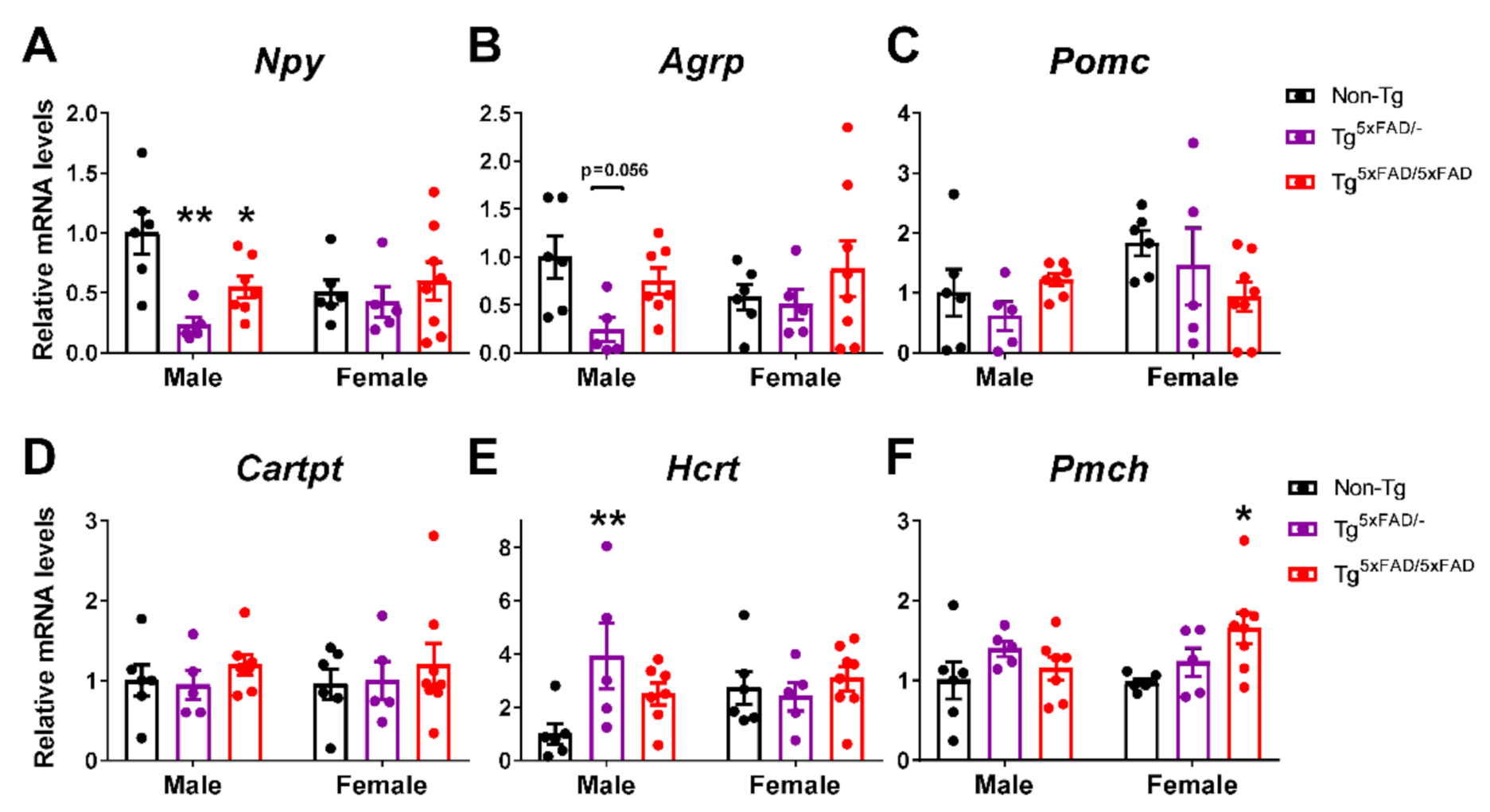
2.6. A Decrease in Orexigenic Neuropeptides Was Observed in Male Transgenic 5xFAD Mice at 6 Months of Age
2.7. Lipid Plasma Profile Was Altered in Female 5xFAD Mice
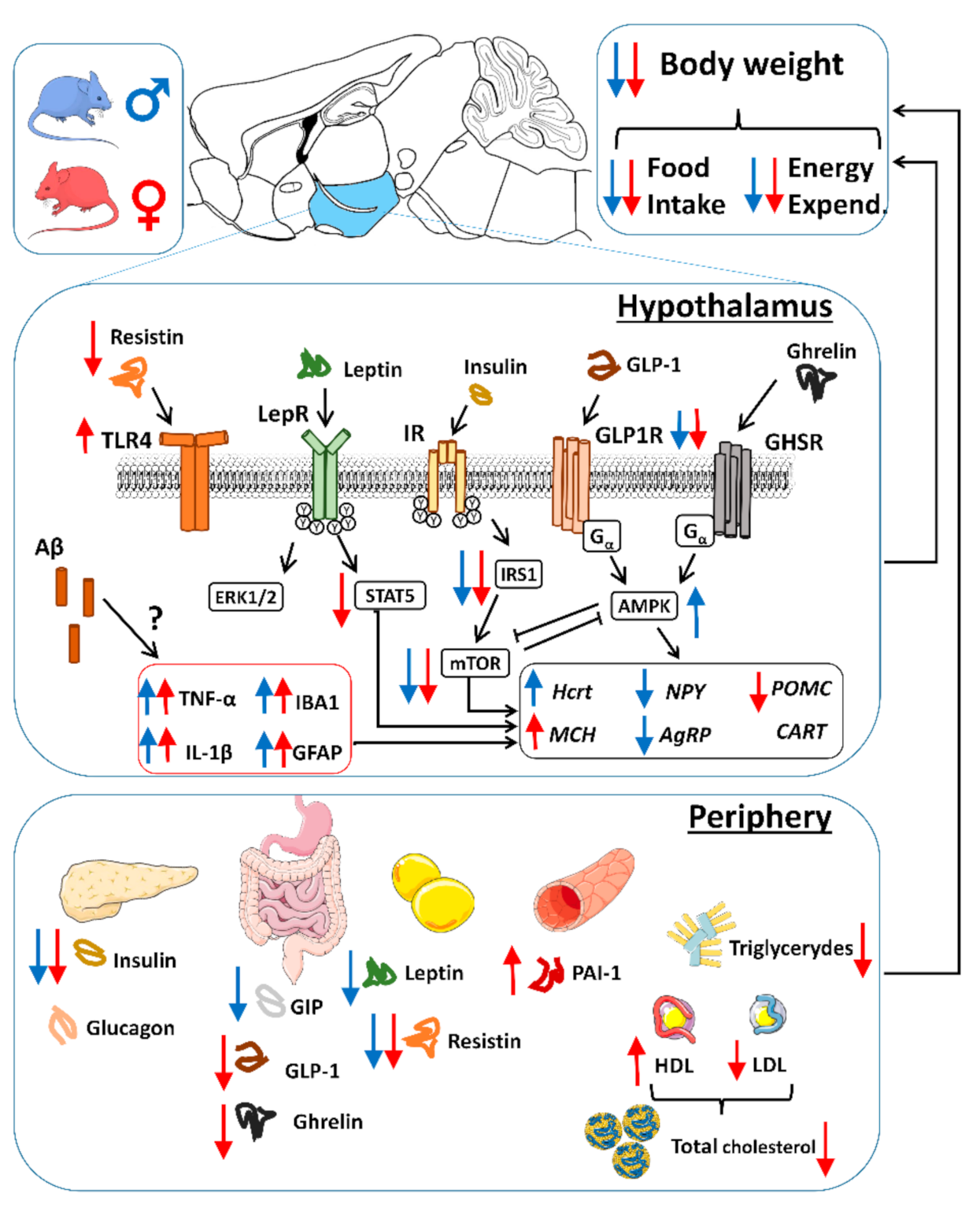
3. Discussion
3.1. Body Weight Deficit in 5xFAD Mice
3.2. Alterations in Food Intake and Energy Expenditure in 5xFAD Mice
3.3. Aβ Pathology and Neuroinflammation in the Hypothalamus of 5xFAD Mice
3.4. Alterations in Plasma Hormones and Hypothalamic Signaling in 5xFAD Mice
3.5. Alterations in Energy Sensors in 5xFAD Mice
3.6. Alterations in Neuropeptides in 5xFAD Mice
3.7. Plasma Lipid Profile and Increased PAI-1 Levels in Female 5xFAD Mice
3.8. Limitations of the Study
4. Conclusions
5. Materials and Methods
5.1. Ethics Statement
5.2. Animals
5.3. Sample Collection
5.4. Measurement of Metabolites in Plasma
5.5. Bio-Plex Pro Multiplex Diabetes Assay
5.6. RNA Isolation and RT-qPCR Analysis
5.7. Protein Extraction and Western Blot Analysis
5.8. Immunohistochemistry
5.9. Measurement of Energy Expenditure and Respiratory Quotient
5.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azzu, V.; Valencak, T.G. Energy Metabolism and Ageing in the Mouse: A Mini-Review. Gerontology 2017, 63, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Milstein, J.L.; Ferris, H.A. The brain as an insulin-sensitive metabolic organ. Mol. Metab. 2021, 101234. [Google Scholar] [CrossRef]
- López-Gambero, A.J.; Sanjuan, C.; Serrano-Castro, P.J.; Suárez, J.; Rodríguez de Fonseca, F. The Biomedical Uses of Inositols: A Nutraceutical Approach to Metabolic Dysfunction in Aging and Neurodegenerative Diseases. Biomedicines 2020, 8, 295. [Google Scholar] [CrossRef]
- Pitt, J.; Thorner, M.; Brautigan, D.; Larner, J.; Klein, W.L. Protection against the synaptic targeting and toxicity of Alzheimer’s-associated Aβ oligomers by insulin mimetic chiro-inositols. FASEB J. 2013, 27, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase-A protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 480–487. [Google Scholar] [CrossRef] [PubMed]
- McGuire, M.J.; Ishii, M. Leptin Dysfunction and Alzheimer’s Disease: Evidence from Cellular, Animal, and Human Studies. Cell. Mol. Neurobiol. 2016, 36, 203–217. [Google Scholar] [CrossRef]
- Johnson, D.K.; Wilkins, C.H.; Morris, J.C. Accelerated weight loss may precede diagnosis in Alzheimer disease. Arch. Neurol. 2006, 63, 1312–1317. [Google Scholar] [CrossRef]
- Vidoni, E.D.; Townley, R.A.; Honea, R.A.; Burns, J.M. Alzheimer disease biomarkers are associated with body mass index. Neurology 2011, 77, 1913–1920. [Google Scholar] [CrossRef]
- Do, K.; Laing, B.T.; Landry, T.; Bunner, W.; Mersaud, N.; Matsubara, T.; Li, P.; Yuan, Y.; Lu, Q.; Huang, H. The effects of exercise on hypothalamic neurodegeneration of Alzheimer’s disease mouse model. PLoS ONE 2018, 13, e0190205. [Google Scholar] [CrossRef] [PubMed]
- Knight, E.M.; Verkhratsky, A.; Luckman, S.M.; Allan, S.M.; Lawrence, C.B. Hypermetabolism in a triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Wang, G.; Racchumi, G.; Dyke, J.P.; Iadecola, C. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 9096–9106. [Google Scholar] [CrossRef]
- Zhao, L.; Mao, Z.; Woody, S.K.; Brinton, R.D. Sex differences in metabolic aging of the brain: Insights into female susceptibility to Alzheimer’s disease. Neurobiol. Aging 2016, 42, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Ohno, M.; Chang, L.; Tseng, W.; Oakley, H.; Citron, M.; Klein, W.L.; Vassar, R.; Disterhoft, J.F. Temporal memory deficits in Alzheimer’s mouse models: Rescue by genetic deletion of BACE1. Eur. J. Neurosci. 2006, 23, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Devi, L.; Ohno, M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J. Neurochem. 2010, 113, 248–261. [Google Scholar] [CrossRef]
- Xiao, N.A.; Zhang, J.; Zhou, M.; Wei, Z.; Wu, X.L.; Dai, X.M.; Zhu, Y.G.; Chen, X.C. Reduction of Glucose Metabolism in Olfactory Bulb is an Earlier Alzheimer’s Disease-related Biomarker in 5XFAD Mice. Chin. Med. J. 2015, 128, 2220–2227. [Google Scholar] [CrossRef]
- López-Gambero, A.J.; Martínez, F.; Salazar, K.; Cifuentes, M.; Nualart, F. Brain Glucose-Sensing Mechanism and Energy Homeostasis. Mol. Neurobiol. 2019, 56, 769–796. [Google Scholar] [CrossRef]
- Kimura, T.; Ishiguro, K.; Hisanaga, S.-I. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef]
- Van Gool, B.; Storck, S.E.; Reekmans, S.M.; Lechat, B.; Gordts, P.L.S.M.; Pradier, L.; Pietrzik, C.U.; Roebroek, A.J.M. LRP1 Has a Predominant Role in Production over Clearance of Aβ in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7234–7245. [Google Scholar] [CrossRef] [PubMed]
- Kulas, J.A.; Puig, K.L.; Combs, C.K. Amyloid precursor protein in pancreatic islets. J. Endocrinol. 2017, 235, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Meloni, A.R.; DeYoung, M.B.; Lowe, C.; Parkes, D.G. GLP-1 receptor activated insulin secretion from pancreatic β-cells: Mechanism and glucose dependence. Diabetes Obes. Metab. 2013, 15, 15–27. [Google Scholar] [CrossRef] [PubMed]
- de Luis, D.A.; Gonzalez Sagrado, M.; Conde, R.; Aller, R.; Izaola, O. Decreased basal levels of glucagon-like peptide-1 after weight loss in obese subjects. Ann. Nutr. Metab. 2007, 51, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Nakazato, M. Is ghrelin a glucagon-like peptide-1 secretagogue? J. Diabetes Investig. 2016, 7, 466–467. [Google Scholar] [CrossRef]
- Gui, Y.; Silha, J.V.; Murphy, L.J. Sexual dimorphism and regulation of resistin, adiponectin, and leptin expression in the mouse. Obes. Res. 2004, 12, 1481–1491. [Google Scholar] [CrossRef]
- Benomar, Y.; Taouis, M. Molecular Mechanisms Underlying Obesity-Induced Hypothalamic Inflammation and Insulin Resistance: Pivotal Role of Resistin/TLR4 Pathways. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Singhal, N.S.; Lazar, M.A.; Ahima, R.S. Central Resistin Induces Hepatic Insulin Resistance via Neuropeptide Y. J. Neurosci. 2007, 27, 12924–12932. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. CMLS 2016, 73, 1457–1477. [Google Scholar] [CrossRef]
- Gong, Y.; Ishida-Takahashi, R.; Villanueva, E.C.; Fingar, D.C.; Münzberg, H.; Myers, M.G., Jr. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J. Biol. Chem. 2007, 282, 31019–31027. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Courchet, J.; Viollet, B.; Brenman, J.E.; Polleux, F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc. Natl. Acad. Sci. USA 2011, 108, 5849–5854. [Google Scholar] [CrossRef]
- Hua, Z.; Gu, X.; Dong, Y.; Tan, F.; Liu, Z.; Thiele, C.J.; Li, Z. PI3K and MAPK pathways mediate the BDNF/TrkB-increased metastasis in neuroblastoma. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Woods, S.C.; Porte, D.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.; Cowley, M.A.; Butler, A.; Fan, W.; Marks, D.; Low, M. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int. J. Obes. 2001, 25, S63–S67. [Google Scholar] [CrossRef]
- Barson, J.R.; Morganstern, I.; Leibowitz, S.F. Complementary roles of orexin and melanin-concentrating hormone in feeding behavior. Int. J. Endocrinol. 2013, 2013, 983964. [Google Scholar] [CrossRef]
- Reitz, C.; Tang, M.X.; Luchsinger, J.; Mayeux, R. Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch. Neurol. 2004, 61, 705–714. [Google Scholar] [CrossRef]
- Notkola, I.L.; Sulkava, R.; Pekkanen, J.; Erkinjuntti, T.; Ehnholm, C.; Kivinen, P.; Tuomilehto, J.; Nissinen, A. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology 1998, 17, 14–20. [Google Scholar] [CrossRef]
- Gu, Y.; Scarmeas, N.; Cosentino, S.; Brandt, J.; Albert, M.; Blacker, D.; Dubois, B.; Stern, Y. Change in body mass index before and after Alzheimer’s disease onset. Curr. Alzheimer Res. 2014, 11, 349–356. [Google Scholar] [CrossRef]
- Cova, I.; Clerici, F.; Rossi, A.; Cucumo, V.; Ghiretti, R.; Maggiore, L.; Pomati, S.; Galimberti, D.; Scarpini, E.; Mariani, C.; et al. Weight Loss Predicts Progression of Mild Cognitive Impairment to Alzheimer’s Disease. PLoS ONE 2016, 11, e0151710. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Olloquequi, J.; Ettcheto, M.; Busquets, O.; Sánchez-López, E.; Cano, A.; Espinosa-Jiménez, T.; García, M.L.; Beas-Zarate, C.; Casadesús, G.; et al. The Involvement of Peripheral and Brain Insulin Resistance in Late Onset Alzheimer’s Dementia. Front. Aging Neurosci. 2019, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Laws, K.R.; Irvine, K.; Gale, T.M. Sex differences in Alzheimer’s disease. Curr. Opin. Psychiatry 2018, 31, 133–139. [Google Scholar] [CrossRef]
- Reilly, A.M.; Tsai, A.P.; Lin, P.B.; Ericsson, A.C.; Oblak, A.L.; Ren, H. Metabolic Defects Caused by High-Fat Diet Modify Disease Risk through Inflammatory and Amyloidogenic Pathways in a Mouse Model of Alzheimer’s Disease. Nutrients 2020, 12, 2977. [Google Scholar] [CrossRef]
- Gendron, W.H.; Fertan, E.; Pelletier, S.; Roddick, K.M.; O’Leary, T.P.; Anini, Y.; Brown, R.E. Age Related Weight Loss in Female 5xFAD Mice from 3 to 12 Months of Age. Behav. Brain Res. 2021, 113214. [Google Scholar] [CrossRef]
- Robison, L.S.; Gannon, O.J.; Thomas, M.A.; Salinero, A.E.; Abi-Ghanem, C.; Poitelon, Y.; Belin, S.; Zuloaga, K.L. Role of sex and high-fat diet in metabolic and hypothalamic disturbances in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 285. [Google Scholar] [CrossRef]
- White, H.; Pieper, C.; Schmader, K. The association of weight change in Alzheimer’s disease with severity of disease and mortality: A longitudinal analysis. J. Am. Geriatr. Soc. 1998, 46, 1223–1227. [Google Scholar] [CrossRef]
- Gillette Guyonnet, S.; Abellan Van Kan, G.; Alix, E.; Andrieu, S.; Belmin, J.; Berrut, G.; Bonnefoy, M.; Brocker, P.; Constans, T.; Ferry, M.; et al. IANA (International Academy on Nutrition and Aging) Expert Group: Weight loss and Alzheimer’s disease. J. Nutr. Health Aging 2007, 11, 38–48. [Google Scholar]
- Soto, M.E.; Secher, M.; Gillette-Guyonnet, S.; Abellan van Kan, G.; Andrieu, S.; Nourhashemi, F.; Rolland, Y.; Vellas, B. Weight loss and rapid cognitive decline in community-dwelling patients with Alzheimer’s disease. J. Alzheimer Dis. JAD 2012, 28, 647–654. [Google Scholar] [CrossRef]
- Müller, S.; Preische, O.; Sohrabi, H.R.; Gräber, S.; Jucker, M.; Dietzsch, J.; Ringman, J.M.; Martins, R.N.; McDade, E.; Schofield, P.R.; et al. Decreased body mass index in the preclinical stage of autosomal dominant Alzheimer’s disease. Sci. Rep. 2017, 7, 1225. [Google Scholar] [CrossRef]
- Volicer, L.; Frijters, D.H.; van der Steen, J.T. Apathy and weight loss in nursing home residents: Longitudinal study. J. Am. Med. Dir. Assoc. 2013, 14, 417–420. [Google Scholar] [CrossRef]
- Doorduijn, A.S.; Visser, M.; van de Rest, O.; Kester, M.I.; de Leeuw, F.A.; Boesveldt, S.; Fieldhouse, J.L.P.; van den Heuvel, E.; Teunissen, C.E.; Scheltens, P.; et al. Associations of AD Biomarkers and Cognitive Performance with Nutritional Status: The NUDAD Project. Nutrients 2019, 11, 1161. [Google Scholar] [CrossRef] [PubMed]
- Doorduijn, A.S.; de van der Schueren, M.A.E.; van de Rest, O.; de Leeuw, F.A.; Hendriksen, H.M.A.; Teunissen, C.E.; Scheltens, P.; van der Flier, W.M.; Visser, M. Energy intake and expenditure in patients with Alzheimer’s disease and mild cognitive impairment: The NUDAD project. Alzheimer Res. Ther. 2020, 12, 116. [Google Scholar] [CrossRef]
- Poehlman, E.T.; Dvorak, R.V. Energy expenditure, energy intake, and weight loss in Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 650S–655S. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Serraneau, K.S.; Brownlow, M.; Marín de Evsikova, C.; Speakman, J.R.; Gordon, M.N.; Morgan, D. Metabolic changes over the course of aging in a mouse model of tau deposition. Neurobiol. Aging 2016, 44, 62–73. [Google Scholar] [CrossRef]
- O’Leary, T.P.; Mantolino, H.M.; Stover, K.R.; Brown, R.E. Age-related deterioration of motor function in male and female 5xFAD mice from 3 to 16 months of age. Genes Brain Behav. 2020, 19, e12538. [Google Scholar] [CrossRef]
- Jul, P.; Volbracht, C.; de Jong, I.E.; Helboe, L.; Elvang, A.B.; Pedersen, J.T. Hyperactivity with Agitative-Like Behavior in a Mouse Tauopathy Model. J. Alzheimer Dis. JAD 2016, 49, 783–795. [Google Scholar] [CrossRef]
- Huebbe, P.; Dose, J.; Schloesser, A.; Campbell, G.; Glüer, C.C.; Gupta, Y.; Ibrahim, S.; Minihane, A.M.; Baines, J.F.; Nebel, A.; et al. Apolipoprotein E (APOE) genotype regulates body weight and fatty acid utilization-Studies in gene-targeted replacement mice. Mol. Nutr. Food Res. 2015, 59, 334–343. [Google Scholar] [CrossRef]
- Loskutova, N.; Honea, R.A.; Brooks, W.M.; Burns, J.M. Reduced limbic and hypothalamic volumes correlate with bone density in early Alzheimer’s disease. J. Alzheimer Dis. JAD 2010, 20, 313–322. [Google Scholar] [CrossRef]
- Callen, D.J.; Black, S.E.; Gao, F.; Caldwell, C.B.; Szalai, J.P. Beyond the hippocampus: MRI volumetry confirms widespread limbic atrophy in AD. Neurology 2001, 57, 1669–1674. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Cano, A.; Busquets, O.; Manzine, P.R.; Sánchez-López, E.; Castro-Torres, R.D.; Beas-Zarate, C.; Verdaguer, E.; García, M.L.; Olloquequi, J.; et al. A metabolic perspective of late onset Alzheimer’s disease. Pharmacol. Res. 2019, 145, 104255. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Cortés, N.; Andrade, V.; Guzmán-Martínez, L.; Estrella, M.; Maccioni, R.B. Neuroimmune Tau Mechanisms: Their Role in the Progression of Neuronal Degeneration. Int. J. Mol. Sci. 2018, 19, 956. [Google Scholar] [CrossRef] [PubMed]
- Chaskiel, L.; Bristow, A.D.; Bluthé, R.M.; Dantzer, R.; Blomqvist, A.; Konsman, J.P. Interleukin-1 reduces food intake and body weight in rat by acting in the arcuate hypothalamus. Brain Behav. Immun. 2019, 81, 560–573. [Google Scholar] [CrossRef]
- Arruda, A.P.; Milanski, M.; Coope, A.; Torsoni, A.S.; Ropelle, E.; Carvalho, D.P.; Carvalheira, J.B.; Velloso, L.A. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology 2011, 152, 1314–1326. [Google Scholar] [CrossRef]
- Velloso, L.A.; Araújo, E.P.; de Souza, C.T. Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation 2008, 15, 189–193. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Hsu, H.-C.; Kao, P.-C.; Shiao, Y.-J.; Yeh, S.H.-H.; Shie, F.-S.; Hsu, S.-M.; Yeh, C.-W.; Liu, H.-K.; Yang, S.-B.; et al. Augmented Insulin and Leptin Resistance of High Fat Diet-Fed APPswe/PS1dE9 Transgenic Mice Exacerbate Obesity and Glycemic Dysregulation. Int. J. Mol. Sci. 2018, 19, 2333. [Google Scholar] [CrossRef]
- Holscher, C. Diabetes as a risk factor for Alzheimer’s disease: Insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem. Soc. Trans. 2011, 39, 891–897. [Google Scholar] [CrossRef]
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- Wong, C.H.Y.; Wanrooy, B.J.; Bruce, D.G. Chapter 10-Neuroinflammation, Type 2 Diabetes, and Dementia. In Type 2 Diabetes and Dementia; Srikanth, V., Arvanitakis, Z., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 195–209. [Google Scholar] [CrossRef]
- Mehlig, K.; Lapidus, L.; Thelle, D.S.; Waern, M.; Zetterberg, H.; Björkelund, C.; Skoog, I.; Lissner, L. Low fasting serum insulin and dementia in nondiabetic women followed for 34 years. Neurology 2018, 91, e427–e435. [Google Scholar] [CrossRef]
- Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundström, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008, 71, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Peila, R.; Rodriguez, B.L.; White, L.R.; Launer, L.J. Fasting insulin and incident dementia in an elderly population of Japanese-American men. Neurology 2004, 63, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, W.; Wang, H.F.; Wang, Z.X.; Jiang, T.; Tan, M.S.; Yu, J.T.; Tan, L. Peripheral Blood Adipokines and Insulin Levels in Patients with Alzheimer’s Disease: A Replication Study and Meta-Analysis. Curr. Alzheimer Res. 2016, 13, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.R.; Zhang, L.; Dasuri, K.; Fernandez-Kim, S.O.; Bruce-Keller, A.J.; Keller, J.N. Mutant amyloid precursor protein differentially alters adipose biology under obesogenic and non-obesogenic conditions. PLoS ONE 2012, 7, e43193. [Google Scholar] [CrossRef]
- Tramutola, A.; Arena, A.; Cini, C.; Butterfield, D.A.; Barone, E. Modulation of GLP-1 signaling as a novel therapeutic approach in the treatment of Alzheimer’s disease pathology. Expert Rev. Neurother. 2017, 17, 59–75. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Hölscher, C. GIP has neuroprotective effects in Alzheimer and Parkinson’s disease models. Peptides 2020, 125, 170184. [Google Scholar] [CrossRef]
- Hölscher, C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 2018, 136, 251–259. [Google Scholar] [CrossRef]
- Kunath, N.; van Groen, T.; Allison, D.B.; Kumar, A.; Dozier-Sharpe, M.; Kadish, I. Ghrelin agonist does not foster insulin resistance but improves cognition in an Alzheimer’s disease mouse model. Sci. Rep. 2015, 5, 11452. [Google Scholar] [CrossRef]
- Al Massadi, O.; López, M.; Tschöp, M.; Diéguez, C.; Nogueiras, R. Current Understanding of the Hypothalamic Ghrelin Pathways Inducing Appetite and Adiposity. Trends Neurosci. 2017, 40, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.O.; Shin, S.J.; Park, J.Y.; Ku, B.K.; Song, J.S.; Kim, J.J.; Jeon, S.G.; Lee, S.M.; Moon, M. MK-0677, a Ghrelin Agonist, Alleviates Amyloid Beta-Related Pathology in 5XFAD Mice, an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1800. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Ahima, R.S. Resistin in rodents and humans. Diabetes Metab. J. 2013, 37, 404–414. [Google Scholar] [CrossRef]
- Benomar, Y.; Amine, H.; Crépin, D.; Al Rifai, S.; Riffault, L.; Gertler, A.; Taouis, M. Central Resistin/TLR4 Impairs Adiponectin Signaling, Contributing to Insulin and FGF21 Resistance. Diabetes 2016, 65, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Demirci, S.; Aynalı, A.; Demirci, K.; Demirci, S.; Arıdoğan, B.C. The Serum Levels of Resistin and Its Relationship with Other Proinflammatory Cytokines in Patients with Alzheimer’s Disease. Clin. Psychopharmacol. Neurosci. 2017, 15, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.; Wilkinson, D.; Wiesner, G.; Morash, B.; Ur, E. Hypothalamic resistin immunoreactivity is reduced by obesity in the mouse: Co-localization with alpha-melanostimulating hormone. Neuroendocrinology 2005, 81, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- López-Gambero, A.J.; Rodríguez de Fonseca, F.; Suárez, J. Energy sensors in drug addiction: A potential therapeutic target. Addict. Biol. 2021, 26, e12936. [Google Scholar] [CrossRef] [PubMed]
- Cota, D.; Proulx, K.; Smith, K.A.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic mTOR signaling regulates food intake. Science 2006, 312, 927–930. [Google Scholar] [CrossRef]
- Shibuya, Y.; Chang, C.C.; Huang, L.H.; Bryleva, E.Y.; Chang, T.Y. Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Aβ1-42 clearance. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 14484–14501. [Google Scholar] [CrossRef] [PubMed]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225. [Google Scholar] [CrossRef]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: In vivo and in vitro studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, J.; Zhu, Y.; Zhang, J.; Shen, H.; Lu, J.; Pan, X.; Lin, N.; Dai, X.; Zhou, M.; et al. Tripchlorolide improves cognitive deficits by reducing amyloid β and upregulating synapse-related proteins in a transgenic model of Alzheimer’s Disease. J. Neurochem. 2015, 133, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, G.-J.; Hu, H.-X.; Ma, C.; Ma, D.-H.; Liu, X.-L.; Jiang, X.-M. Cerebral mTOR signal and pro-inflammatory cytokines in Alzheimer’s disease rats. Transl. Neurosci. 2016, 7, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Mueed, Z.; Tandon, P.; Maurya, S.K.; Deval, R.; Kamal, M.A.; Poddar, N.K. Tau and mTOR: The Hotspots for Multifarious Diseases in Alzheimer’s Development. Front. Neurosci. 2018, 12, 1017. [Google Scholar] [CrossRef]
- Czapski, G.A.; Gąssowska, M.; Wilkaniec, A.; Chalimoniuk, M.; Strosznajder, J.B.; Adamczyk, A. The mechanisms regulating cyclin-dependent kinase 5 in hippocampus during systemic inflammatory response: The effect on inflammatory gene expression. Neurochem. Int. 2016, 93, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Hiller, A.J.; Pham, L.; McGuire, M.J.; Iadecola, C.; Wang, G. Amyloid-Beta Modulates Low-Threshold Activated Voltage-Gated L-Type Calcium Channels of Arcuate Neuropeptide Y Neurons Leading to Calcium Dysregulation and Hypothalamic Dysfunction. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 8816–8825. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.Y.; Acuna-Goycolea, C.; van den Pol, A.N. Neuropeptide Y inhibits hypocretin/orexin neurons by multiple presynaptic and postsynaptic mechanisms: Tonic depression of the hypothalamic arousal system. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 8741–8751. [Google Scholar] [CrossRef][Green Version]
- Sethi, M.; Joshi, S.S.; Webb, R.L.; Beckett, T.L.; Donohue, K.D.; Murphy, M.P.; O’Hara, B.F.; Duncan, M.J. Increased fragmentation of sleep-wake cycles in the 5XFAD mouse model of Alzheimer’s disease. Neuroscience 2015, 290, 80–89. [Google Scholar] [CrossRef]
- Liu, R.M.; van Groen, T.; Katre, A.; Cao, D.; Kadisha, I.; Ballinger, C.; Wang, L.; Carroll, S.L.; Li, L. Knockout of plasminogen activator inhibitor 1 gene reduces amyloid beta peptide burden in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Akhter, H.; Huang, W.T.; van Groen, T.; Kuo, H.C.; Miyata, T.; Liu, R.M. A Small Molecule Inhibitor of Plasminogen Activator Inhibitor-1 Reduces Brain Amyloid-β Load and Improves Memory in an Animal Model of Alzheimer’s Disease. J. Alzheimer Dis. JAD 2018, 64, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.S.; Comery, T.A.; Martone, R.L.; Elokdah, H.; Crandall, D.L.; Oganesian, A.; Aschmies, S.; Kirksey, Y.; Gonzales, C.; Xu, J.; et al. Enhanced clearance of Aβ in brain by sustaining the plasmin proteolysis cascade. Proc. Natl. Acad. Sci. USA 2008, 105, 8754–8759. [Google Scholar] [CrossRef]
- de Oliveira, F.F.; Chen, E.S.; Smith, M.C.; Bertolucci, P.H.F. Longitudinal lipid profile variations and clinical change in Alzheimer’s disease dementia. Neurosci. Lett. 2017, 646, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, C.L.; Kokjohn, T.A.; Whiteside, C.M.; Macias, M.P.; Kalback, W.M.; Sabbagh, M.N.; Beach, T.G.; Vassar, R.; Roher, A.E. Molecular Differences and Similarities Between Alzheimer’s Disease and the 5XFAD Transgenic Mouse Model of Amyloidosis. Biochem. Insights 2013, 6, 1–10. [Google Scholar] [CrossRef]
- Rivera, P.; Ramírez-López, M.T.; Vargas, A.; Decara, J.; Vázquez, M.; Arco, R.; Gómez de Heras, R.; Argente, J.; Rodríguez de Fonseca, F.; Chowen, J.A.; et al. Perinatal free-choice of a high-calorie low-protein diet affects leptin signaling through IRS1 and AMPK dephosphorylation in the hypothalami of female rat offspring in adulthood. Acta Physiol. 2019, 226, e13244. [Google Scholar] [CrossRef]
- Rivera, P.; Guerra-Cantera, S.; Vargas, A.; Díaz, F.; García-Úbeda, R.; Tovar, R.; Ramírez-López, M.T.; Argente, J.; de Fonseca, F.R.; Suárez, J.; et al. Maternal hypercaloric diet affects factors involved in lipid metabolism and the endogenous cannabinoid systems in the hypothalamus of adult offspring: Sex-specific response of astrocytes to palmitic acid and anandamide. Nutr. Neurosci. 2020, 1–14. [Google Scholar] [CrossRef]
- Navarro, J.A.; Decara, J.; Medina-Vera, D.; Tovar, R.; Suarez, J.; Pavón, J.; Serrano, A.; Vida, M.; Gutierrez-Adan, A.; Sanjuan, C.; et al. D-Pinitol from Ceratonia siliqua Is an Orally Active Natural Inositol that Reduces Pancreas Insulin Secretion and Increases Circulating Ghrelin Levels in Wistar Rats. Nutrients 2020, 12, 2030. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Medina-Vera, D.; Rosell-Valle, C.; López-Gambero, A.J.; Navarro, J.A.; Zambrana-Infantes, E.N.; Rivera, P.; Santín, L.J.; Suarez, J.; Rodríguez de Fonseca, F. Imbalance of Endocannabinoid/Lysophosphatidylinositol Receptors Marks the Severity of Alzheimer’s Disease in a Preclinical Model: A Therapeutic Opportunity. Biology 2020, 9, 377. [Google Scholar] [CrossRef]
- Suárez, J.; Rivera, P.; Arrabal, S.; Crespillo, A.; Serrano, A.; Baixeras, E.; Pavón, F.J.; Cifuentes, M.; Nogueiras, R.; Ballesteros, J.; et al. Oleoylethanolamide enhances β-adrenergic-mediated thermogenesis and white-to-brown adipocyte phenotype in epididymal white adipose tissue in rat. Dis. Models Mech. 2014, 7, 129–141. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Males | Females | |||||
|---|---|---|---|---|---|---|
| Body Weight Versus: | Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD | Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD |
| Il-1β (hyp) | ns 1 | ns | ns | ns | R2 = 0.820 p = 0.034 | R2 = 0.593 p = 0.025 |
| TNF-α (hyp) | ns | ns | ns | R2 = 0.799 p = 0.016 | R2 = 0.713 p = 0.071 | R2 = 0.548 p = 0.035 |
| GFAP (hyp) | ns | ns | ns | ns | ns | ns |
| IBA1 (hyp) | ns | ns | ns | R2 = 0.769 p = 0.021 | R2 = 0.783 p = 0.046 | R2 = 0.783 p = 0.019 |
| Males | Females | |||||
|---|---|---|---|---|---|---|
| Body Weight Versus: | Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD | Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD |
| Resistin (hyp) | R2 = 0.584 p = 0.076 | ns 1 | ns | ns | R2 = 0.920 p = 0.009 | R2 = 0.546 p = 0.036 |
| Ghrelin (hyp) | ns | ns | ns | ns | ns | ns |
| Resistin (plasma) | R2 = 0.594 p = 0.072 | ns | R2 = 0.704 p = 0.018 | ns | R2 = 0.894 p = 0.001 | R2 = 0.755 p = 0.011 |
| Ghrelin (plasma) | ns | ns | ns | ns | ns | R2 = 0.580 p = 0.046 |
| Insulin (plasma) | ns | R2 = 0.760 p = 0.023 | R2 = 0.851 p = 0.003 | ns | R2 = 0.708 p = 0.017 | R2 = 0.578 p = 0.047 |
| Leptin (plasma) | ns | R2 = 0.942 p =0.001 | R2 = 0.612 p = 0.037 | ns | ns | ns |
| GLP-1 (plasma) | ns | ns | ns | ns | ns | R2 = 0.622 p = 0.034 |
| GIP (plasma) | ns | R2 = 0.813 p = 0.014 | R2 = 0.644 p = 0.029 | ns | ns | ns |
| PAI-1 (plasma) | ns | ns | ns | ns | R2 = 0.628 p = 0.033 | R2 = 0.708 p = 0.017 |
| Glucagon (plasma) | ns | ns | ns | ns | ns | ns |
| Males | Females | |||||
|---|---|---|---|---|---|---|
| Insulin (Plasma) Versus: | Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD | Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD |
| GIP | ns 1 | R2 = 0.712 p = 0.034 | R2 = 0.572 p = 0.048 | R2 = 0.659 p = 0.049 | ns | ns |
| GLP-1 | ns | ns | ns | ns | ns | R2 = 0.846 p = 0.003 |
| Ghrelin | ns | ns | ns | ns | ns | ns |
| Glucagon | ns | ns | ns | R2 = 0.582 p = 0.077 | ns | R2 = 0.495 p = 0.077 |
| Males | Females | |||||
|---|---|---|---|---|---|---|
| Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD | Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD | |
| Il-1β versus | ||||||
| NPY | ns 1 | ns | ns | ns | ns | ns |
| AgRP | ns | ns | ns | ns | ns | ns |
| POMC | ns | ns | ns | ns | R2 = 0.211 p = 0.009 | R2 = 0.546 p = 0.036 |
| CART | ns | ns | R2 = 0.566 p = 0.051 | ns | ns | ns |
| HCRT | ns | ns | ns | ns | R2 = 0.765 p = 0.052 | ns |
| MCH | R2 = 0.868 p = 0.006 | ns | ns | ns | ns | ns |
| Resistin (hyp) | ns | ns | ns | ns | R2 = 0.947 p = 0.005 | R2 = 0.513 p = 0.045 |
| Ghrelin (hyp) | ns | ns | ns | ns | ns | ns |
| TNF-α versus | ||||||
| NPY | ns | ns | ns | R2 = 0.831 p = 0.011 | ns | ns |
| AgRP | ns | ns | ns | ns | ns | ns |
| POMC | ns | ns | ns | ns | ns | ns |
| CART | ns | R2 = 0.829 p = 0.031 | ns | ns | ns | ns |
| HCRT | ns | ns | ns | ns | ns | ns |
| MCH | ns | ns | ns | ns | ns | R2 = 0.436 p = 0.074 |
| Resistin (hyp) | ns | R2 = 0.902 p = 0.013 | R2 = 0.833 p = 0.004 | ns | ns | R2 = 0.724 p = 0.007 |
| Ghrelin (hyp) | ns | ns | ns | ns | ns | ns |
| NPY versus | ||||||
| AgRP | ns | R2 = 0.945 p = 0.005 | R2 = 0.896 p = 0.001 | R2 = 0.707 p = 0.035 | R2 = 0.952 p = 0.004 | R2 = 0.985 p < 0.0001 |
| POMC | ns | R2 = 0.707 p = 0.074 | ns | ns | R2 = 0.797 p = 0.041 | R2 = 0.640 p = 0.017 |
| CART | ns | ns | ns | ns | R2 = 0.7987 p = 0.0409 | ns |
| HCRT | ns | R2 = 0.780 p = 0.046 | R2 = 0.692 p = 0.020 | ns | ns | R2 = 0.511 p = 0.046 |
| MCH | ns | R2 = 0.790 p = 0.043 | R2 = 0.701 p = 0.018 | ns | ns | R2 = 0.672 p = 0.012 |
| Males | Females | |||||
|---|---|---|---|---|---|---|
| Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD | Non-Tg | Tg5xFAD/- | Tg5xFAD/5xFAD | |
| Triglycerides versus | ||||||
| Total cholesterol | ns 1 | ns | ns | R2 = 0.912 p = 0.0002 | R2 = 0.737 p = 0.013 | R2 = 0.582 p = 0.010 |
| HDL | ns | ns | ns | ns | R2 = 0.746 p = 0.012 | R2 = 0.570 p = 0.011 |
| LDL | ns | ns | ns | ns | ns | R2 = 0.761 p = 0.001 |
| GOT versus | ||||||
| Total cholesterol | ns | ns | ns | ns | ns | ns |
| HDL | ns | ns | ns | ns | ns | ns |
| LDL | ns | ns | ns | ns | ns | R2 = 0.426 p = 0.040 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Gambero, A.J.; Rosell-Valle, C.; Medina-Vera, D.; Navarro, J.A.; Vargas, A.; Rivera, P.; Sanjuan, C.; Rodríguez de Fonseca, F.; Suárez, J. A Negative Energy Balance Is Associated with Metabolic Dysfunctions in the Hypothalamus of a Humanized Preclinical Model of Alzheimer’s Disease, the 5XFAD Mouse. Int. J. Mol. Sci. 2021, 22, 5365. https://doi.org/10.3390/ijms22105365
López-Gambero AJ, Rosell-Valle C, Medina-Vera D, Navarro JA, Vargas A, Rivera P, Sanjuan C, Rodríguez de Fonseca F, Suárez J. A Negative Energy Balance Is Associated with Metabolic Dysfunctions in the Hypothalamus of a Humanized Preclinical Model of Alzheimer’s Disease, the 5XFAD Mouse. International Journal of Molecular Sciences. 2021; 22(10):5365. https://doi.org/10.3390/ijms22105365
Chicago/Turabian StyleLópez-Gambero, Antonio J., Cristina Rosell-Valle, Dina Medina-Vera, Juan Antonio Navarro, Antonio Vargas, Patricia Rivera, Carlos Sanjuan, Fernando Rodríguez de Fonseca, and Juan Suárez. 2021. "A Negative Energy Balance Is Associated with Metabolic Dysfunctions in the Hypothalamus of a Humanized Preclinical Model of Alzheimer’s Disease, the 5XFAD Mouse" International Journal of Molecular Sciences 22, no. 10: 5365. https://doi.org/10.3390/ijms22105365
APA StyleLópez-Gambero, A. J., Rosell-Valle, C., Medina-Vera, D., Navarro, J. A., Vargas, A., Rivera, P., Sanjuan, C., Rodríguez de Fonseca, F., & Suárez, J. (2021). A Negative Energy Balance Is Associated with Metabolic Dysfunctions in the Hypothalamus of a Humanized Preclinical Model of Alzheimer’s Disease, the 5XFAD Mouse. International Journal of Molecular Sciences, 22(10), 5365. https://doi.org/10.3390/ijms22105365