
Sexual Dimorphism of Corticosteroid Signaling during Kidney Development

 and
and
Abstract
1. Introduction
2. Mineralocorticoid Signaling Pathway
2.1. Regulation of Aldosterone Synthesis
2.2. The Mineralocorticoid Receptor (MR)
2.2.1. Gene, Transcripts, and Protein Variants
2.2.2. Mechanisms of Regulation of MR Expression and Activity
3. Glucocorticoid Signaling Pathway
3.1. Glucocorticoid Hormones and the Hypothalamic–Pituitary–Adrenal Axis
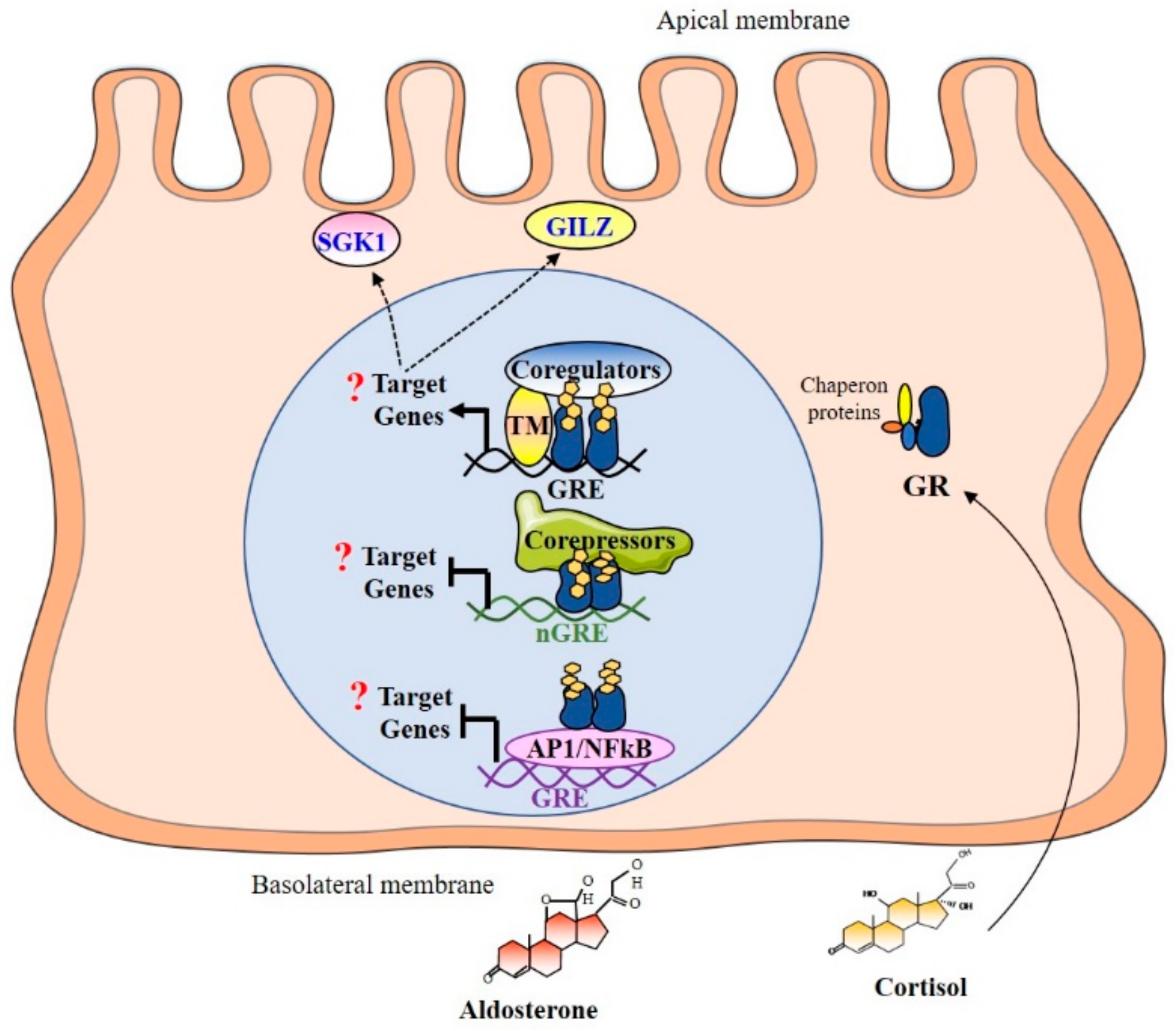
3.2. The Glucocorticoid Receptor
3.2.1. Gene, Transcripts, and Protein Variants
3.2.2. Mechanisms of Regulation of GR Expression and Activity
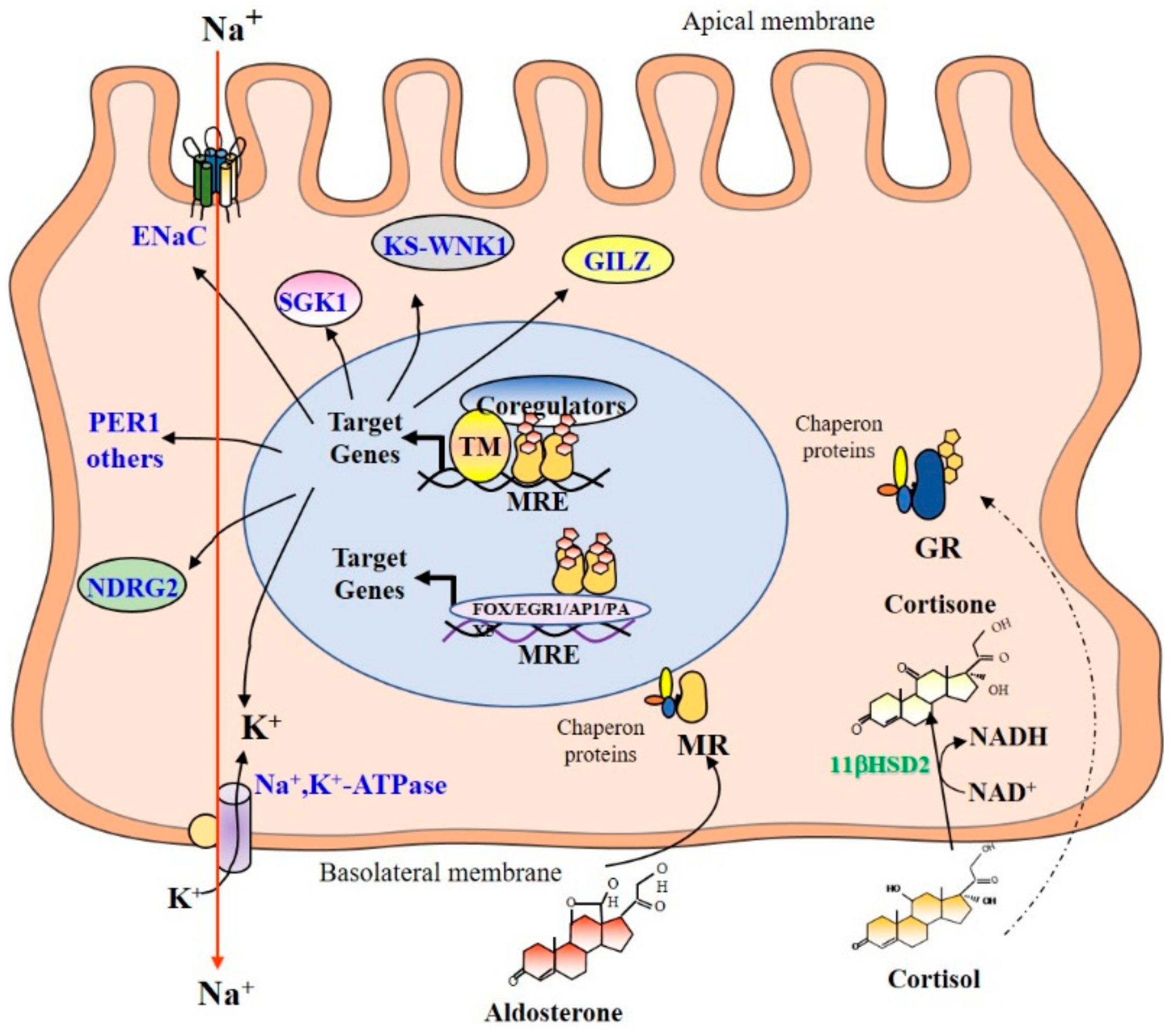
4. Mechanism of Corticosteroids Action in Renal Principal Cells
4.1. Subcellular Distribution
4.2. Mineralocorticoid Selectivity
4.3. Promoter Binding and Recruitment of Coregulators
4.4. MR and GR Target Genes
5. Sexual Dimorphism of Corticosteroid Signaling Aside from the Kidney
6. Gender Differences in Kidney Development and Organogenesis
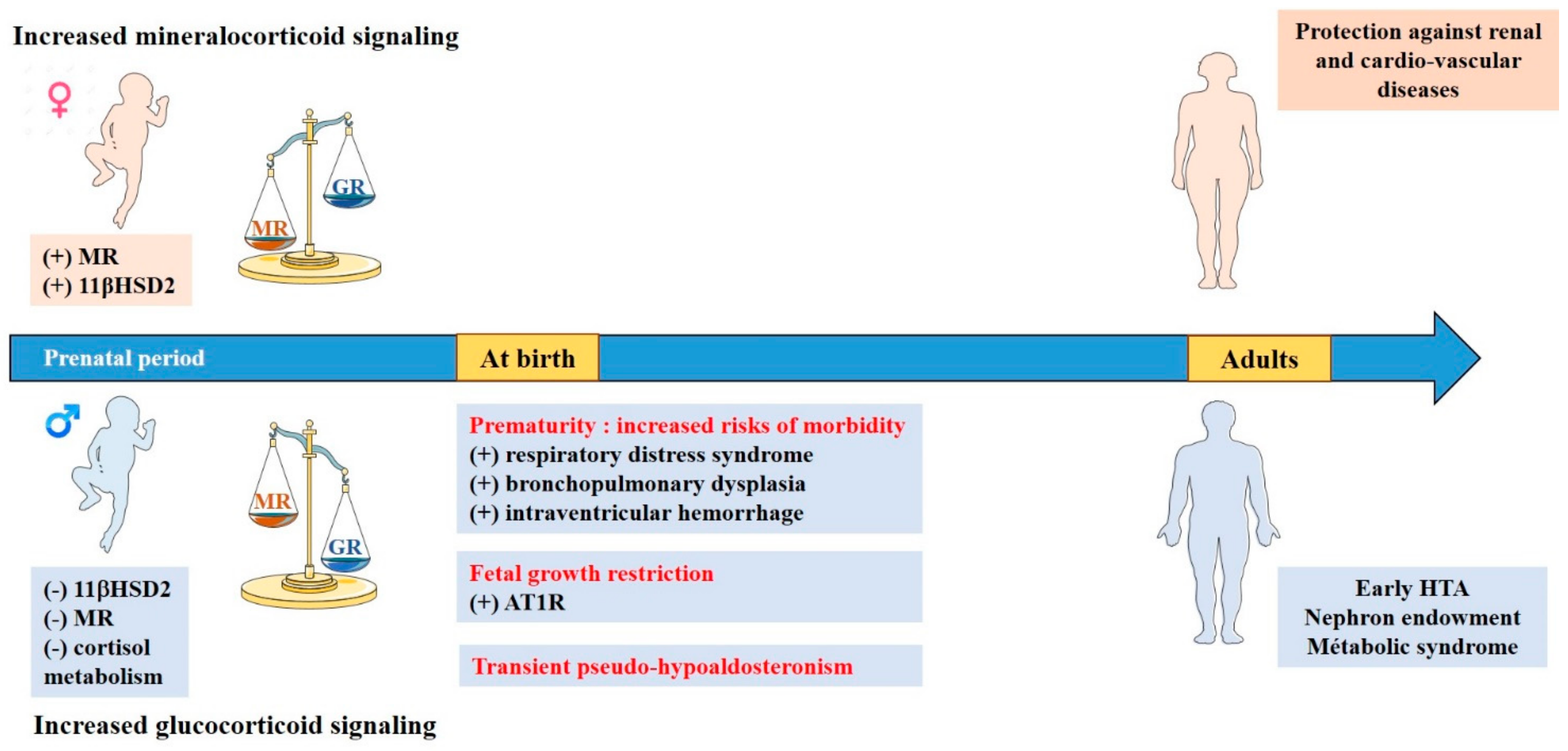
7. Particularities of Mineralocorticoid and Glucocorticoid Signalings during Renal Development
8. Sexual Dimorphism in the Equilibrium between Renal Mineralocorticoid and Glucocorticoid Signaling
9. Consequences in Pathophysiology
Fetal Growth Restriction
- Prematurity
- Transient Pseudo-Hypoaldosteronism
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seccia, T.M.; Caroccia, B.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E.; Rossi, G.P. The Biology of Normal Zona Glomerulosa and Aldosterone-Producing Adenoma: Pathological Implications. Endocr. Rev. 2018, 39, 1029–1056. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Steroidogenesis: Unanswered Questions. Trends Endocrinol. Metab. 2017, 28, 771–793. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, H.; Duparc, C.; Naccache, A.; Lopez, A.-G.; Castanet, M.; Louiset, E. Paracrine Regulation of Aldosterone Secretion in Physiological and Pathophysiological Conditions. Vitam. Horm. 2019, 109, 303–339. [Google Scholar] [CrossRef] [PubMed]
- Wils, J.; Duparc, C.; Cailleux, A.-F.; Lopez, A.-G.; Guiheneuf, C.; Boutelet, I.; Boyer, H.-G.; Dubessy, C.; Cherifi, S.; Cauliez, B.; et al. The Neuropeptide Substance P Regulates Aldosterone Secretion in Human Adrenals. Nat. Commun. 2020, 11, 2673. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.M.; van Kralingen, J.C.; Davies, E. Regulation of Aldosterone Secretion. Vitam. Horm. 2019, 109, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.J. ACTH Action on StAR Biology. Front. Neurosci. 2016, 10, 547. [Google Scholar] [CrossRef] [PubMed]
- Melau, C.; Nielsen, J.E.; Frederiksen, H.; Kilcoyne, K.; Perlman, S.; Lundvall, L.; Langhoff Thuesen, L.; Juul Hare, K.; Andersson, A.-M.; Mitchell, R.T.; et al. Characterization of Human Adrenal Steroidogenesis During Fetal Development. J. Clin. Endocrinol. Metab. 2019, 104, 1802–1812. [Google Scholar] [CrossRef]
- Naccache, A.; Louiset, E.; Duparc, C.; Laquerrière, A.; Patrier, S.; Renouf, S.; Gomez-Sanchez, C.E.; Mukai, K.; Lefebvre, H.; Castanet, M. Temporal and Spatial Distribution of Mast Cells and Steroidogenic Enzymes in the Human Fetal Adrenal. Mol. Cell. Endocrinol. 2016, 434, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Johnston, Z.C.; Bellingham, M.; Filis, P.; Soffientini, U.; Hough, D.; Bhattacharya, S.; Simard, M.; Hammond, G.L.; King, P.; O’Shaughnessy, P.J.; et al. The Human Fetal Adrenal Produces Cortisol but No Detectable Aldosterone throughout the Second Trimester. BMC Med. 2018, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Martinerie, L.; Pussard, E.; Yousef, N.; Cosson, C.; Lema, I.; Husseini, K.; Mur, S.; Lombès, M.; Boileau, P. Aldosterone-Signaling Defect Exacerbates Sodium Wasting in Very Preterm Neonates: The Premaldo Study. J. Clin. Endocrinol. Metab. 2015, 100, 4074–4081. [Google Scholar] [CrossRef]
- Ishimoto, H.; Jaffe, R.B. Development and Function of the Human Fetal Adrenal Cortex: A Key Component in the Feto-Placental Unit. Endocr. Rev. 2011, 32, 317–355. [Google Scholar] [CrossRef]
- Abdel Mohsen, A.H.; Taha, G.; Kamel, B.A.; Maksood, M.A. Evaluation of Aldosterone Excretion in Very Low Birth Weight Infants. Saudi J. Kidney Dis. Transpl. 2016, 27, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Viengchareun, S.; Le Menuet, D.; Martinerie, L.; Munier, M.; Pascual-Le Tallec, L.; Lombès, M. The Mineralocorticoid Receptor: Insights into Its Molecular and (Patho)Physiological Biology. Nucl. Recept. Signal. 2007, 5, e012. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.S.; Eddy, R.L.; Byers, M.G.; Haley, L.L.; Henry, W.M.; Nowak, N.J.; Shows, T.B. The Human Mineralocorticoid Receptor Gene (MLR) Is Located on Chromosome 4 at Q31.2. Cytogenet. Cell Genet. 1989, 52, 83–84. [Google Scholar] [CrossRef]
- Morrison, N.; Harrap, S.B.; Arriza, J.L.; Boyd, E.; Connor, J.M. Regional Chromosomal Assignment of the Human Mineralocorticoid Receptor Gene to 4q31.1. Hum. Genet. 1990, 85, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Keightley, M.C.; Kotelevtsev, Y.; Conway, G.S.; Soubrier, F.; Fuller, P.J. Human Mineralocorticoid Receptor Genomic Structure and Identification of Expressed Isoforms. J. Biol. Chem. 1995, 270, 21016–21020. [Google Scholar] [CrossRef] [PubMed]
- Lema, I.; Amazit, L.; Lamribet, K.; Fagart, J.; Blanchard, A.; Lombès, M.; Cherradi, N.; Viengchareun, S. HuR-Dependent Editing of a New Mineralocorticoid Receptor Splice Variant Reveals an Osmoregulatory Loop for Sodium Homeostasis. Sci. Rep. 2017, 7, 4835. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Souque, A.; Viengchareun, S.; Poisson, E.; Lombès, M. A New Human MR Splice Variant Is a Ligand-Independent Transactivator Modulating Corticosteroid Action. Mol. Endocrinol. 2001, 15, 1586–1598. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Le Tallec, L.; Demange, C.; Lombès, M. Human Mineralocorticoid Receptor A and B Protein Forms Produced by Alternative Translation Sites Display Different Transcriptional Activities. Eur. J. Endocrinol. 2004, 150, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Le Menuet, D.; Lombès, M. Characterization of the Human Mineralocorticoid Receptor Gene 5′-Regulatory Region: Evidence for Differential Hormonal Regulation of Two Alternative Promoters via Nonclassical Mechanisms. Mol. Endocrinol. 1996, 10, 1549–1560. [Google Scholar] [CrossRef]
- Le Menuet, D.; Zennaro, M.C.; Viengchareun, S.; Lombès, M. Transgenic Mouse Models to Study Human Mineralocorticoid Receptor Function in Vivo. Kidney Int. 2000, 57, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Viengchareun, S.; Kamenicky, P.; Teixeira, M.; Butlen, D.; Meduri, G.; Blanchard-Gutton, N.; Kurschat, C.; Lanel, A.; Martinerie, L.; Sztal-Mazer, S.; et al. Osmotic Stress Regulates Mineralocorticoid Receptor Expression in a Novel Aldosterone-Sensitive Cortical Collecting Duct Cell Line. Mol. Endocrinol. 2009, 23, 1948–1962. [Google Scholar] [CrossRef] [PubMed]
- Viengchareun, S.; Lema, I.; Lamribet, K.; Keo, V.; Blanchard, A.; Cherradi, N.; Lombès, M. Hypertonicity Compromises Renal Mineralocorticoid Receptor Signaling through Tis11b-Mediated Post-Transcriptional Control. J. Am. Soc. Nephrol. 2014, 25, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Lema, I.; Amazit, L.; Lamribet, K.; Fagart, J.; Blanchard, A.; Lombès, M.; Cherradi, N.; Viengchareun, S. RNA-Binding Protein HuR Enhances Mineralocorticoid Signaling in Renal KC3AC1 Cells under Hypotonicity. Cell. Mol. Life Sci. 2017, 74, 4587–4597. [Google Scholar] [CrossRef]
- Frederick, M.I.; Heinemann, I.U. Regulation of RNA Stability at the 3′ End. Biol. Chem. 2021, 402, 425–431. [Google Scholar] [CrossRef]
- Ozbaki-Yagan, N.; Liu, X.; Bodnar, A.J.; Ho, J.; Butterworth, M.B. Aldosterone-Induced MicroRNAs Act as Feedback Regulators of Mineralocorticoid Receptor Signaling in Kidney Epithelia. FASEB J. 2020, 34, 11714–11728. [Google Scholar] [CrossRef] [PubMed]
- Faresse, N. Post-Translational Modifications of the Mineralocorticoid Receptor: How to Dress the Receptor According to the Circumstances? J. Steroid Biochem. Mol. Biol. 2014, 143, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Busada, J.T.; Cidlowski, J.A. Mechanisms of Glucocorticoid Action During Development. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 125, pp. 147–170. ISBN 978-0-12-802172-9. [Google Scholar]
- Morsi, A.; DeFranco, D.; Witchel, S.F. The Hypothalamic-Pituitary-Adrenal Axis and the Fetus. Horm. Res. Paediatr. 2018, 89, 380–387. [Google Scholar] [CrossRef]
- Van der Voorn, B.; Hollanders, J.J.; Ket, J.C.F.; Rotteveel, J.; Finken, M.J.J. Gender-Specific Differences in Hypothalamus-Pituitary-Adrenal Axis Activity during Childhood: A Systematic Review and Meta-Analysis. Biol. Sex Differ. 2017, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Van Keulen, B.J.; Dolan, C.V.; van der Voorn, B.; Andrew, R.; Walker, B.R.; Hulshoff Pol, H.; Boomsma, D.I.; Rotteveel, J.; Finken, M.J.J. Sexual Dimorphism in Cortisol Metabolism throughout Pubertal Development: A Longitudinal Study. Endocr. Connect. 2020, 9, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.; Grecian, S.M.; Reynolds, R.M. Sex Differences in Early-Life Programming of the Hypothalamic-Pituitary-Adrenal Axis in Humans Suggest Increased Vulnerability in Females: A Systematic Review. J. Dev. Orig. Health Dis. 2017, 8, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Van Keulen, B.J.; Romijn, M.; van der Voorn, B.; de Waard, M.; Hartmann, M.F.; van Goudoever, J.B.; Wudy, S.A.; Rotteveel, J.; Finken, M.J.J. Sex-Specific Differences in HPA Axis Activity in VLBW Preterm Newborns. Endocr. Connect. 2021, 10, 214–219. [Google Scholar] [CrossRef]
- Finken, M.J.; Andrews, R.C.; Andrew, R.; Walker, B.R. Cortisol Metabolism in Healthy Young Adults: Sexual Dimorphism in Activities of A-Ring Reductases, but Not 11beta-Hydroxysteroid Dehydrogenases. J. Clin. Endocrinol. Metab. 1999, 84, 3316–3321. [Google Scholar] [CrossRef] [PubMed]
- Wudy, S.A.; Hartmann, M.F.; Remer, T. Sexual Dimorphism in Cortisol Secretion Starts after Age 10 in Healthy Children: Urinary Cortisol Metabolite Excretion Rates during Growth. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E970–E976. [Google Scholar] [CrossRef] [PubMed]
- Toogood, A.A.; Taylor, N.F.; Shalet, S.M.; Monson, J.P. Sexual Dimorphism of Cortisol Metabolism Is Maintained in Elderly Subjects and Is Not Oestrogen Dependent. Clin. Endocrinol. 2000, 52, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Vitellius, G.; Trabado, S.; Bouligand, J.; Delemer, B.; Lombès, M. Pathophysiology of Glucocorticoid Signaling. Ann. d’Endocrinol. 2018, 79, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid Receptor Signaling in Health and Disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Ramírez, P.; Tliba, O. Glucocorticoid Receptor β (GRβ): Beyond Its Dominant-Negative Function. Int. J. Mol. Sci. 2021, 22, 3649. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Charmandari, E. Chrousos Syndrome: From Molecular Pathogenesis to Therapeutic Management. Eur. J. Clin. Investig. 2015, 45, 504–514. [Google Scholar] [CrossRef]
- Turner, J.D.; Muller, C.P. Structure of the Glucocorticoid Receptor (NR3C1) Gene 5′ Untranslated Region: Identification, and Tissue Distribution of Multiple New Human Exon 1. J. Mol. Endocrinol. 2005, 35, 283–292. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, N.Z.; Cidlowski, J.A. Translational Regulatory Mechanisms Generate N-Terminal Glucocorticoid Receptor Isoforms with Unique Transcriptional Target Genes. Mol. Cell 2005, 18, 331–342. [Google Scholar] [CrossRef]
- Binart, N.; Lombes, M.; Rafestin-Oblin, M.E.; Baulieu, E.E. Characterization of Human Mineralocorticosteroid Receptor Expressed in the Baculovirus System. Proc. Natl. Acad. Sci. USA 1991, 88, 10681–10685. [Google Scholar] [CrossRef] [PubMed]
- Lombès, M.; Binart, N.; Delahaye, F.; Baulieu, E.E.; Rafestin-Oblin, M.E. Differential Intracellular Localization of Human Mineralocorticosteroid Receptor on Binding of Agonists and Antagonists. Biochem. J. 1994, 302, 191–197. [Google Scholar] [CrossRef]
- Cheung, J.; Smith, D.F. Molecular Chaperone Interactions with Steroid Receptors: An Update. Mol. Endocrinol. 2000, 14, 939–946. [Google Scholar] [CrossRef]
- Suren, T.; Rutz, D.; Mößmer, P.; Merkel, U.; Buchner, J.; Rief, M. Single-Molecule Force Spectroscopy Reveals Folding Steps Associated with Hormone Binding and Activation of the Glucocorticoid Receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 11688–11693. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of Human Mineralocorticoid Receptor Complementary DNA: Structural and Functional Kinship with the Glucocorticoid Receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W.; Pearce, P.T.; Smith, R.; Smith, A.I. Mineralocorticoid Action: Target Tissue Specificity Is Enzyme, Not Receptor, Mediated. Science 1988, 242, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.R.; Stewart, P.M.; Burt, D.; Brett, L.; McIntyre, M.A.; Sutanto, W.S.; de Kloet, E.R.; Monder, C. Localisation of 11 Beta-Hydroxysteroid Dehydrogenase—Tissue Specific Protector of the Mineralocorticoid Receptor. Lancet 1988, 2, 986–989. [Google Scholar] [CrossRef]
- White, P.C. Alterations of Cortisol Metabolism in Human Disorders. Horm. Res. Paediatr. 2018, 89, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Bianchetti, L.; Sinar, D.; Depenveiller, C.; Dejaegere, A. Insights into Mineralocorticoid Receptor Homodimerization from a Combined Molecular Modeling and Bioinformatics Study. Proteins 2021. [Google Scholar] [CrossRef]
- Le Billan, F.; Amazit, L.; Bleakley, K.; Xue, Q.-Y.; Pussard, E.; Lhadj, C.; Kolkhof, P.; Viengchareun, S.; Fagart, J.; Lombès, M. Corticosteroid Receptors Adopt Distinct Cyclical Transcriptional Signatures. FASEB J. 2018, 32, 5626–5639. [Google Scholar] [CrossRef] [PubMed]
- Le Billan, F.; Khan, J.A.; Lamribet, K.; Viengchareun, S.; Bouligand, J.; Fagart, J.; Lombès, M. Cistrome of the Aldosterone-Activated Mineralocorticoid Receptor in Human Renal Cells. FASEB J. 2015, 29, 3977–3989. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Yang, J.; Young, M.J. 30 Years of the Mineralocorticoid Receptor: Coregulators as Mediators of Mineralocorticoid Receptor Signalling Diversity. J. Endocrinol. 2017, 234, T23–T34. [Google Scholar] [CrossRef] [PubMed]
- Na, W.; Shin, J.Y.; Lee, J.Y.; Jeong, S.; Kim, W.-S.; Yune, T.Y.; Ju, B.-G. Dexamethasone Suppresses JMJD3 Gene Activation via a Putative Negative Glucocorticoid Response Element and Maintains Integrity of Tight Junctions in Brain Microvascular Endothelial Cells. J. Cereb. Blood Flow Metab. 2017, 37, 3695–3708. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How Glucocorticoid Receptors Modulate the Activity of Other Transcription Factors: A Scope beyond Tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, X.; Zhang, W. Regulation of AENaC Transcription. Vitam. Horm. 2015, 98, 101–135. [Google Scholar] [CrossRef]
- Feraille, E.; Dizin, E. Coordinated Control of ENaC and Na+,K+-ATPase in Renal Collecting Duct. J. Am. Soc. Nephrol. 2016, 27, 2554–2563. [Google Scholar] [CrossRef]
- Lou, Y.; Zhang, F.; Luo, Y.; Wang, L.; Huang, S.; Jin, F. Serum and Glucocorticoid Regulated Kinase 1 in Sodium Homeostasis. Int. J. Mol. Sci. 2016, 17, 1307. [Google Scholar] [CrossRef]
- Roy, A.; Al-Qusairi, L.; Donnelly, B.F.; Ronzaud, C.; Marciszyn, A.L.; Gong, F.; Chang, Y.P.C.; Butterworth, M.B.; Pastor-Soler, N.M.; Hallows, K.R.; et al. Alternatively Spliced Proline-Rich Cassettes Link WNK1 to Aldosterone Action. J. Clin. Investig. 2015, 125, 3433–3448. [Google Scholar] [CrossRef]
- Boulkroun, S.; Fay, M.; Zennaro, M.-C.; Escoubet, B.; Jaisser, F.; Blot-Chabaud, M.; Farman, N.; Courtois-Coutry, N. Characterization of Rat NDRG2 (N-Myc Downstream Regulated Gene 2), a Novel Early Mineralocorticoid-Specific Induced Gene. J. Biol. Chem. 2002, 277, 31506–31515. [Google Scholar] [CrossRef]
- Robert-Nicoud, M.; Flahaut, M.; Elalouf, J.M.; Nicod, M.; Salinas, M.; Bens, M.; Doucet, A.; Wincker, P.; Artiguenave, F.; Horisberger, J.D.; et al. Transcriptome of a Mouse Kidney Cortical Collecting Duct Cell Line: Effects of Aldosterone and Vasopressin. Proc. Natl. Acad. Sci. USA 2001, 98, 2712–2716. [Google Scholar] [CrossRef] [PubMed]
- Gumz, M.L.; Stow, L.R.; Lynch, I.J.; Greenlee, M.M.; Rudin, A.; Cain, B.D.; Weaver, D.R.; Wingo, C.S. The Circadian Clock Protein Period 1 Regulates Expression of the Renal Epithelial Sodium Channel in Mice. J. Clin. Investig. 2009, 119, 2423–2434. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Ouvrard-Pascaud, A.; Tronche, F.; Clemessy, M.; Gonzalez-Nunez, D.; Farman, N.; Jaisser, F. Conditional Transgenic Mice for Studying the Role of the Glucocorticoid Receptor in the Renal Collecting Duct. Endocrinology 2009, 150, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Martinerie, L.; Viengchareun, S.; Delezoide, A.-L.; Jaubert, F.; Sinico, M.; Prevot, S.; Boileau, P.; Meduri, G.; Lombès, M. Low Renal Mineralocorticoid Receptor Expression at Birth Contributes to Partial Aldosterone Resistance in Neonates. Endocrinology 2009, 150, 4414–4424. [Google Scholar] [CrossRef] [PubMed]
- Martinerie, L.; Pussard, E.; Meduri, G.; Delezoide, A.-L.; Boileau, P.; Lombès, M. Lack of Renal 11 Beta-Hydroxysteroid Dehydrogenase Type 2 at Birth, a Targeted Temporal Window for Neonatal Glucocorticoid Action in Human and Mice. PLoS ONE 2012, 7, e31949. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Fujiki, K.; Shirahige, K.; Gomez-Sanchez, C.E.; Fujita, T.; Nangaku, M.; Nagase, M. Genome-Wide Analysis of Murine Renal Distal Convoluted Tubular Cells for the Target Genes of Mineralocorticoid Receptor. Biochem. Biophys. Res. Commun. 2014, 445, 132–137. [Google Scholar] [CrossRef]
- Liu, L.; Li, A.; Matthews, S.G. Maternal Glucocorticoid Treatment Programs HPA Regulation in Adult Offspring: Sex-Specific Effects. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E729–E739. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.; Matthews, S.G. Glucocorticoids and Sex-Dependent Development of Brain Glucocorticoid and Mineralocorticoid Receptors. Endocrinology 2003, 144, 2775–2784. [Google Scholar] [CrossRef]
- Karandrea, D.; Kittas, C.; Kitraki, E. Contribution of Sex and Cellular Context in the Regulation of Brain Corticosteroid Receptors Following Restraint Stress. Neuroendocrinology 2000, 71, 343–353. [Google Scholar] [CrossRef]
- Kanashiro-Takeuchi, R.M.; Heidecker, B.; Lamirault, G.; Dharamsi, J.W.; Hare, J.M. Sex-Specific Impact of Aldosterone Receptor Antagonism on Ventricular Remodeling and Gene Expression after Myocardial Infarction. Clin. Transl. Sci. 2009, 2, 134–142. [Google Scholar] [CrossRef]
- Mihailidou, A.S.; Ashton, A.W. Cardiac Effects of Aldosterone: Does Gender Matter? Steroids 2014, 91, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Barrett Mueller, K.; Lu, Q.; Mohammad, N.N.; Luu, V.; McCurley, A.; Williams, G.H.; Adler, G.K.; Karas, R.H.; Jaffe, I.Z. Estrogen Receptor Inhibits Mineralocorticoid Receptor Transcriptional Regulatory Function. Endocrinology 2014, 155, 4461–4472. [Google Scholar] [CrossRef]
- Duma, D.; Collins, J.B.; Chou, J.W.; Cidlowski, J.A. Sexually Dimorphic Actions of Glucocorticoids Provide a Link to Inflammatory Diseases with Gender Differences in Prevalence. Sci. Signal. 2010, 3, ra74. [Google Scholar] [CrossRef] [PubMed]
- Moisan, M.-P. Sexual Dimorphism in Glucocorticoid Stress Response. Int. J. Mol. Sci. 2021, 22, 3139. [Google Scholar] [CrossRef]
- Ayyar, V.S.; DuBois, D.C.; Almon, R.R.; Jusko, W.J. Modeling Corticosteroid Pharmacokinetics and Pharmacodynamics, Part III: Estrous Cycle and Estrogen Receptor-Dependent Antagonism of Glucocorticoid-Induced Leucine Zipper (GILZ) Enhancement by Corticosteroids. J. Pharmacol. Exp. Ther. 2019, 370, 337–349. [Google Scholar] [CrossRef]
- Dumeige, L.; Storey, C.; Decourtye, L.; Nehlich, M.; Lhadj, C.; Viengchareun, S.; Kappeler, L.; Lombès, M.; Martinerie, L. Sex-Specificity of Mineralocorticoid Target Gene Expression during Renal Development, and Long-Term Consequences. Int. J. Mol. Sci. 2017, 18, 457. [Google Scholar] [CrossRef]
- Woolf, A.S.; Winyard, P.J.D. Molecular Mechanisms of Human Embryogenesis: Developmental Pathogenesis of Renal Tract Malformations. Pediatr. Dev. Pathol. 2002, 5, 108–129. [Google Scholar] [CrossRef] [PubMed]
- Gueutin, V.; Deray, G.; Isnard-Bagnis, C. [Renal physiology]. Bull. Cancer 2012, 99, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F. Renal Branching Morphogenesis: Concepts, Questions, and Recent Advances. Differentiation 2006, 74, 402–421. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Sakurai, H.; Makino, H. Single Adult Kidney Stem/Progenitor Cells Reconstitute Three-Dimensional Nephron Structures in Vitro. Stem Cells 2015, 33, 774–784. [Google Scholar] [CrossRef]
- Michos, O. Kidney Development: From Ureteric Bud Formation to Branching Morphogenesis. Curr. Opin. Genet. Dev. 2009, 19, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Dressler, G.R. The Genetics and Epigenetics of Kidney Development. Semin. Nephrol. 2013, 33, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Dressler, G.R.; Patel, S.R. Epigenetics in Kidney Development and Renal Disease. Transl. Res. 2015, 165, 166–176. [Google Scholar] [CrossRef]
- Iosipiv, I.V.; Schroeder, M. A Role for Angiotensin II AT1 Receptors in Ureteric Bud Cell Branching. Am. J. Physiol. Renal Physiol. 2003, 285, F199–F207. [Google Scholar] [CrossRef]
- Wolf, G. Angiotensin II and Tubular Development. Nephrol. Dial. Transplant. 2002, 17 (Suppl. S9), 48–51. [Google Scholar] [CrossRef] [PubMed]
- Haycock, G.B. Development of Glomerular Filtration and Tubular Sodium Reabsorption in the Human Fetus and Newborn. Br. J. Urol. 1998, 81 (Suppl. S2), 33–38. [Google Scholar] [CrossRef]
- Nyengaard, J.R.; Bendtsen, T.F. Glomerular Number and Size in Relation to Age, Kidney Weight, and Body Surface in Normal Man. Anat. Rec. 1992, 232, 194–201. [Google Scholar] [CrossRef]
- Kalucki, S.A.; Lardi, C.; Garessus, J.; Kfoury, A.; Grabherr, S.; Burnier, M.; Pruijm, M. Reference Values and Sex Differences in Absolute and Relative Kidney Size. A Swiss Autopsy Study. BMC Nephrol. 2020, 21, 289. [Google Scholar] [CrossRef]
- Denic, A.; Mathew, J.; Lerman, L.O.; Lieske, J.C.; Larson, J.J.; Alexander, M.P.; Poggio, E.; Glassock, R.J.; Rule, A.D. Single-Nephron Glomerular Filtration Rate in Healthy Adults. N. Engl. J. Med. 2017, 376, 2349–2357. [Google Scholar] [CrossRef]
- Ferdous, F.; Ma, E.; Raqib, R.; Wagatsuma, Y. Birth Weight Influences the Kidney Size and Function of Bangladeshi Children. J. Dev. Orig. Health Dis. 2018, 9, 386–394. [Google Scholar] [CrossRef]
- Geelhoed, J.J.M.; Taal, H.R.; Steegers, E.A.P.; Arends, L.R.; Lequin, M.; Moll, H.A.; Hofman, A.; van der Heijden, A.J.; Jaddoe, V.W.V. Kidney Growth Curves in Healthy Children from the Third Trimester of Pregnancy until the Age of Two Years. The Generation R Study. Pediatr. Nephrol. 2010, 25, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, I.M.; Main, K.M.; Damgaard, I.N.; Mau, C.; Haavisto, A.-M.; Chellakooty, M.; Boisen, K.A.; Petersen, J.H.; Scheike, T.; Olgaard, K. Kidney Growth in 717 Healthy Children Aged 0-18 Months: A Longitudinal Cohort Study. Pediatr. Nephrol. 2004, 19, 992–1003. [Google Scholar] [CrossRef]
- Mañalich, R.; Reyes, L.; Herrera, M.; Melendi, C.; Fundora, I. Relationship between Weight at Birth and the Number and Size of Renal Glomeruli in Humans: A Histomorphometric Study. Kidney Int. 2000, 58, 770–773. [Google Scholar] [CrossRef]
- Okada, A.; Yabuki, A.; Matsumoto, M.; Suzuki, S. Development of Gender Differences in DBA/2Cr Mouse Kidney Morphology during Maturation. J. Vet. Med. Sci. 2005, 67, 877–882. [Google Scholar] [CrossRef]
- Shortliffe, L.M.D.; Ye, Y.; Behr, B.; Wang, B. Testosterone Changes Bladder and Kidney Structure in Juvenile Male Rats. J. Urol. 2014, 191, 1913–1919. [Google Scholar] [CrossRef]
- Gagliano-Jucá, T.; Tang, E.R.; Bhasin, S.; Pencina, K.M.; Anderson, S.; Jara, H.; Li, Z.; Melamud, K.; Coleman, S.L.; Aakil, A.; et al. Effects of Testosterone Administration (and Its 5-Alpha-Reduction) on Parenchymal Organ Volumes in Healthy Young Men: Findings from a Dose-Response Trial. Andrology 2017, 5, 889–897. [Google Scholar] [CrossRef]
- Forest, M.G.; de Peretti, E.; Lecoq, A.; Cadillon, E.; Zabot, M.T.; Thoulon, J.M. Concentration of 14 Steroid Hormones in Human Amniotic Fluid of Midpregnancy. J. Clin. Endocrinol. Metab. 1980, 51, 816–822. [Google Scholar] [CrossRef]
- Bábíčková, J.; Borbélyová, V.; Tóthová, L.; Kubišová, K.; Janega, P.; Hodosy, J.; Celec, P. The Renal Effects of Prenatal Testosterone in Rats. J. Urol. 2015, 193, 1700–1708. [Google Scholar] [CrossRef]
- Martinerie, L.; Munier, M.; Le Menuet, D.; Meduri, G.; Viengchareun, S.; Lombès, M. The Mineralocorticoid Signaling Pathway throughout Development: Expression, Regulation and Pathophysiological Implications. Biochimie 2013, 95, 148–157. [Google Scholar] [CrossRef]
- Martinerie, L.; Viengchareun, S.; Meduri, G.; Kim, H.-S.; Luther, J.M.; Lombès, M. Aldosterone Postnatally, but Not at Birth, Is Required for Optimal Induction of Renal Mineralocorticoid Receptor Expression and Sodium Reabsorption. Endocrinology 2011, 152, 2483–2491. [Google Scholar] [CrossRef]
- Wiinberg, N.; Høegholm, A.; Christensen, H.R.; Bang, L.E.; Mikkelsen, K.L.; Nielsen, P.E.; Svendsen, T.L.; Kampmann, J.P.; Madsen, N.H.; Bentzon, M.W. 24-h Ambulatory Blood Pressure in 352 Normal Danish Subjects, Related to Age and Gender. Am. J. Hypertens. 1995, 8, 978–986. [Google Scholar] [CrossRef]
- Hilliard, L.M.; Sampson, A.K.; Brown, R.D.; Denton, K.M. The “His and Hers” of the Renin-Angiotensin System. Curr. Hypertens. Rep. 2013, 15, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Meng, Q.C. Sexual Dimorphism of Blood Pressure in Spontaneously Hypertensive Rats Is Androgen Dependent. Life Sci. 1991, 48, 85–96. [Google Scholar] [CrossRef]
- Rowland, N.E.; Fregly, M.J. Role of Gonadal Hormones in Hypertension in the Dahl Salt-Sensitive Rat. Clin. Exp. Hypertens. A 1992, 14, 367–375. [Google Scholar] [CrossRef]
- Veiras, L.C.; Girardi, A.C.C.; Curry, J.; Pei, L.; Ralph, D.L.; Tran, A.; Castelo-Branco, R.C.; Pastor-Soler, N.; Arranz, C.T.; Yu, A.S.L.; et al. Sexual Dimorphic Pattern of Renal Transporters and Electrolyte Homeostasis. J. Am. Soc. Nephrol. 2017, 28, 3504–3517. [Google Scholar] [CrossRef]
- Zheng, W.; Shi, M.; You, S.-E.; Ji, H.; Roesch, D.M. Estrogens Contribute to a Sex Difference in Plasma Potassium Concentration: A Mechanism for Regulation of Adrenal Angiotensin Receptors. Gend. Med. 2006, 3, 43–53. [Google Scholar] [CrossRef]
- Condon, J.; Ricketts, M.L.; Whorwood, C.B.; Stewart, P.M. Ontogeny and Sexual Dimorphic Expression of Mouse Type 2 11beta-Hydroxysteroid Dehydrogenase. Mol. Cell. Endocrinol. 1997, 127, 121–128. [Google Scholar] [CrossRef]
- Steen, E.E.; Källén, K.; Maršál, K.; Norman, M.; Hellström-Westas, L. Impact of Sex on Perinatal Mortality and Morbidity in Twins. J. Perinat. Med. 2014, 42, 225–231. [Google Scholar] [CrossRef]
- Sandberg, K.; Ji, H. Sex Differences in Primary Hypertension. Biol. Sex Differ. 2012, 3, 7. [Google Scholar] [CrossRef]
- Ojeda, N.B.; Intapad, S.; Alexander, B.T. Sex Differences in the Developmental Programming of Hypertension. Acta Physiol. 2014, 210, 307–316. [Google Scholar] [CrossRef]
- Barker, D.J.; Bull, A.R.; Osmond, C.; Simmonds, S.J. Fetal and Placental Size and Risk of Hypertension in Adult Life. BMJ 1990, 301, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Winter, P.D.; Osmond, C.; Margetts, B.; Simmonds, S.J. Weight in Infancy and Death from Ischaemic Heart Disease. Lancet 1989, 2, 577–580. [Google Scholar] [CrossRef]
- Moritz, K.M.; Cuffe, J.S.M.; Wilson, L.B.; Dickinson, H.; Wlodek, M.E.; Simmons, D.G.; Denton, K.M. Review: Sex Specific Programming: A Critical Role for the Renal Renin-Angiotensin System. Placenta 2010, 31, S40–S46. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, E.C.; Holmes, M.C.; Livingstone, D.E.; Kenyon, C.J.; Seckl, J.R. Reconciling the Nutritional and Glucocorticoid Hypotheses of Fetal Programming. FASEB J. 2012, 26, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Moritz, K.M.; Wintour, E.M.; Black, M.J.; Bertram, J.F.; Caruana, G. Factors Influencing Mammalian Kidney Development: Implications for Health in Adult Life. Adv. Anat. Embryol. Cell Biol. 2008, 196, 1–78. [Google Scholar] [CrossRef] [PubMed]
- Hughson, M.; Farris, A.B.; Douglas-Denton, R.; Hoy, W.E.; Bertram, J.F. Glomerular Number and Size in Autopsy Kidneys: The Relationship to Birth Weight. Kidney Int. 2003, 63, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Grigore, D.; Ojeda, N.B.; Alexander, B.T. Sex Differences in the Fetal Programming of Hypertension. Gend. Med. 2008, 5 (Suppl. S1), S121–S132. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.S.; Goldenberg, R.L. Global Burden of Prematurity. Semin. Fetal Neonatal. Med. 2016, 21, 74–79. [Google Scholar] [CrossRef]
- Boghossian, N.S.; Geraci, M.; Edwards, E.M.; Horbar, J.D. Sex Differences in Mortality and Morbidity of Infants Born at Less Than 30 Weeks’ Gestation. Pediatrics 2018, 142. [Google Scholar] [CrossRef]
- Verloove-Vanhorick, S.P.; Veen, S.; Ens-Dokkum, M.H.; Schreuder, A.M.; Brand, R.; Ruys, J.H. Sex Difference in Disability and Handicap at Five Years of Age in Children Born at Very Short Gestation. Pediatrics 1994, 93, 576–579. [Google Scholar]
- De Jong, F.; Monuteaux, M.C.; van Elburg, R.M.; Gillman, M.W.; Belfort, M.B. Systematic Review and Meta-Analysis of Preterm Birth and Later Systolic Blood Pressure. Hypertension 2012, 59, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Staub, E.; Urfer-Maurer, N.; Lemola, S.; Risch, L.; Evers, K.S.; Welzel, T.; Pfister, M. Comparison of Blood Pressure and Kidney Markers between Adolescent Former Preterm Infants and Term Controls. Children 2020, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Battarbee, A.N.; Glover, A.V.; Vladutiu, C.J.; Gyamfi-Bannerman, C.; Aliaga, S.; Manuck, T.A.; Boggess, K.A. Sex-Specific Differences in Late Preterm Neonatal Outcomes. Am. J. Perinatol. 2019, 36, 1223–1228. [Google Scholar] [CrossRef]
- Dumeige, L.; Nehlich, M.; Viengchareun, S.; Perrot, J.; Pussard, E.; Lombès, M.; Martinerie, L. Preterm Birth Is Associated with Epigenetic Programming of Transgenerational Hypertension in Mice. Exp. Mol. Med. 2020, 52, 152–165. [Google Scholar] [CrossRef]
- Barker, D.J. The Fetal and Infant Origins of Adult Disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef]
- Brenner, B.M.; Mackenzie, H.S. Nephron Mass as a Risk Factor for Progression of Renal Disease. Kidney Int. Suppl. 1997, 63, S124–S127. [Google Scholar]
- Stelloh, C.; Allen, K.P.; Mattson, D.L.; Lerch-Gaggl, A.; Reddy, S.; El-Meanawy, A. Prematurity in Mice Leads to Reduction in Nephron Number, Hypertension, and Proteinuria. Transl. Res. 2012, 159, 80–89. [Google Scholar] [CrossRef]
- Mathai, S.; Derraik, J.G.B.; Cutfield, W.S.; Dalziel, S.R.; Harding, J.E.; Biggs, J.B.; Jefferies, C.; Hofman, P.L. Blood Pressure Abnormalities in Adults Born Moderately Preterm and Their Children. Int. J. Cardiol. 2015, 181, 152–154. [Google Scholar] [CrossRef]
- Bogdanović, R.; Stajić, N.; Putnik, J.; Paripović, A. Transient Type 1 Pseudo-Hypoaldosteronism: Report on an Eight-Patient Series and Literature Review. Pediatr. Nephrol. 2009, 24, 2167–2175. [Google Scholar] [CrossRef]
- De Seigneux, S.; Leroy, V.; Ghzili, H.; Rousselot, M.; Nielsen, S.; Rossier, B.C.; Martin, P.-Y.; Féraille, E. NF-KappaB Inhibits Sodium Transport via down-Regulation of SGK1 in Renal Collecting Duct Principal Cells. J. Biol. Chem. 2008, 283, 25671–25681. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Target Genes | Nuclear Receptors | Functions | References |
|---|---|---|---|
| αENAC subunit | MR | Na+ transport | [57] |
| Na+, K+-ATPase | MR | Na+ transport | [58] |
| KS-WNK1 | MR | Na+ transport | [60] |
| PER1 | MR, GR | Circadian rhythm | [63] |
| NDRG2 | MR | Cell differentiation | [61] |
| SGK1 | GR, MR | Ser/Thr protein kinase ENAC trafficking/Na+ reabsorption | [59] |
| GILZ | GR, MR | ENAC trafficking/Na+ reabsorption | [62] |
| FKBP5 | GR, MR | Chaperone protein | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laulhé, M.; Dumeige, L.; Vu, T.A.; Hani, I.; Pussard, E.; Lombès, M.; Viengchareun, S.; Martinerie, L. Sexual Dimorphism of Corticosteroid Signaling during Kidney Development. Int. J. Mol. Sci. 2021, 22, 5275. https://doi.org/10.3390/ijms22105275
Laulhé M, Dumeige L, Vu TA, Hani I, Pussard E, Lombès M, Viengchareun S, Martinerie L. Sexual Dimorphism of Corticosteroid Signaling during Kidney Development. International Journal of Molecular Sciences. 2021; 22(10):5275. https://doi.org/10.3390/ijms22105275
Chicago/Turabian StyleLaulhé, Margaux, Laurence Dumeige, Thi An Vu, Imene Hani, Eric Pussard, Marc Lombès, Say Viengchareun, and Laetitia Martinerie. 2021. "Sexual Dimorphism of Corticosteroid Signaling during Kidney Development" International Journal of Molecular Sciences 22, no. 10: 5275. https://doi.org/10.3390/ijms22105275
APA StyleLaulhé, M., Dumeige, L., Vu, T. A., Hani, I., Pussard, E., Lombès, M., Viengchareun, S., & Martinerie, L. (2021). Sexual Dimorphism of Corticosteroid Signaling during Kidney Development. International Journal of Molecular Sciences, 22(10), 5275. https://doi.org/10.3390/ijms22105275