
Protein Kinase C Activation Drives a Differentiation Program in an Oligodendroglial Precursor Model through the Modulation of Specific Biological Networks

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
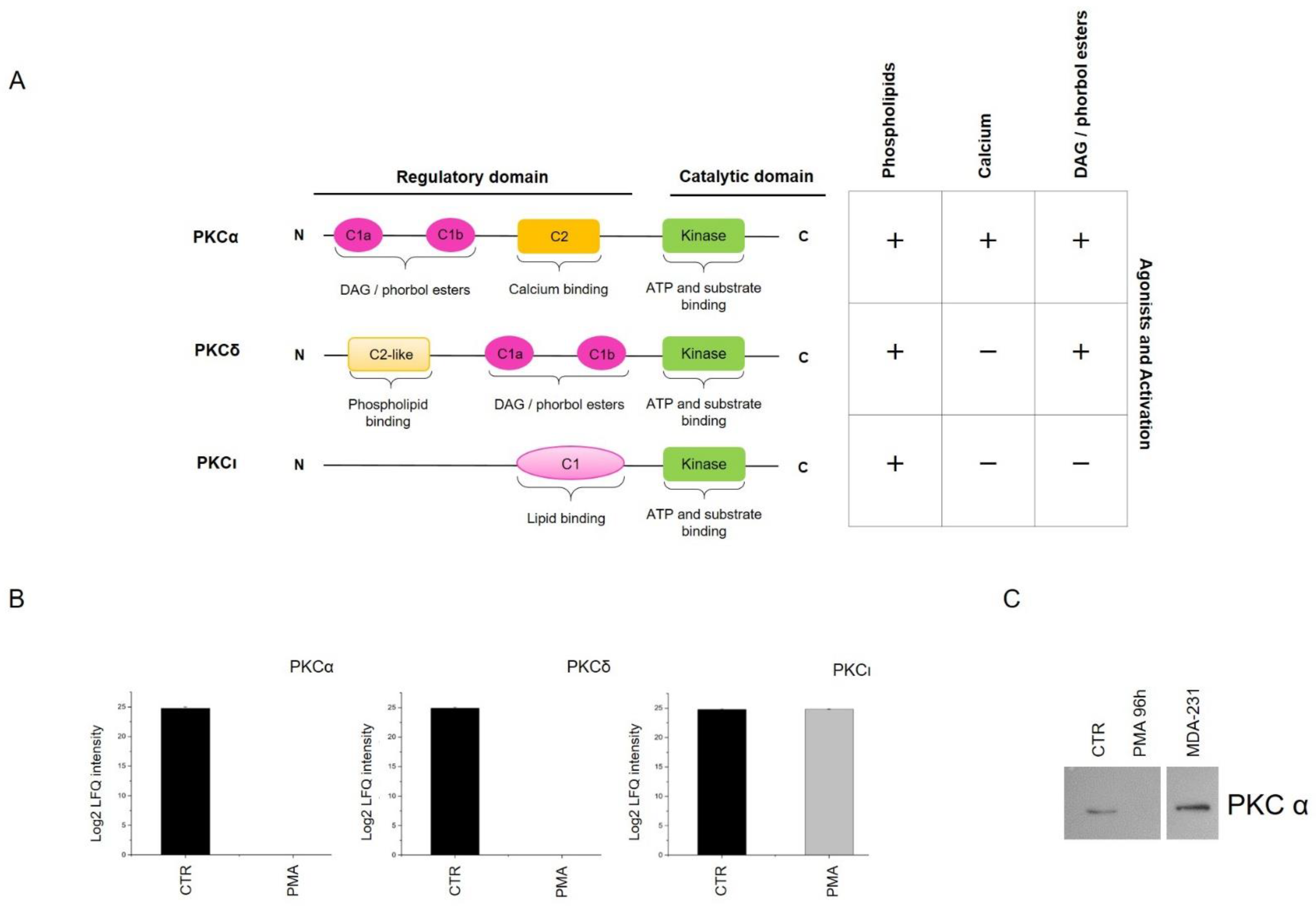
2.1. Phorbol 12-Myristate 13-Acetate (PMA) Treatment Induces Phospho-PKC Activation and Regulates MO3.13 Proliferation and Differentiation
2.2. Networks Modulated after PMA Treatment
2.3. Signaling Pathways Modulated by PKC Activation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Viability Assay
4.3. Sample Preparation and Mass Spectrometry Analysis
4.4. Mass Spectrometry Analysis and Database Searching
4.5. Experimental Design and Statistical Rationale
4.6. Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Merrell, A.J.; Stanger, B.Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Fagnocchi, L.; Mazzoleni, S.; Zippo, A. Integration of Signaling Pathways with the Epigenetic Machinery in the Maintenance of Stem Cells. Stem Cells Int. 2016, 2016, 8652748. [Google Scholar] [CrossRef]
- Hass, R.; Bartels, H.; Topley, N.; Hadam, M.; Köhler, L.; Goppelt-Strübe, M.; Resch, K. TPA-induced differentiation and adhesion of U937 cells: Changes in ultrastructure, cytoskeletal organization and expression of cell surface antigens. Eur. J. Cell Biol. 1989, 48, 282–293. [Google Scholar] [PubMed]
- Leli, U.; Cataldo, A.; Shea, T.B.; Nixon, R.A.; Hauser, G. Distinct Mechanisms of Differentiation of SH-SY5Y Neuroblastoma Cells by Protein Kinase C Activators and Inhibitors. J. Neurochem. 1992, 58, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, F.; Benito-Muñoz, M.; Panicker, M.; Matute, C. NMDA modulates oligodendrocyte differentiation of subventricular zone cells through PKC activation. Front. Cell. Neurosci. 2013, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.; Alkon, D. Neuroprotective versus tumorigenic protein kinase C activators. Trends Biochem. Sci. 2009, 34, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.B.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.C.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef]
- Lin, R.; Taylor, B.V.; Simpson, S., Jr.; Charlesworth, J.; Ponsonby, A.-L.; Pittas, F.; Dwyer, T.; van der Mei, I.A. Novel modulating effects of PKC family genes on the relationship between serum vitamin D and relapse in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 85, 399–404. [Google Scholar] [CrossRef]
- Garrido, J.L.; Godoy, J.A.; Alvarez, A.; Bronfman, M.; Inestrosa, N.C. Protein kinase C inhibits amyloid β-peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 2002, 16, 1982–1984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Neuroprotective Effect of Protein Kinase C delta Inhibitor Rottlerin in Cell Culture and Animal Models of Parkinson’s Disease. J. Pharmacol. Exp. Ther. 2007, 322, 913–922. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Chari, D.M. Remyelination in Multiple Sclerosis. Int. Rev. Neurobiol. 2007, 79, 589–620. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—From mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Altelaar, A.F.M.; Munoz, J.; Heck, A.J. Next-generation proteomics: Towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2012, 14, 35–48. [Google Scholar] [CrossRef]
- Sabidó, E.; Selevsek, N.; Aebersold, R. Mass spectrometry-based proteomics for systems biology. Curr. Opin. Biotechnol. 2012, 23, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Café-Mendes, C.C.; Schmitt, A.; Steiner, J.; Manabe, T.; Matsuzaki, H.; Falkai, P.; Turck, C.W.; Martins-De-Souza, D. The human oligodendrocyte proteome. Proteomics 2013, 13, 3548–3553. [Google Scholar] [CrossRef]
- Schoor, C.; Brocke-Ahmadinejad, N.; Gieselmann, V.; Winter, D. Investigation of Oligodendrocyte Precursor Cell Differentiation by Quantitative Proteomics. Proteomics 2019, 19, e1900057. [Google Scholar] [CrossRef]
- Bribián, A.; Medina-Rodríguez, E.M.; Josa-Prado, F.; García-Álvarez, I.; Machín-Díaz, I.; Esteban, P.F.; Murcia-Belmonte, V.; Vega-Zelaya, L.; Pastor, J.; Garrido, L.; et al. Functional Heterogeneity of Mouse and Human Brain OPCs: Relevance for Preclinical Studies in Multiple Sclerosis. J. Clin. Med. 2020, 9, 1681. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Trudel, G.C.; Shaw, I.T.; Antel, J.P.; Cashman, N.R. A human glial hybrid cell line differentially expressing genes subserving oligodendrocyte and astrocyte phenotype. J. Neurobiol. 1995, 26, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Boscia, F.; D’Avanzo, C.; Pannaccione, A.; Secondo, A.; Casamassa, A.; Formisano, L.; Guida, N.; Annunziato, L. Silencing or knocking out the Na+/Ca2+ exchanger-3 (NCX3) impairs oligodendrocyte differentiation. Cell Death Differ. 2011, 19, 562–572. [Google Scholar] [CrossRef]
- Accetta, R.; Damiano, S.; Morano, A.; Mondola, P.; Paternò, R.; Avvedimento, E.V.; Santillo, M. Reactive Oxygen Species Derived from NOX3 and NOX5 Drive Differentiation of Human Oligodendrocytes. Front. Cell. Neurosci. 2016, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Secondo, A.; Pannaccione, A.; Ciccone, R.; Formisano, L.; Guida, N.; Crispino, R.; Fico, A.; Polishchuk, R.; D’Aniello, A.; et al. D-Aspartate treatment attenuates myelin damage and stimulates myelin repair. EMBO Mol. Med. 2019, 11, e9278. [Google Scholar] [CrossRef] [PubMed]
- Damiano, S.; La Rosa, G.; Sozio, C.; Cavaliere, G.; Trinchese, G.; Raia, M.; Paternò, R.; Mollica, M.; Avvedimento, V.; Santillo, M. 5-Hydroxytryptamine Modulates Maturation and Mitochondria Function of Human Oligodendrocyte Progenitor M03-13 Cells. Int. J. Mol. Sci. 2021, 22, 2621. [Google Scholar] [CrossRef] [PubMed]
- Abbastabar, M.; Kheyrollah, M.; Azizian, K.; Bagherlou, N.; Tehrani, S.S.; Maniati, M.; Karimian, A. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA Repair 2018, 69, 63–72. [Google Scholar] [CrossRef]
- Nguyen, L.; Besson, A.; Heng, J.I.-T.; Schuurmans, C.; Teboul, L.; Parras, C.; Philpott, A.; Roberts, J.M.; Guillemot, F. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006, 20, 1511–1524. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Pedone, E.; Marucci, L. Role of β-Catenin Activation Levels and Fluctuations in Controlling Cell Fate. Genes 2019, 10, 176. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Frese, C.K.; Mikhaylova, M.; Stucchi, R.; Gautier, V.; Liu, Q.; Mohammed, S.; Heck, A.J.; Altelaar, A.M.; Hoogenraad, C.C. Quantitative Map of Proteome Dynamics during Neuronal Differentiation. Cell Rep. 2017, 18, 1527–1542. [Google Scholar] [CrossRef]
- Tawk, M.; Makoukji, J.; Belle, M.; Fonte, C.; Trousson, A.; Hawkins, T.; Li, H.; Ghandour, S.; Schumacher, M.; Massaad, C. Wnt/beta-Catenin Signaling Is an Essential and Direct Driver of Myelin Gene Expression and Myelinogenesis. J. Neurosci. 2011, 31, 3729–3742. [Google Scholar] [CrossRef]
- Lee, J.; Gravel, M.; Zhang, R.; Thibault, P.; Braun, P.E. Process outgrowth in oligodendrocytes is mediated by CNP, a novel microtubule assembly myelin protein. J. Cell Biol. 2005, 170, 661–673. [Google Scholar] [CrossRef]
- Shao, Z.; Lee, X.; Huang, G.; Sheng, G.; Henderson, C.E.; Louvard, D.; Sohn, J.; Pepinsky, B.; Mi, S. LINGO-1 Regulates Oligodendrocyte Differentiation through the Cytoplasmic Gelsolin Signaling Pathway. J. Neurosci. 2017, 37, 3127–3137. [Google Scholar] [CrossRef]
- Ehrman, L.A.; Nardini, D.; Ehrman, S.; Rizvi, T.A.; Gulick, J.; Krenz, M.; Dasgupta, B.; Robbins, J.; Ratner, N.; Nakafuku, M.; et al. The protein tyrosine phosphatase Shp2 is required for the generation of oligodendrocyte progenitor cells and myelination in the mouse telencephalon. J. Neurosci. 2014, 34, 3767–3778. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.N.; Appel, B. Oligodendrocytes express synaptic proteins that modulate myelin sheath formation. Nat. Commun. 2019, 10, 4125. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, D.; Hornia, A.; Devonish, W.; Pagano, M.; Foster, D.A. Activation of Protein Kinase C Triggers Its Ubiquitination and Degradation. Mol. Cell. Biol. 1998, 18, 839–845. [Google Scholar] [CrossRef]
- Zhimin, L.; Tony, H. Degradation of Activated Protein Kinases by Ubiquitination. Annu. Rev. Biochem. 2009, 78, 435–475. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Mayer, J.A.; Duncan, I.D. Autophagy Promotes Oligodendrocyte Survival and Function following Dysmyelination in a Long-Lived Myelin Mutant. J. Neurosci. 2013, 33, 8088–8100. [Google Scholar] [CrossRef]
- Bankston, A.N.; Forston, M.D.; Howard, R.M.; Andres, K.R.; Smith, A.E.; Ohri, S.S.; Bates, M.L.; Bunge, M.B.; Whittemore, S.R. Autophagy is essential for oligodendrocyte differentiation, survival, and proper myelination. Glia 2019, 67, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Pooyan, P.; Karamzadeh, R.; Mirzaei, M.; Meyfour, A.; Amirkhan, A.; Wu, Y.; Gupta, V.; Baharvand, H.; Javan, M.; Salekdeh, G.H. The Dynamic Proteome of Oligodendrocyte Lineage Differentiation Features Planar Cell Polarity and Macroautophagy Pathways. GigaScience 2020, 9, giaa116. [Google Scholar] [CrossRef]
- Hogrebe, N.J.; Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 460–470. [Google Scholar] [CrossRef]
- Ambriz, X.; De Lanerolle, P.; Ambrosio, J.R. The Mechanobiology of the Actin Cytoskeleton in Stem Cells during Differentiation and Interaction with Biomaterials. Stem Cells Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dobretsova, A.; Johnson, J.W.; Jones, R.C.; Edmondson, R.D.; Wight, P.A. Proteomic analysis of nuclear factors binding to an intronic enhancer in the myelin proteolipid protein gene. J. Neurochem. 2008, 105, 1979–1995. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Thatikunta, P.; Steplewski, A.; Johnson, E.M.; Khalili, K.; Amini, S. A 39-kD DNA-binding protein from mouse brain stimulates transcription of myelin basic protein gene in oligodendrocytic cells. J. Cell Biol. 1995, 130, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Daniel, D.C.; Johnson, E.M. PURA, the gene encoding Pur-alpha, member of an ancient nucleic acid-binding protein family with mammalian neurological functions. Gene 2018, 643, 133–143. [Google Scholar] [CrossRef]
- Larocque, D.; Pilotte, J.; Chen, T.; Cloutier, F.; Massie, B.; Pedraza, L.; Couture, R.; Lasko, P.; Almazan, G.; Richard, S. Nuclear Retention of MBP mRNAs in the Quaking Viable Mice. Neuron 2002, 36, 815–829. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Z.; Ku, L.; Chen, Y.; Wang, H.; Feng, Y. Tyrosine phosphorylation of QKI mediates developmental signals to regulate mRNA metabolism. EMBO J. 2003, 22, 1801–1810. [Google Scholar] [CrossRef]
- Wu, J.I.; Reed, R.B.; Grabowski, P.J.; Artzt, K. Function of quaking in myelination: Regulation of alternative splicing. Proc. Natl. Acad. Sci. USA 2002, 99, 4233–4238. [Google Scholar] [CrossRef]
- Aberg, K.; Saetre, P.; Jareborg, N.; Jazin, E. Human QKI, a potential regulator of mRNA expression of human oligodendrocyte-related genes involved in schizophrenia. Proc. Natl. Acad. Sci. USA 2006, 103, 7482–7487. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.A.; Lauriat, T.L. RNA metabolism and dysmyelination in schizophrenia. Neurosci. Biobehav. Rev. 2006, 30, 551–561. [Google Scholar] [CrossRef]
- Zhou, X.; He, C.; Ren, J.; Dai, C.; Stevens, S.; Wang, Q.; Zamler, D.; Shingu, T.; Yuan, L.; Chandregowda, C.R.; et al. Mature myelin maintenance requires Qki to coactivate PPARβ-RXRα–mediated lipid metabolism. J. Clin. Investig. 2020, 130, 2220–2236. [Google Scholar] [CrossRef]
- Heng, B.C.; Zhang, X.; Aubel, D.; Bai, Y.; Li, X.; Wei, Y.; Fussenegger, M.; Deng, X. Role of YAP/TAZ in Cell Lineage Fate Determination and Related Signaling Pathways. Front. Cell Dev. Biol. 2020, 8, 735. [Google Scholar] [CrossRef]
- Shimizu, T.; Osanai, Y.; Tanaka, K.F.; Abe, M.; Natsume, R.; Sakimura, K.; Ikenaka, K. YAP functions as a mechanotransducer in oligodendrocyte morphogenesis and maturation. Glia 2017, 65, 360–374. [Google Scholar] [CrossRef]
- Swire, M.; Kotelevtsev, Y.; Webb, D.J.; Lyons, D.A.; Ffrench-Constant, C. Endothelin signalling mediates experience-dependent myelination in the CNS. eLife 2019, 8, e49493. [Google Scholar] [CrossRef] [PubMed]
- Pintado-Sierra, M.; García-Álvarez, I.; Bribián, A.; Medina-Rodríguez, E.; Lebrón-Aguilar, R.; Garrido, L.; de Castro, F.; Fernández-Mayoralas, A.; Quintanilla-López, J. A comprehensive profiling of sulfatides in myelin from mouse brain using liquid chromatography coupled to high-resolution accurate tandem mass spectrometry. Anal. Chim. Acta 2017, 951, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-L.; Lin, J.-Y.; Shieh, J.-C.; Yeh, H.-F.; Hsieh, Y.-H.; Cheng, Y.-C.; Lee, H.-J.; Shen, C.-Y.; Cheng, C.-W. Induction of G2/M Phase Arrest by Diosgenin via Activation of Chk1 Kinase and Cdc25C Regulatory Pathways to Promote Apoptosis in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 21, 172. [Google Scholar] [CrossRef]
- Magri, L.; Gacias, M.; Wu, M.; Swiss, V.; Janssen, W.; Casaccia, P. c-Myc-dependent transcriptional regulation of cell cycle and nucleosomal histones during oligodendrocyte differentiation. Neuroscience 2014, 276, 72–86. [Google Scholar] [CrossRef]
- León, J.; Ferrandiz, N.; Acosta, J.C.; Delgado, M.D. Inhibition of cell differentiation: A critical mechanism for MYC-mediated carcinogenesis? Cell Cycle 2009, 8, 1148–1157. [Google Scholar] [CrossRef]
- Pernet, V.; Joly, S.; Christ, F.; Dimou, L.; Schwab, M.E. Nogo-A and Myelin-Associated Glycoprotein Differently Regulate Oligodendrocyte Maturation and Myelin Formation. J. Neurosci. 2008, 28, 7435–7444. [Google Scholar] [CrossRef]
- Ahrendsen, J.T.; Harlow, D.E.; Finseth, L.T.; Bourne, J.N.; Hickey, S.P.; Gould, E.A.; Culp, C.M.; Macklin, W.B. The Protein Tyrosine Phosphatase Shp2 Regulates Oligodendrocyte Differentiation and Early Myelination and Contributes to Timely Remyelination. J. Neurosci. 2018, 38, 787–802. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Fu, M.-M.; Sloan, S.A.; Ibrahim, A.; Olson, A.; Zaremba, A.; Dugas, J.C.; Wienbar, S.; Caprariello, A.V.; Kantor, C.; et al. CNS Myelin Wrapping Is Driven by Actin Disassembly. Dev. Cell 2015, 34, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.; Sánchez, P.; Schmitt, S.; Snaidero, N.; Mitkovski, M.; Velte, C.; Brückner, B.R.; Alexopoulos, I.; Czopka, T.; Jung, S.Y.; et al. Actin Filament Turnover Drives Leading Edge Growth during Myelin Sheath Formation in the Central Nervous System. Dev. Cell 2015, 34, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dai, F.; Tang, L.; Le, Y.; Yao, W. Macrophage differentiation induced by PMA is mediated by activation of RhoA/ROCK signaling. J. Toxicol. Sci. 2017, 42, 763–771. [Google Scholar] [CrossRef]
- Takahashi, N.; Nobusue, H.; Shimizu, T.; Sugihara, E.; Yamaguchi-Iwai, S.; Onishi, N.; Kunitomi, H.; Kuroda, T.; Saya, H. ROCK Inhibition Induces Terminal Adipocyte Differentiation and Suppresses Tumorigenesis in Chemoresistant Osteosarcoma Cells. Cancer Res. 2019, 79, 3088–3099. [Google Scholar] [CrossRef] [PubMed]
- McMullan, R.; Lax, S.; Robertson, V.H.; Radford, D.J.; Broad, S.; Watt, F.M.; Rowles, A.; Croft, D.R.; Olson, M.F.; Hotchin, N.A. Keratinocyte Differentiation Is Regulated by the Rho and ROCK Signaling Pathway. Curr. Biol. 2003, 13, 2185–2189. [Google Scholar] [CrossRef]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell Shape, Cytoskeletal Tension, and RhoA Regulate Stem Cell Lineage Commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef]
- Pedraza, C.E.; Taylor, C.; Pereira, A.; Seng, M.; Tham, C.-S.; Izrael, M.; Webb, M. Induction of Oligodendrocyte Differentiation and In Vitro Myelination by Inhibition of Rho-Associated Kinase. ASN Neuro 2014, 6, 1759091414538134. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damato, M.; Cardon, T.; Wisztorski, M.; Fournier, I.; Pieragostino, D.; Cicalini, I.; Salzet, M.; Vergara, D.; Maffia, M. Protein Kinase C Activation Drives a Differentiation Program in an Oligodendroglial Precursor Model through the Modulation of Specific Biological Networks. Int. J. Mol. Sci. 2021, 22, 5245. https://doi.org/10.3390/ijms22105245
Damato M, Cardon T, Wisztorski M, Fournier I, Pieragostino D, Cicalini I, Salzet M, Vergara D, Maffia M. Protein Kinase C Activation Drives a Differentiation Program in an Oligodendroglial Precursor Model through the Modulation of Specific Biological Networks. International Journal of Molecular Sciences. 2021; 22(10):5245. https://doi.org/10.3390/ijms22105245
Chicago/Turabian StyleDamato, Marina, Tristan Cardon, Maxence Wisztorski, Isabelle Fournier, Damiana Pieragostino, Ilaria Cicalini, Michel Salzet, Daniele Vergara, and Michele Maffia. 2021. "Protein Kinase C Activation Drives a Differentiation Program in an Oligodendroglial Precursor Model through the Modulation of Specific Biological Networks" International Journal of Molecular Sciences 22, no. 10: 5245. https://doi.org/10.3390/ijms22105245
APA StyleDamato, M., Cardon, T., Wisztorski, M., Fournier, I., Pieragostino, D., Cicalini, I., Salzet, M., Vergara, D., & Maffia, M. (2021). Protein Kinase C Activation Drives a Differentiation Program in an Oligodendroglial Precursor Model through the Modulation of Specific Biological Networks. International Journal of Molecular Sciences, 22(10), 5245. https://doi.org/10.3390/ijms22105245