
The Role of GSK-3β in the Regulation of Protein Turnover, Myosin Phenotype, and Oxidative Capacity in Skeletal Muscle under Disuse Conditions



Abstract
1. Introduction
2. Regulation of GSK-3β Activity
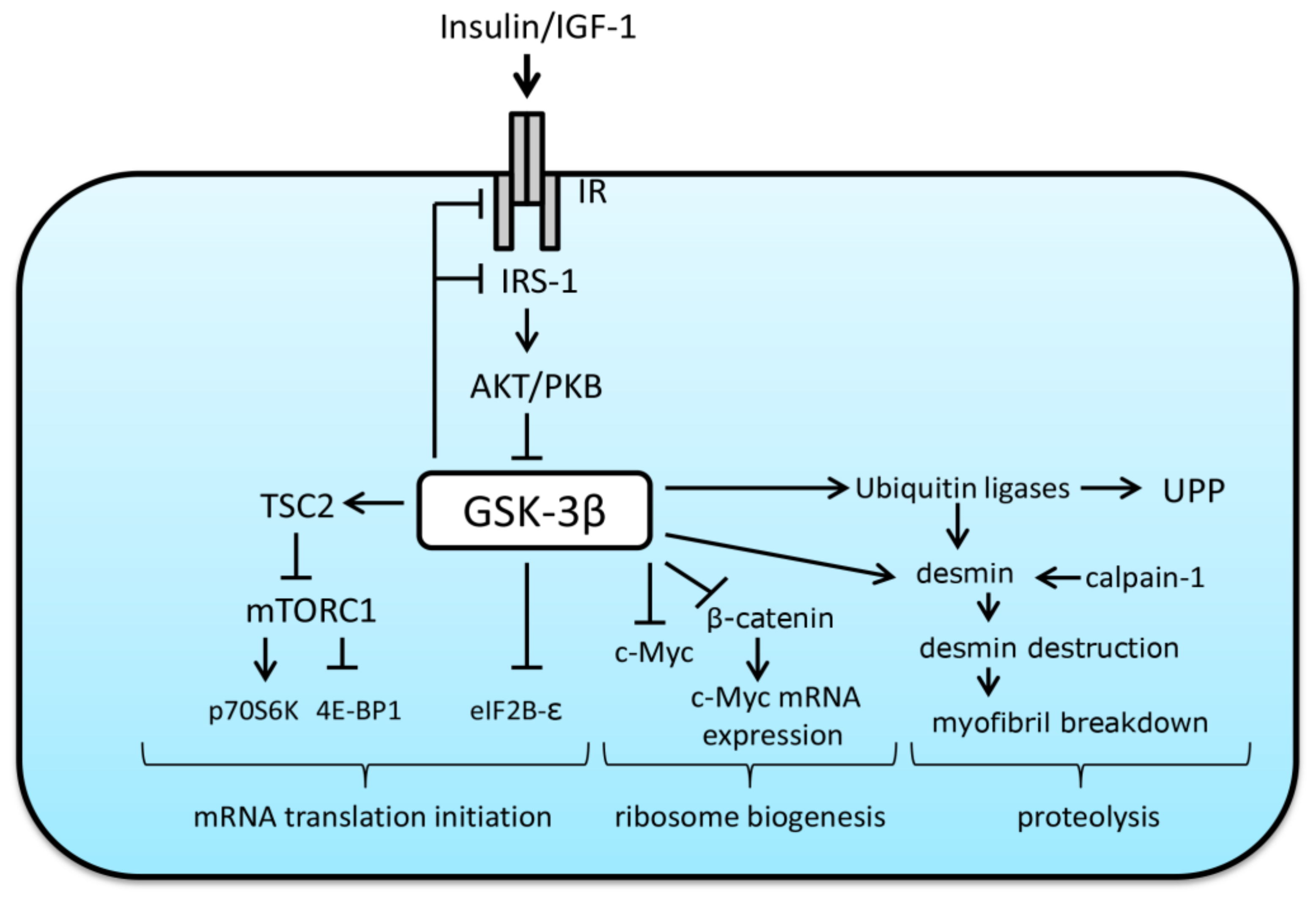
3. The Role of GSK-3β in the Regulation of Protein Synthesis and Breakdown
4. Impact of Mechanical Unloading on GSK-3β Activity and Possible Roles of this Enzyme in the Regulation of Protein Turnover in Mammalian Skeletal Muscles under Disuse Conditions and Subsequent Recovery
5. Impact of GSK-3β Activity on Fiber-Type Transitions and Oxidative Capacity in Skeletal Muscles under Mechanical Unloading and Subsequent Reloading
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4E-BP1 | eukaryotic initiation factor 4E binding protein |
| AKT | protein kinase B |
| AMPK | AMP-activated protein kinase |
| CaN | calcineurin |
| cGMP | cyclic guanosine monophosphate |
| c-Myc | c-myelocytomatosis oncogene (transcription factor) |
| CSA | cross-sectional area |
| eIF2B | eukaryotic initiation factor 2B |
| FoxO | forkhead box O protein |
| GC | guanylate cyclase |
| GS1 | glycogen synthase-1 |
| GSK-3β | glycogen synthase kinase-3β |
| GSK-3β KO | mice lacking muscle GSK-3β |
| HS | hindlimb suspension |
| HU | hindlimb unloading |
| IGF-1 | insulin-like growth factor 1 |
| ILK | integrin-linked kinase |
| IR | insulin/insulin-like growth factor 1 receptor |
| IRS-1 | insulin receptor substrate 1 |
| LC3 | microtubule-associated proteins 1A/1B light chain 3B |
| LiCl | lithium chloride |
| L-NAME | N(gamma)-nitro-L-arginine methyl ester |
| MAFbx | muscle atrophy F-box protein/atrogin-1 |
| MAPK | mitogen-activated protein kinase |
| MCIP 1.4 | modulatory calcineurin-interaction protein 1.4 |
| MEF-2 | myocyte enhancer factor-2 |
| mir-208 | micro-RNA 208 |
| mTORC1 | mammalian/mechanistic target of rapamycin complex 1 |
| MuRF1 | muscle RING finger protein |
| MyHC | myosin heavy chain |
| NES | nuclear export signal |
| NFAT | nuclear factor of activated T-cells |
| NO | nitric oxide |
| NRF | nuclear respiratory factor |
| p70S6K | ribosomal protein S6 kinase p70 |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | phosphatidylinositol 3-kinase |
| PKA | protein kinase A |
| PKB | protein kinase B (AKT) |
| PKC | protein kinase C |
| PKD1 | protein kinase D1 |
| PKG | protein kinase G |
| PMS | plantar mechanical stimulation |
| PP1 | protein phosphatase 1 |
| PP2A | protein phosphatase 2A |
| PuRA | purine Rich Element Binding Protein A |
| PuRB | purine Rich Element Binding Protein B |
| rRNA | ribosomal RNA |
| SOX-6 | transcription factor |
| SP3 | transcription factor |
| TDZD-8 | thiadiazolidinone-8 |
| TFAM | Mitochondrial transcription factor A |
| TFEB | transcription factor EB |
| THRAP | mediator of RNA polymerase II transcription subunit 13 |
| TNNI1 | slow isoform of troponin |
| TSC2 | tuberous sclerosis complex 2 |
| UPP | ubiquitin–proteasome pathway |
| WT | wild-type mice |
References
- Thomason, D.B.; Booth, F.W. Atrophy of the soleus muscle by hindlimb unweighting. J. Appl. Physiol. 1990, 68, 1–12. [Google Scholar] [CrossRef]
- Fitts, R.H.; Riley, D.R.; Widrick, J.J. Functional and structural adaptations of skeletal muscle to microgravity. J. Exp. Biol. 2001, 204, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Loughna, P.; Goldspink, G.; Goldspink, D.F. Effect of inactivity and passive stretch on protein turnover in phasic and postural rat muscles. J. Appl. Physiol. 1986, 61, 173–179. [Google Scholar] [PubMed]
- Wang, S.B.; Venkatraman, V.; Crowgey, E.L.; Liu, T.; Fu, Z.; Holewinski, R.; Ranek, M.; Kass, D.A.; O’Rourke, B.; Van Eyk, J.E. Protein S-Nitrosylation Controls Glycogen Synthase Kinase 3beta Function Independent of Its Phosphorylation State. Circ. Res. 2018, 122, 1517–1531. [Google Scholar] [CrossRef]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Verhees, K.J.; Pansters, N.A.; Schols, A.M.; Langen, R.C. Regulation of skeletal muscle plasticity by glycogen synthase kinase-3beta: A potential target for the treatment of muscle wasting. Curr. Pharm. Des. 2013, 19, 3276–3298. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Van Weeren, P.C.; de Bruyn, K.M.; de Vries-Smits, A.M.; van Lint, J.; Burgering, B.M. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J. Biol. Chem. 1998, 273, 13150–13156. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C., Jr.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Z.; Zhang, R.; Hu, Y.; Jiang, Y.; Cao, T.; Wang, J.; Gong, L.; Ji, L.; Mu, H.; et al. PKCgamma promotes axonal remodeling in the cortico-spinal tract via GSK3beta/beta-catenin signaling after traumatic brain injury. Sci. Rep. 2019, 9, 17078. [Google Scholar] [CrossRef] [PubMed]
- Kim Do, Y.; Park, E.Y.; Chang, E.; Kang, H.G.; Koo, Y.; Lee, E.J.; Ko, J.Y.; Kong, H.K.; Chun, K.H.; Park, J.H. A novel miR-34a target, protein kinase D1, stimulates cancer stemness and drug resistance through GSK3/beta-catenin signaling in breast cancer. Oncotarget 2016, 7, 14791–14802. [Google Scholar] [CrossRef]
- Zhao, X.; Zhuang, S.; Chen, Y.; Boss, G.R.; Pilz, R.B. Cyclic GMP-dependent protein kinase regulates CCAAT enhancer-binding protein beta functions through inhibition of glycogen synthase kinase-3. J. Biol. Chem. 2005, 280, 32683–32692. [Google Scholar] [CrossRef]
- Birschmann, I.; Walter, U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3’,5’-cyclic monophosphate-dependent protein kinase. Acta Biochim. Pol. 2004, 51, 397–404. [Google Scholar] [CrossRef]
- Drenning, J.A.; Lira, V.A.; Simmons, C.G.; Soltow, Q.A.; Sellman, J.E.; Criswell, D.S. Nitric oxide facilitates NFAT-dependent transcription in mouse myotubes. Am. J. Physiol. Cell Physiol. 2008, 294, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Bonavaud, S.M.; Toole, B.J.; Yeaman, S.J. Regulation of glycogen synthesis by amino acids in cultured human muscle cells. J. Biol. Chem. 2001, 276, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Delcommenne, M.; Tan, C.; Gray, V.; Rue, L.; Woodgett, J.; Dedhar, S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 11211–11216. [Google Scholar] [CrossRef]
- Hughes, K.; Nikolakaki, E.; Plyte, S.E.; Totty, N.F.; Woodgett, J.R. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993, 12, 803–808. [Google Scholar]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal structure of glycogen synthase kinase 3 beta: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 2001, 105, 721–732. [Google Scholar] [CrossRef]
- Bhat, R.V.; Shanley, J.; Correll, M.P.; Fieles, W.E.; Keith, R.A.; Scott, C.W.; Lee, C.M. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 11074–11079. [Google Scholar] [CrossRef]
- Zhang, F.; Phiel, C.J.; Spece, L.; Gurvich, N.; Klein, P.S. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J. Biol. Chem. 2003, 278, 33067–33077. [Google Scholar] [CrossRef]
- Ivaska, J.; Nissinen, L.; Immonen, N.; Eriksson, J.E.; Kahari, V.M.; Heino, J. Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol. Cell. Biol. 2002, 22, 1352–1359. [Google Scholar] [CrossRef][Green Version]
- Goni-Oliver, P.; Lucas, J.J.; Avila, J.; Hernandez, F. N-terminal cleavage of GSK-3 by calpain: A new form of GSK-3 regulation. J. Biol. Chem. 2007, 282, 22406–22413. [Google Scholar] [CrossRef]
- Kandasamy, A.D.; Schulz, R. Glycogen synthase kinase-3beta is activated by matrix metalloproteinase-2 mediated proteolysis in cardiomyoblasts. Cardiovasc. Res. 2009, 83, 698–706. [Google Scholar] [CrossRef]
- Figueiredo, V.C.; McCarthy, J.J. Regulation of Ribosome Biogenesis in Skeletal Muscle Hypertrophy. Physiology 2019, 34, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, H.W.; Powers, S.K. The Role of Calpains in Skeletal Muscle Remodeling with Exercise and Inactivity-induced Atrophy. Int. J. Sports Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.L.; Proud, C.G. Eukaryotic initiation factor 2B (eIF2B). Int. J. Biochem. Cell Biol. 1997, 29, 1127–1131. [Google Scholar] [CrossRef]
- Welsh, G.I.; Miller, C.M.; Loughlin, A.J.; Price, N.T.; Proud, C.G. Regulation of eukaryotic initiation factor eIF2B: Glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 1998, 421, 125–130. [Google Scholar] [CrossRef]
- Jefferson, L.S.; Fabian, J.R.; Kimball, S.R. Glycogen synthase kinase-3 is the predominant insulin-regulated eukaryotic initiation factor 2B kinase in skeletal muscle. Int. J. Biochem. Cell Biol. 1999, 31, 191–200. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef]
- Kelly, G.M.; Buckley, D.A.; Kiely, P.A.; Adams, D.R.; O’Connor, R. Serine phosphorylation of the insulin-like growth factor I (IGF-1) receptor C-terminal tail restrains kinase activity and cell growth. J. Biol. Chem. 2012, 287, 28180–28194. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Krebs, E.G. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc. Natl. Acad. Sci. USA 1997, 94, 9660–9664. [Google Scholar] [CrossRef] [PubMed]
- Liberman, Z.; Plotkin, B.; Tennenbaum, T.; Eldar-Finkelman, H. Coordinated phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 and protein kinase C betaII in the diabetic fat tissue. Am. J. Physiol. Endocrinol. Metab. 2008, 294, 1169–1177. [Google Scholar] [CrossRef]
- Chaillou, T.; Kirby, T.J.; McCarthy, J.J. Ribosome biogenesis: Emerging evidence for a central role in the regulation of skeletal muscle mass. J. Cell. Physiol. 2014, 229, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.D.; Esser, K.A. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2005, 289, C853–C859. [Google Scholar] [CrossRef]
- Mei, Z.; Zhang, D.; Hu, B.; Wang, J.; Shen, X.; Xiao, W. FBXO32 Targets c-Myc for Proteasomal Degradation and Inhibits c-Myc Activity. J. Biol. Chem. 2015, 290, 16202–16214. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates beta-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, S.; Lang, C.H.; Vary, T.C. Inhibition of glycogen synthase kinase 3[beta] activity with lithium in vitro attenuates sepsis-induced changes in muscle protein turnover. Shock 2011, 35, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Choukroun, G.; Kang, Z.B.; Ranu, H.; Matsui, T.; Rosenzweig, A.; Molkentin, J.D.; Alessandrini, A.; Woodgett, J.; Hajjar, R.; et al. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol. 2000, 151, 117–130. [Google Scholar] [CrossRef]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Evenson, A.R.; Fareed, M.U.; Menconi, M.J.; Mitchell, J.C.; Hasselgren, P.O. GSK-3beta inhibitors reduce protein degradation in muscles from septic rats and in dexamethasone-treated myotubes. Int. J. Biochem. Cell Biol. 2005, 37, 2226–2238. [Google Scholar] [CrossRef]
- Li, B.G.; Hasselgren, P.O.; Fang, C.H. Insulin-like growth factor-I inhibits dexamethasone-induced proteolysis in cultured L6 myotubes through PI3K/Akt/GSK-3beta and PI3K/Akt/mTOR-dependent mechanisms. Int. J. Biochem. Cell Biol. 2005, 37, 2207–2216. [Google Scholar] [CrossRef]
- Verhees, K.J.; Schols, A.M.; Kelders, M.C.; Op den Kamp, C.M.; van der Velden, J.L.; Langen, R.C. Glycogen synthase kinase-3beta is required for the induction of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 2011, 301, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.H.; Li, B.G.; James, J.H.; King, J.K.; Evenson, A.R.; Warden, G.D.; Hasselgren, P.O. Protein breakdown in muscle from burned rats is blocked by insulin-like growth factor i and glycogen synthase kinase-3beta inhibitors. Endocrinology 2005, 146, 3141–3149. [Google Scholar] [CrossRef]
- Aweida, D.; Rudesky, I.; Volodin, A.; Shimko, E.; Cohen, S. GSK3-beta promotes calpain-1-mediated desmin filament depolymerization and myofibril loss in atrophy. J. Cell Biol. 2018, 217, 3698–3714. [Google Scholar] [CrossRef] [PubMed]
- Sharlo, K.; Paramonova, I.; Turtikova, O.; Tyganov, S.; Shenkman, B. Plantar mechanical stimulation prevents calcineurin-NFATc1 inactivation and slow-to-fast fiber type shift in rat soleus muscle under hindlimb unloading. J. Appl. Physiol. 2019, 126, 1769–1781. [Google Scholar] [CrossRef]
- Mirzoev, T.; Tyganov, S.; Vilchinskaya, N.; Lomonosova, Y.; Shenkman, B. Key Markers of mTORC1-Dependent and mTORC1-Independent Signaling Pathways Regulating Protein Synthesis in Rat Soleus Muscle During Early Stages of Hindlimb Unloading. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 39, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Dupont, E.; Cieniewski-Bernard, C.; Bastide, B.; Stevens, L. Electrostimulation during hindlimb unloading modulates PI3K-AKT downstream targets without preventing soleus atrophy and restores slow phenotype through ERK. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, 408–417. [Google Scholar] [CrossRef]
- Lomonosova, Y.N.; Turtikova, O.V.; Shenkman, B.S. Reduced expression of MyHC slow isoform in rat soleus during unloading is accompanied by alterations of endogenous inhibitors of calcineurin/NFAT signaling pathway. J. Muscle Res. Cell Motil. 2016, 37, 7–16. [Google Scholar] [CrossRef]
- Baehr, L.M.; West, D.W.; Marcotte, G.; Marshall, A.G.; De Sousa, L.G.; Baar, K.; Bodine, S.C. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging 2016, 8, 127–146. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Confides, A.L.; Moore-Reed, S.; Hoch, J.M.; Dupont-Versteegden, E.E. Regrowth after skeletal muscle atrophy is impaired in aged rats, despite similar responses in signaling pathways. Exp. Gerontol. 2015, 64, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, J.L.; Langen, R.C.; Kelders, M.C.; Willems, J.; Wouters, E.F.; Janssen-Heininger, Y.M.; Schols, A.M. Myogenic differentiation during regrowth of atrophied skeletal muscle is associated with inactivation of GSK-3beta. Am. J. Physiol. Cell Physiol. 2007, 292, 1636–1644. [Google Scholar] [CrossRef]
- Mirzoev, T.M.; Tyganov, S.A.; Petrova, I.O.; Shenkman, B.S. Signaling pathways of protein synthesis regulation in rat postural muscle during recovery after hindlimb unloading. Aerosp. Environ. Med. 2017, 51, 99–105. [Google Scholar]
- Mirzoev, T.M.; Tyganov, S.A.; Shenkman, B.S. Akt-dependent and Akt-independent pathways are involved in protein synthesis activation during reloading of disused soleus muscle. Muscle Nerve 2017, 55, 393–399. [Google Scholar] [CrossRef]
- Childs, T.E.; Spangenburg, E.E.; Vyas, D.R.; Booth, F.W. Temporal alterations in protein signaling cascades during recovery from muscle atrophy. Am. J. Physiol. Cell Physiol. 2003, 285, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Hilder, T.L.; Tou, J.C.; Grindeland, R.E.; Wade, C.E.; Graves, L.M. Phosphorylation of insulin receptor substrate-1 serine 307 correlates with JNK activity in atrophic skeletal muscle. FEBS Lett. 2003, 553, 63–67. [Google Scholar] [CrossRef]
- Tyganov, S.A.; Mochalova, E.P.; Belova, S.P.; Sharlo, K.A.; Rozhkov, S.V.; Vilchinskaya, N.A.; Paramonova, I.I.; Mirzoev, T.M.; Shenkman, B.S. Effects of Plantar Mechanical Stimulation on Anabolic and Catabolic Signaling in Rat Postural Muscle Under Short-Term Simulated Gravitational Unloading. Front. Physiol. 2019, 10, 1252. [Google Scholar] [CrossRef] [PubMed]
- Mochalova, E.P.; Belova, S.P.; Mirzoev, T.M.; Shenkman, B.S.; Nemirovskaya, T.L. Atrogin-1/MAFbx mRNA expression is regulated by histone deacetylase 1 in rat soleus muscle under hindlimb unloading. Sci. Rep. 2019, 9, 10263. [Google Scholar] [CrossRef] [PubMed]
- Pierno, S.; Desaphy, J.F.; Liantonio, A.; De Luca, A.; Zarrilli, A.; Mastrofrancesco, L.; Procino, G.; Valenti, G.; Conte Camerino, D. Disuse of rat muscle in vivo reduces protein kinase C activity controlling the sarcolemma chloride conductance. J. Physiol. 2007, 584, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Chibalin, A.V.; Benziane, B.; Zakyrjanova, G.F.; Kravtsova, V.V.; Krivoi, I.I. Early endplate remodeling and skeletal muscle signaling events following rat hindlimb suspension. J. Cell. Physiol. 2018, 233, 6329–6336. [Google Scholar] [CrossRef]
- Lomonosova, Y.N.; Kalamkarov, G.R.; Bugrova, A.E.; Shevchenko, T.F.; Kartashkina, N.L.; Lysenko, E.A.; Shvets, V.I.; Nemirovskaya, T.L. Protective effect of L-Arginine administration on proteins of unloaded m. soleus. Biochem. Biokhimiia 2011, 76, 571–580. [Google Scholar] [CrossRef]
- Sharlo, K.A.; Paramonova, I.I.; Lvova, I.D.; Mochalova, E.P.; Kalashnikov, V.E.; Vilchinskaya, N.A.; Tyganov, S.A.; Konstantinova, T.S.; Shevchenko, T.F.; Kalamkarov, G.R.; et al. Plantar Mechanical Stimulation Maintains Slow Myosin Expression in Disused Rat Soleus Muscle via NO-Dependent Signaling. Int. J. Mol. Sci. 2021, 22, 1372. [Google Scholar] [CrossRef]
- Kozlovskaya, I.B.; Sayenko, I.V.; Sayenko, D.G.; Miller, T.F.; Khusnutdinova, D.R.; Melnik, K.A. Role of support afferentation in control of the tonic muscle activity. Acta Astronaut. 2007, 60, 285–294. [Google Scholar] [CrossRef]
- Pansters, N.A.; Schols, A.M.; Verhees, K.J.; de Theije, C.C.; Snepvangers, F.J.; Kelders, M.C.; Ubags, N.D.; Haegens, A.; Langen, R.C. Muscle-specific GSK-3beta ablation accelerates regeneration of disuse-atrophied skeletal muscle. Biochim. Biophys. Acta 2015, 1852, 490–506. [Google Scholar] [CrossRef] [PubMed]
- Urso, M.L.; Scrimgeour, A.G.; Chen, Y.W.; Thompson, P.D.; Clarkson, P.M. Analysis of human skeletal muscle after 48 h immobilization reveals alterations in mRNA and protein for extracellular matrix components. J. Appl. Physiol. 2006, 101, 1136–1148. [Google Scholar] [CrossRef]
- Glover, E.I.; Phillips, S.M.; Oates, B.R.; Tang, J.E.; Tarnopolsky, M.A.; Selby, A.; Smith, K.; Rennie, M.J. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J. Physiol. 2008, 586, 6049–6061. [Google Scholar] [CrossRef]
- Theeuwes, W.F.; Pansters, N.A.M.; Gosker, H.R.; Schols, A.; Verhees, K.J.P.; de Theije, C.C.; van Gorp, R.H.P.; Kelders, M.; Ronda, O.; Haegens, A.; et al. Recovery of muscle mass and muscle oxidative phenotype following disuse does not require GSK-3 inactivation. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165740. [Google Scholar] [CrossRef]
- Wroblewski, R.; Jansson, E. Fine structure of single fibres of human skeletal muscle. Cell Tissue Res. 1975, 161, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.; Stevens, J.; Binder-Macleod, S.A. Human skeletal muscle fiber type classifications. Phys. Ther. 2001, 81, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Myosin isoforms in mammalian skeletal muscle. J. Appl. Physiol. 1994, 77, 493–501. [Google Scholar] [CrossRef]
- Hamalainen, N.; Pette, D. The histochemical profiles of fast fiber types IIB, IID, and IIA in skeletal muscles of mouse, rat, and rabbit. J. Histochem. Cytochem. 1993, 41, 733–743. [Google Scholar] [CrossRef]
- Herbison, G.J.; Jaweed, M.M.; Ditunno, J.F. Muscle fiber types. Arch. Phys. Med. Rehabil. 1982, 63, 227–230. [Google Scholar] [PubMed]
- Medler, S. Mixing it up: The biological significance of hybrid skeletal muscle fibers. J. Exp. Biol. 2019, 222. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Chen, X.; Chen, D.; Yu, B.; Li, M.; He, J.; Huang, Z. MicroRNA-499-5p regulates skeletal myofiber specification via NFATc1/MEF2C pathway and Thrap1/MEF2C axis. Life Sci. 2018, 215, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, X.; Zhou, D.; Lai, L.; Xiao, L.; Liu, L.; Fu, T.; Kong, Y.; Zhou, Q.; Vega, R.B.; et al. Coupling of mitochondrial function and skeletal muscle fiber type by a miR-499/Fnip1/AMPK circuit. EMBO Mol. Med. 2016, 8, 1212–1228. [Google Scholar] [CrossRef]
- Hagiwara, N.; Ma, B.; Ly, A. Slow and fast fiber isoform gene expression is systematically altered in skeletal muscle of the Sox6 mutant, p100H. Dev. Dyn. 2005, 234, 301–311. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J. The MyomiR network in skeletal muscle plasticity. Exerc. Sport Sci. Rev. 2011, 39, 150–154. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Esser, K.A.; Peterson, C.A.; Dupont-Versteegden, E.E. Evidence of MyomiR network regulation of beta-myosin heavy chain gene expression during skeletal muscle atrophy. Physiol. Genomics 2009, 39, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Alford, E.K.; Roy, R.R.; Hodgson, J.A.; Edgerton, V.R. Electromyography of rat soleus, medial gastrocnemius, and tibialis anterior during hind limb suspension. Exp. Neurol. 1987, 96, 635–649. [Google Scholar] [CrossRef]
- Desplanches, D.; Mayet, M.H.; Ilyina-Kakueva, E.I.; Frutoso, J.; Flandrois, R. Structural and metabolic properties of rat muscle exposed to weightlessness aboard Cosmos 1887. Eur. J. Appl. Physiol. Occup. Physiol. 1991, 63, 288–292. [Google Scholar] [CrossRef]
- Desplanches, D.; Mayet, M.H.; Sempore, B.; Flandrois, R. Structural and functional responses to prolonged hindlimb suspension in rat muscle. J. Appl. Physiol. 1987, 63, 558–563. [Google Scholar] [CrossRef]
- Fitts, R.H.; Trappe, S.W.; Costill, D.L.; Gallagher, P.M.; Creer, A.C.; Colloton, P.A.; Peters, J.R.; Romatowski, J.G.; Bain, J.L.; Riley, D.A. Prolonged space flight-induced alterations in the structure and function of human skeletal muscle fibres. J. Physiol. 2010, 588, 3567–3592. [Google Scholar] [CrossRef]
- Martin, T.P.; Edgerton, V.R.; Grindeland, R.E. Influence of spaceflight on rat skeletal muscle. J. Appl. Physiol. 1988, 65, 2318–2325. [Google Scholar] [CrossRef]
- Shenkman, B.S.; Nemirovskaya, T.L. Calcium-dependent signaling mechanisms and soleus fiber remodeling under gravitational unloading. J. Muscle Res. Cell Motil. 2008, 29, 221–230. [Google Scholar] [CrossRef]
- Templeton, G.H.; Sweeney, H.L.; Timson, B.F.; Padalino, M.; Dudenhoeffer, G.A. Changes in fiber composition of soleus muscle during rat hindlimb suspension. J. Appl. Physiol. 1988, 65, 1191–1195. [Google Scholar] [CrossRef]
- De-Doncker, L.; Picquet, F.; Falempin, M. Effects of cutaneous receptor stimulation on muscular atrophy developed in hindlimb unloading condition. J. Appl. Physiol. 2000, 89, 2344–2351. [Google Scholar] [CrossRef]
- Gazenko, O.G.; Grigoriev, A.I.; Kozlovskaya, I.B. Mechanisms of acute and chronic effects of microgravity. Physiologist 1987, 30, 1–5. [Google Scholar]
- Giger, J.M.; Bodell, P.W.; Zeng, M.; Baldwin, K.M.; Haddad, F. Rapid muscle atrophy response to unloading: Pretranslational processes involving MHC and actin. J. Appl. Physiol. 2009, 107, 1204–1212. [Google Scholar] [CrossRef]
- Vilchinskaya, N.A.; Mochalova, E.P.; Nemirovskaya, T.L.; Mirzoev, T.M.; Turtikova, O.V.; Shenkman, B.S. Rapid decline in MyHC I(beta) mRNA expression in rat soleus during hindlimb unloading is associated with AMPK dephosphorylation. J. Physiol. 2017, 595, 7123–7134. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.; Gohlsch, B.; Mounier, Y.; Pette, D. Changes in myosin heavy chain mRNA and protein isoforms in single fibers of unloaded rat soleus muscle. FEBS Lett. 1999, 463, 15–18. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, X.; Fan, M. Signaling mechanisms involved in disuse muscle atrophy. Med. Hypotheses 2007, 69, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Shenkman, B.S. From Slow to Fast: Hypogravity-Induced Remodeling of Muscle Fiber Myosin Phenotype. Acta Naturae 2016, 8, 47–59. [Google Scholar] [CrossRef]
- Grigor’ev, A.I.; Kozlovskaia, I.B.; Shenkman, B.S. The role of support afferents in organisation of the tonic muscle system. Rossiiskii Fiziologicheskii Zhurnal Imeni IM Sechenova 2004, 90, 508–521. [Google Scholar]
- Standley, R.A.; Distefano, G.; Trevino, M.B.; Chen, E.; Narain, N.R.; Greenwood, B.; Kondakci, G.; Tolstikov, V.V.; Kiebish, M.A.; Yu, G.; et al. Skeletal Muscle Energetics and Mitochondrial Function Are Impaired Following 10 Days of Bed Rest in Older Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1744–1753. [Google Scholar] [CrossRef]
- Shenkman, B.S.; Nemirovskaya, T.L.; Belozerova, I.N.; Mazin, M.G.; Matveeva, O.A. Mitochondrial adaptations in skeletal muscle cells in mammals exposed to gravitational unloading. J. Gravit. Physiol. J. Int. Soc. Gravit. Physiol. 2002, 9, 159–162. [Google Scholar]
- Buso, A.; Comelli, M.; Picco, R.; Isola, M.; Magnesa, B.; Pisot, R.; Rittweger, J.; Salvadego, D.; Simunic, B.; Grassi, B.; et al. Mitochondrial Adaptations in Elderly and Young Men Skeletal Muscle Following 2 Weeks of Bed Rest and Rehabilitation. Front Physiol. 2019, 10, 474. [Google Scholar] [CrossRef]
- Knoll, R.; Linke, W.A.; Zou, P.; Miocic, S.; Kostin, S.; Buyandelger, B.; Ku, C.H.; Neef, S.; Bug, M.; Schafer, K.; et al. Telethonin deficiency is associated with maladaptation to biomechanical stress in the mammalian heart. Circ. Res. 2011, 109, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.; Shaw, K.T.; Carew, J.; Viola, J.P.; Luo, C.; Perrino, B.A.; Rao, A. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J. Biol. Chem. 1996, 271, 10884–10891. [Google Scholar] [CrossRef]
- Chin, E.R.; Olson, E.N.; Richardson, J.A.; Yang, Q.; Humphries, C.; Shelton, J.M.; Wu, H.; Zhu, W.; Bassel-Duby, R.; Williams, R.S. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes. Dev. 1998, 12, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.D.; Umeda, P.K.; Chang, K.C.; Gros, G.; Scheibe, R.J. Activation of the beta myosin heavy chain promoter by MEF-2D, MyoD, p300, and the calcineurin/NFATc1 pathway. J. Cell Physiol. 2007, 211, 138–148. [Google Scholar] [CrossRef]
- Wu, H.; Rothermel, B.; Kanatous, S.; Rosenberg, P.; Naya, F.J.; Shelton, J.M.; Hutcheson, K.A.; DiMaio, J.M.; Olson, E.N.; Bassel-Duby, R.; et al. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J. 2001, 20, 6414–6423. [Google Scholar] [CrossRef]
- Beals, C.R.; Sheridan, C.M.; Turck, C.W.; Gardner, P.; Crabtree, G.R. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 1997, 275, 1930–1934. [Google Scholar] [CrossRef]
- Shen, T.; Cseresnyes, Z.; Liu, Y.; Randall, W.R.; Schneider, M.F. Regulation of the nuclear export of the transcription factor NFATc1 by protein kinases after slow fibre type electrical stimulation of adult mouse skeletal muscle fibres. J. Physiol. 2007, 579, 535–551. [Google Scholar] [CrossRef]
- Martins, K.J.; St-Louis, M.; Murdoch, G.K.; MacLean, I.M.; McDonald, P.; Dixon, W.T.; Putman, C.T.; Michel, R.N. Nitric oxide synthase inhibition prevents activity-induced calcineurin-NFATc1 signalling and fast-to-slow skeletal muscle fibre type conversions. J. Physiol. 2012, 590, 1427–1442. [Google Scholar] [CrossRef]
- Shigueva, T.A.; Zakirova, A.Z.; Tomilovskaya, E.S.; Kozlovskaya, I.B. Effect of support deprivation on the sequence of motor units recruiting. Aviakosm. Ekolog. Med. 2013, 47, 50–53. [Google Scholar] [PubMed]
- Moukhina, A.; Shenkman, B.; Blottner, D.; Nemirovskaya, T.; Lemesheva, Y.; Puttmann, B.; Kozlovskaya, I. Effects of support stimulation on human soleus fiber characteristics during exposure to “dry” immersion. J. Gravit. Physiol. J. Int. Soc. Gravit. Physiol. 2004, 11, 137–138. [Google Scholar]
- Layne, C.S.; Mulavara, A.P.; Pruett, C.J.; McDonald, P.V.; Kozlovskaya, I.B.; Bloomberg, J.J. The use of in-flight foot pressure as a countermeasure to neuromuscular degradation. Acta Astronaut. 1998, 42, 231–246. [Google Scholar] [CrossRef]
- Sharlo, K.A.; Lomonosova, Y.N.; Turtikova, O.V.; Mitrofanova, O.V.; Kalamkarov, G.R.; Bugrova, A.E.; Shevchenko, T.F.; Shenkman, B.S. The Role of GSK-3β Phosphorylation in the Regulation of Slow Myosin Expression in Soleus Muscle during Functional Unloading. Biochem. Suppl. Ser. Membr. Cell Biol. 2018, 12, 85–91. [Google Scholar] [CrossRef]
- Sharlo, K.A.; Paramonova, I.I.; Lvova, I.D.; Vilchinskaya, N.A.; Bugrova, A.E.; Shevchenko, T.F.; Kalamkarov, G.R.; Shenkman, B.S. NO-Dependent Mechanisms of Myosin Heavy Chain Transcription Regulation in Rat Soleus Muscle After 7-Days Hindlimb Unloading. Front Physiol. 2020, 11, 814. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, H.; DiMario, J.X. Control of slow myosin heavy chain 2 gene expression by glycogen synthase kinase activity in skeletal muscle fibers. Cell Tissue Res. 2006, 323, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, Y.; Liu, X.; Chao, Z.; Wang, Y.; Zhong, T.; Guo, J.; Zhan, S.; Li, L.; Zhang, H. Glycogen synthase kinase 3beta (GSK3beta) regulates the expression of MyHC2a in goat skeletal muscle satellite cells (SMSCs). Anim. Sci. J. 2019. [Google Scholar] [CrossRef]
- Theeuwes, W.F.; Gosker, H.R.; Langen, R.C.J.; Pansters, N.A.M.; Schols, A.; Remels, A.H.V. Inactivation of glycogen synthase kinase 3beta (GSK-3beta) enhances mitochondrial biogenesis during myogenesis. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 2913–2926. [Google Scholar] [CrossRef] [PubMed]
- Theeuwes, W.F.; Gosker, H.R.; Langen, R.C.J.; Verhees, K.J.P.; Pansters, N.A.M.; Schols, A.; Remels, A.H.V. Inactivation of glycogen synthase kinase-3beta (GSK-3beta) enhances skeletal muscle oxidative metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J., Jr.; Olson, E.N. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Tsika, G.L.; Rindt, H.; Schreiber, K.L.; McCarthy, J.J.; Kelm, R.J., Jr.; Tsika, R. Puralpha and Purbeta collaborate with Sp3 to negatively regulate beta-myosin heavy chain gene expression during skeletal muscle inactivity. Mol. Cell Biol. 2007, 27, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Sharlo, K.A.; Lvova, I.D.; Tyganov, S.A.; Shenkman, B.S. Mechanisms of Slow Myosin Expression Maintainance in Postural Muscle Fibers by Plantar Mechanical Stimulation during Gravitational Unloading. Russ. J. Physiol. 2019, 105, 1561–1570. [Google Scholar]
- Sharlo, K.A.; Mochalova, E.P.; Belova, S.P.; Lvova, I.D.; Nemirovskaya, T.L.; Shenkman, B.S. The role of MAP-kinase p38 in the m. soleus slow myosin mRNA transcription regulation during short-term functional unloading. Arch. Biochem. Biophys. 2020, 695, 108622. [Google Scholar] [CrossRef]
- Theeuwes, W.F.; Gosker, H.R.; Schols, A.; Langen, R.C.J.; Remels, A.H.V. Regulation of PGC-1alpha expression by a GSK-3beta-TFEB signaling axis in skeletal muscle. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118610. [Google Scholar] [CrossRef]
- Dionyssiou, M.G.; Nowacki, N.B.; Hashemi, S.; Zhao, J.; Kerr, A.; Tsushima, R.G.; McDermott, J.C. Cross-talk between glycogen synthase kinase 3beta (GSK3beta) and p38MAPK regulates myocyte enhancer factor 2 (MEF2) activity in skeletal and cardiac muscle. J. Mol. Cell Cardiol. 2013, 54, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef]
- Whitley, K.C.; Hamstra, S.I.; Baranowski, R.W.; Watson, C.J.F.; MacPherson, R.E.K.; MacNeil, A.J.; Roy, B.D.; Vandenboom, R.; Fajardo, V.A. GSK3 inhibition with low dose lithium supplementation augments murine muscle fatigue resistance and specific force production. Physiol. Rep. 2020, 8, 14517. [Google Scholar] [CrossRef]
- Kenny, H.C.; Tascher, G.; Ziemianin, A.; Rudwill, F.; Zahariev, A.; Chery, I.; Gauquelin-Koch, G.; Barielle, M.P.; Heer, M.; Blanc, S.; et al. Effectiveness of Resistive Vibration Exercise and Whey Protein Supplementation Plus Alkaline Salt on the Skeletal Muscle Proteome Following 21 Days of Bed Rest in Healthy Males. J. Proteome. Res. 2020, 19, 3438–3451. [Google Scholar] [CrossRef]
- Trappe, S.; Trappe, T.; Gallagher, P.; Harber, M.; Alkner, B.; Tesch, P. Human single muscle fibre function with 84 day bed-rest and resistance exercise. J. Physiol. 2004, 557, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Borina, E.; Pellegrino, M.A.; D’Antona, G.; Bottinelli, R. Myosin and actin content of human skeletal muscle fibers following 35 days bed rest. Scand. J. Med. Sci. Sports 2010, 20, 65–73. [Google Scholar] [CrossRef]
- Varga, O.E.; Hansen, A.K.; Sandoe, P.; Olsson, I.A. Validating animal models for preclinical research: A scientific and ethical discussion. Altern. Lab. Anim. 2010, 38, 245–248. [Google Scholar] [CrossRef]
- Aitman, T.; Dhillon, P.; Geurts, A.M. A RATional choice for translational research? Dis. Model. Mech. 2016, 9, 1069–1072. [Google Scholar] [CrossRef]
- Animal Models & Translational Medicine: Quality and Reproducibility of Experimental Design|AISAL Symposium. Comp. Med. 2018, 68, 84–94.
- Wei, C.; Stock, L.; Valanejad, L.; Zalewski, Z.A.; Karns, R.; Puymirat, J.; Nelson, D.; Witte, D.; Woodgett, J.; Timchenko, N.A.; et al. Correction of GSK3beta at young age prevents muscle pathology in mice with myotonic dystrophy type 1. FASEB J. 2018, 32, 2073–2085. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weng, W.C.; Stock, L.; Lindquist, D.; Martinez, A.; Gourdon, G.; Timchenko, N.; Snape, M.; Timchenko, L. Correction of Glycogen Synthase Kinase 3beta in Myotonic Dystrophy 1 Reduces the Mutant RNA and Improves Postnatal Survival of DMSXL Mice. Mol. Cell Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhong, Z.; Zheng, Z.; Shi, X.M.; Zhang, W. Inhibition of glycogen synthase kinase-3beta attenuates glucocorticoid-induced suppression of myogenic differentiation in vitro. PLoS ONE 2014, 9, e105528. [Google Scholar] [CrossRef]
- Verhees, K.J.; Pansters, N.A.; Baarsma, H.A.; Remels, A.H.; Haegens, A.; de Theije, C.C.; Schols, A.M.; Gosens, R.; Langen, R.C. Pharmacological inhibition of GSK-3 in a guinea pig model of LPS-induced pulmonary inflammation: II. Effects on skeletal muscle atrophy. Respir. Res. 2013, 14, 117. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unloading Conditions | |||||
|---|---|---|---|---|---|
| Species | Model | Muscle | Time-Point | Changes in GSK-3β | References |
| mouse | HU | soleus, plantaris | 14 days | p-GSK-3β (Ser9) ─ | [56] |
| rat | IM | soleus | 10 days | p-GSK-3β (Ser9) ─ | [59] |
| rat | HU | soleus | 1, 3 days | p-GSK-3β (Ser9) ↓ | [50] |
| rat | HU | soleus | 3, 7 days | p-GSK-3β (Ser9) ↓ | [51,52] |
| rat | HU | soleus | 14 days | p-GSK-3β (Ser9) ↓ | [52,54,55] |
| rat | HU | tibialis anterior | 14 days | p-GSK-3β (Ser9) ─ | [54] |
| rat | HU | soleus | 28 days | p-GSK-3β (Ser9) ↓ | [52] |
| rat | HU | soleus | 38 days | p-GSK-3β (Ser9) ↓ | [60] |
| rat | HU | gastrocnemius | 38 days | p-GSK-3β (Ser9) ─ | [60] |
| human | IM | quadriceps femoris | 2 days | p-GSK-3β (Ser9) ↓ | [69] |
| human | IM | quadriceps femoris | 14 days | p-GSK-3β (Ser9) ─ | [70] |
| Reloading conditions: | |||||
| Species | Model | Muscle | Time-point | Changes in GSK-3β | References |
| mouse | HU | soleus | 3 and 5 days | p-GSK-3β (Ser9) ↑ | [56] |
| mouse | HU | soleus | 7 and 14 days | p-GSK-3β (Ser9) ─ | [56] |
| mouse | HU | plantaris | 3, 5, 7, 14 days | p-GSK-3β (Ser9) ─ | [56] |
| rat | IM | soleus | 6 and 15 days | p-GSK-3β (Ser9) ↑ | [59] |
| rat | HU | soleus | 3 days | p-GSK-3β (Ser9) ↑ | [54,57,58] |
| rat | HU | soleus | 7 and 14 days | p-GSK-3β (Ser9) ─ | [54,57] |
| rat | HU | tibialis anterior | 3, 7,14 days | p-GSK-3β (Ser9) ↑ | [54] |
| rat | HU | soleus | 14 days | p-GSK-3β (Ser9) ─ | [55] |
| rat | HU | soleus | 7 and 14 days | p-GSK-3β (Ser9) ─ | [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirzoev, T.M.; Sharlo, K.A.; Shenkman, B.S. The Role of GSK-3β in the Regulation of Protein Turnover, Myosin Phenotype, and Oxidative Capacity in Skeletal Muscle under Disuse Conditions. Int. J. Mol. Sci. 2021, 22, 5081. https://doi.org/10.3390/ijms22105081
Mirzoev TM, Sharlo KA, Shenkman BS. The Role of GSK-3β in the Regulation of Protein Turnover, Myosin Phenotype, and Oxidative Capacity in Skeletal Muscle under Disuse Conditions. International Journal of Molecular Sciences. 2021; 22(10):5081. https://doi.org/10.3390/ijms22105081
Chicago/Turabian StyleMirzoev, Timur M., Kristina A. Sharlo, and Boris S. Shenkman. 2021. "The Role of GSK-3β in the Regulation of Protein Turnover, Myosin Phenotype, and Oxidative Capacity in Skeletal Muscle under Disuse Conditions" International Journal of Molecular Sciences 22, no. 10: 5081. https://doi.org/10.3390/ijms22105081
APA StyleMirzoev, T. M., Sharlo, K. A., & Shenkman, B. S. (2021). The Role of GSK-3β in the Regulation of Protein Turnover, Myosin Phenotype, and Oxidative Capacity in Skeletal Muscle under Disuse Conditions. International Journal of Molecular Sciences, 22(10), 5081. https://doi.org/10.3390/ijms22105081