
Modified Gold Nanoparticles for Efficient Delivery of Betulinic Acid to Cancer Cell Mitochondria



Abstract
1. Introduction
2. Results
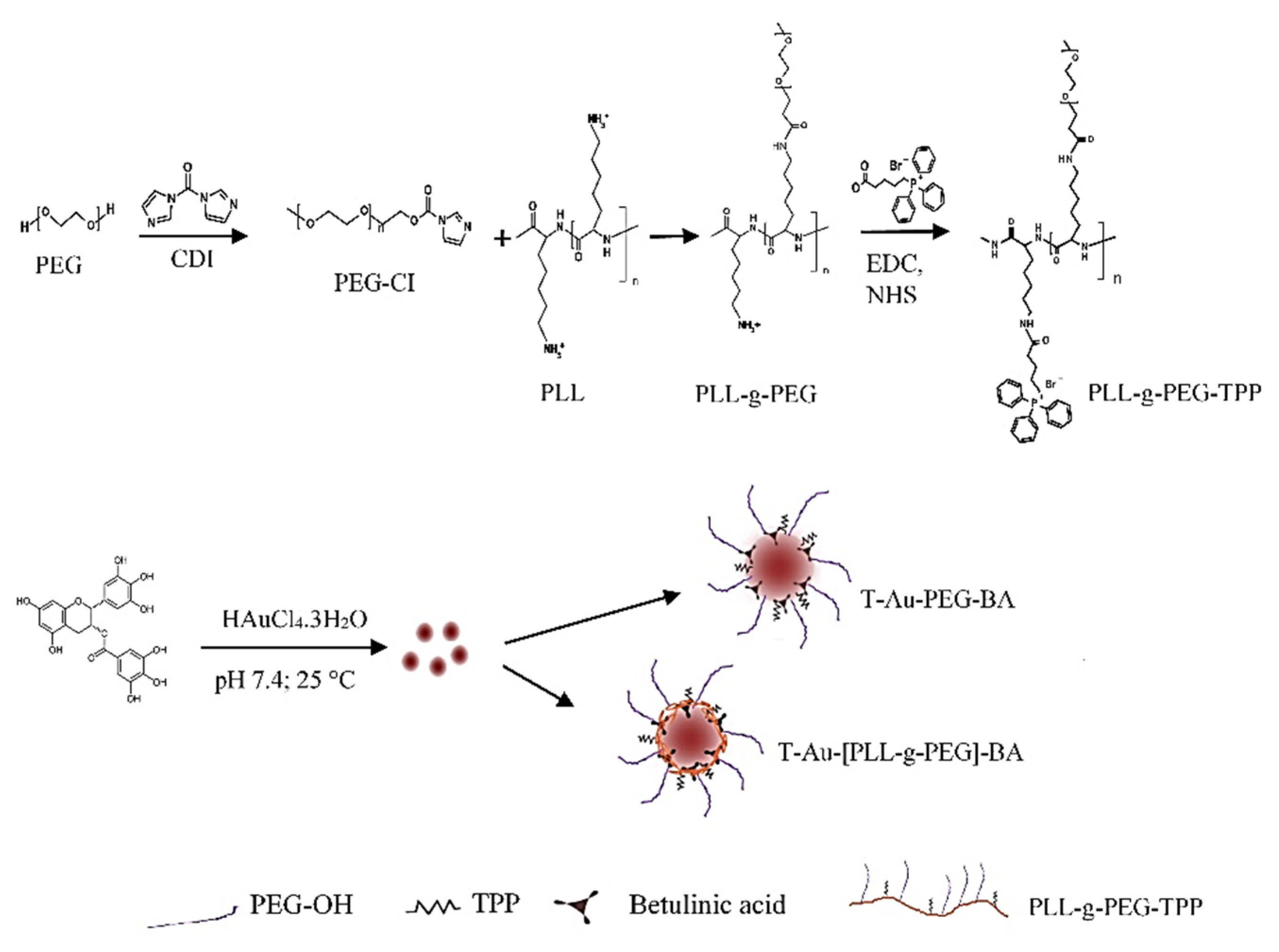
2.1. Synthesis and Functionalization of EGCG-Capped AuNPs
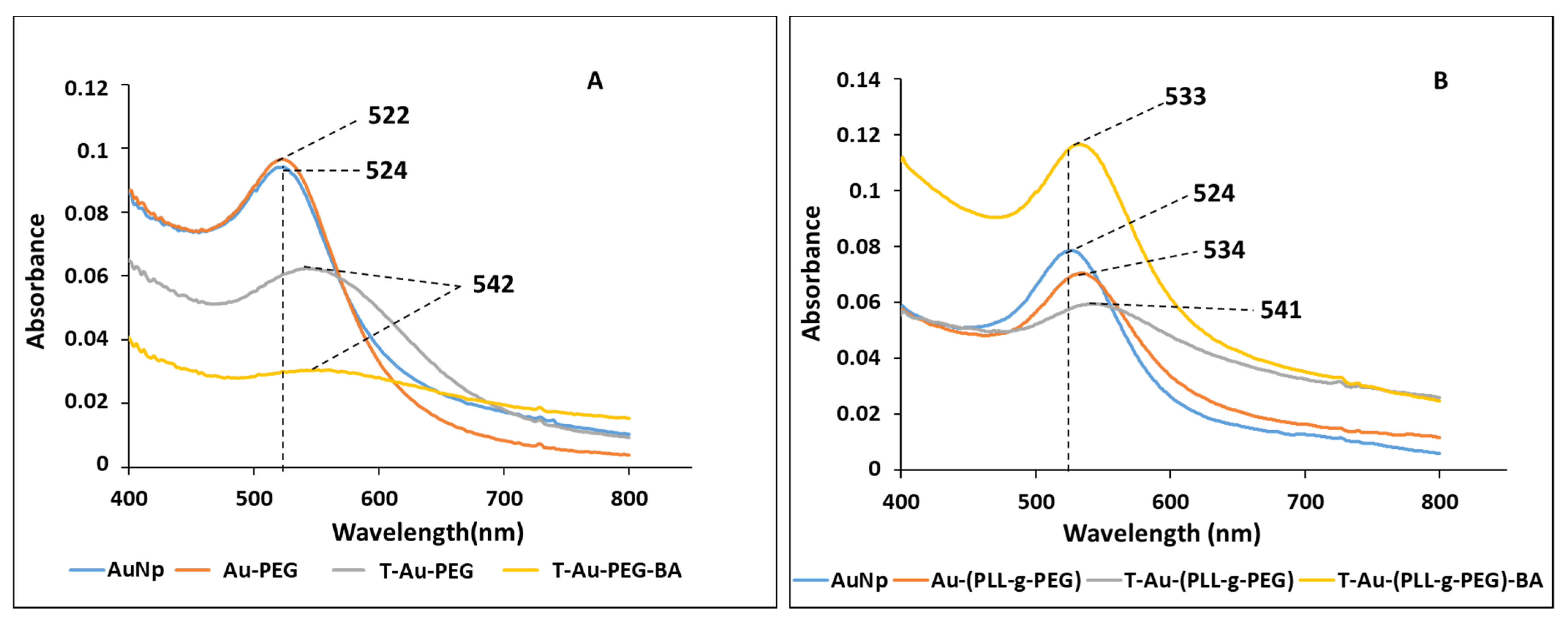
2.2. UV-Vis Spectroscopy
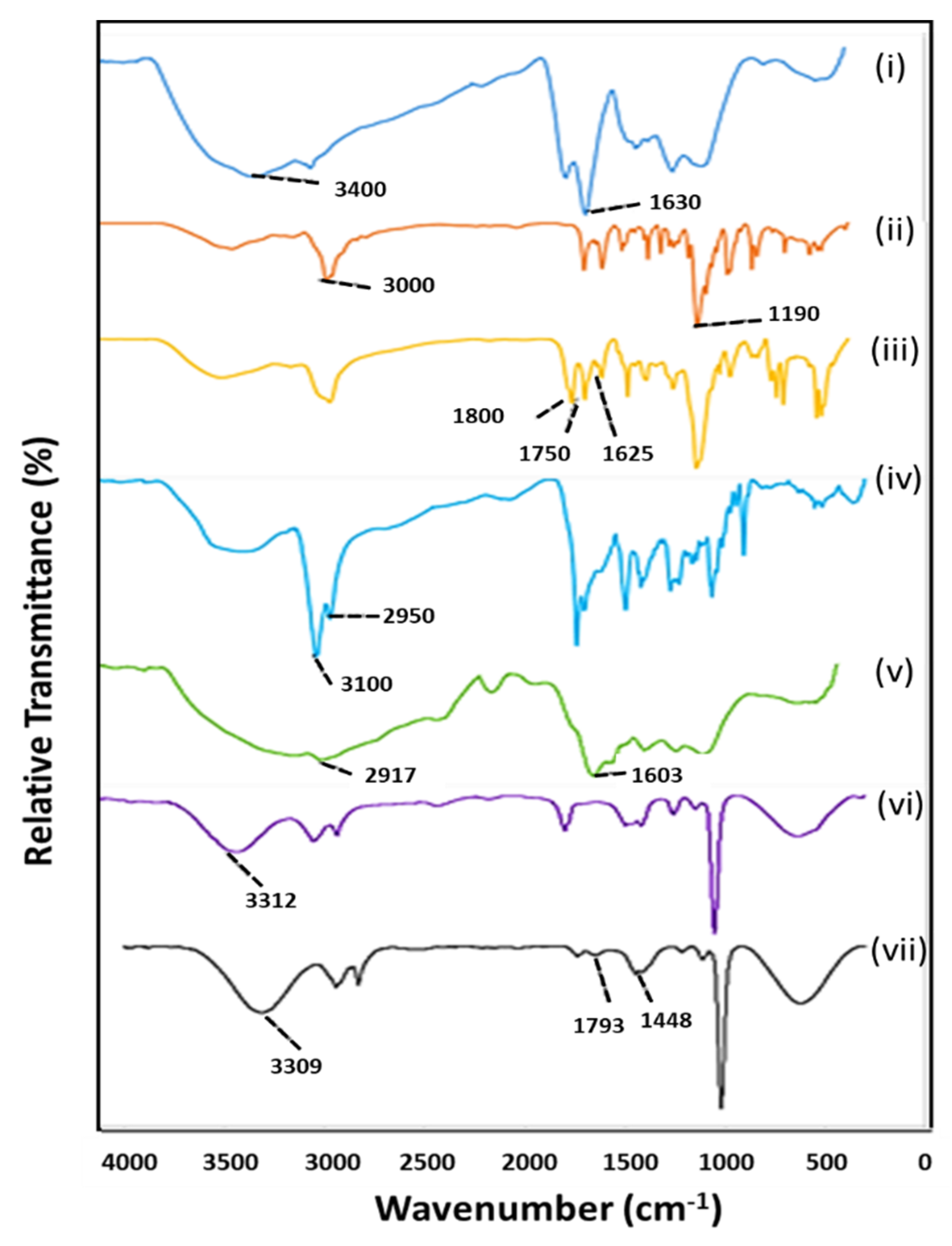
2.3. Fourier-Transform Infrared Spectroscopy (FTIR)
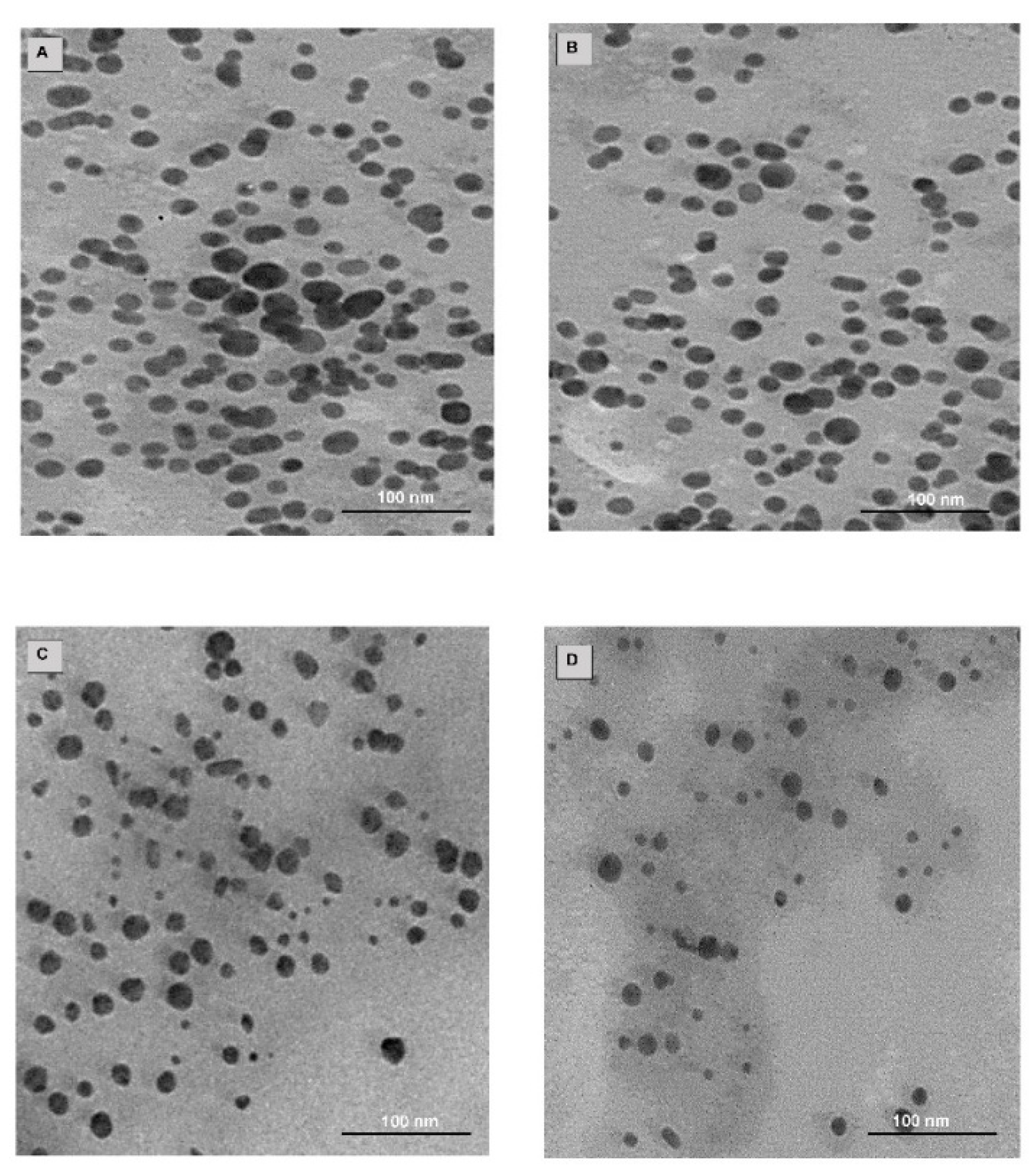
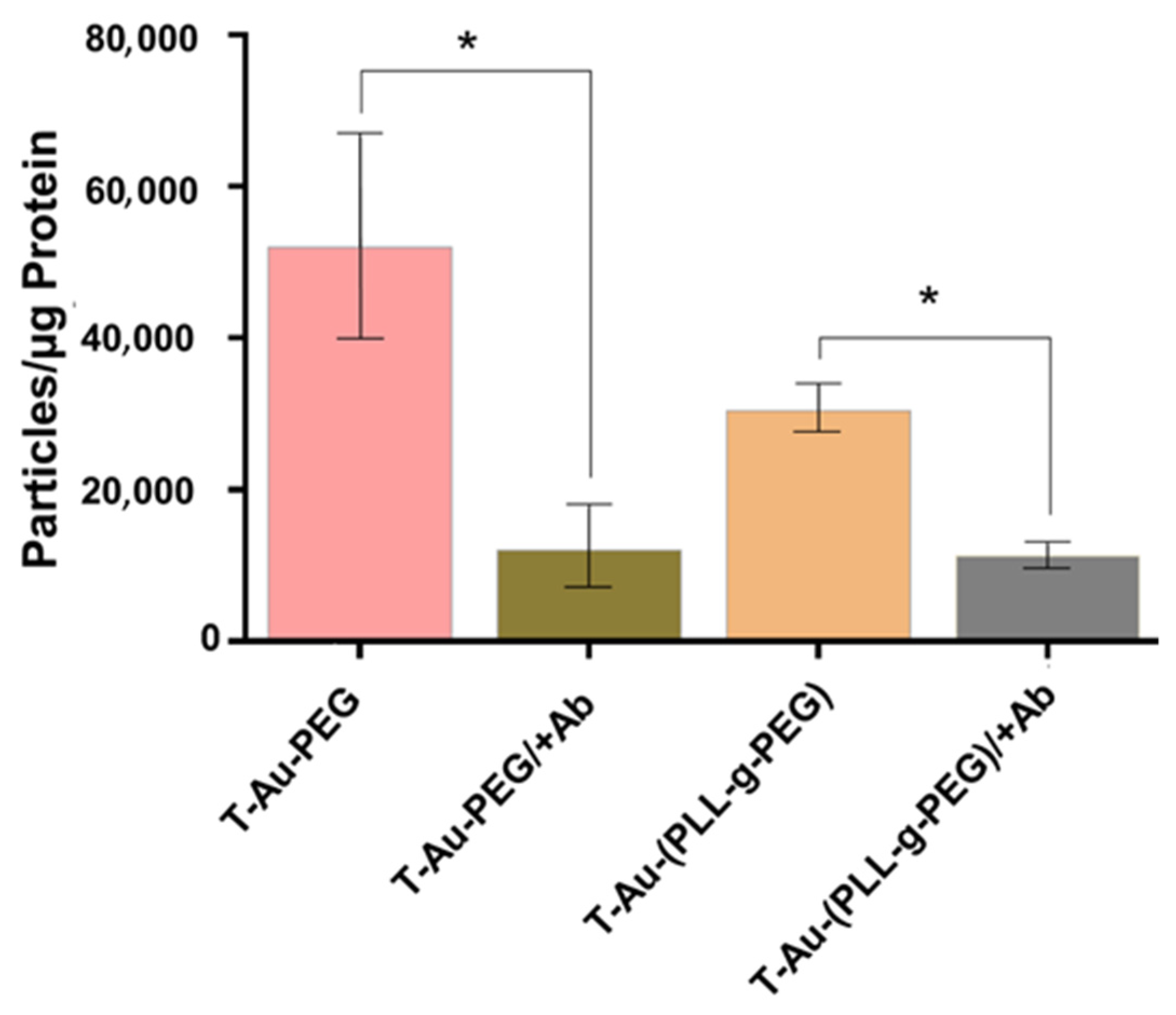
2.4. Drug Loading, TEM and NTA
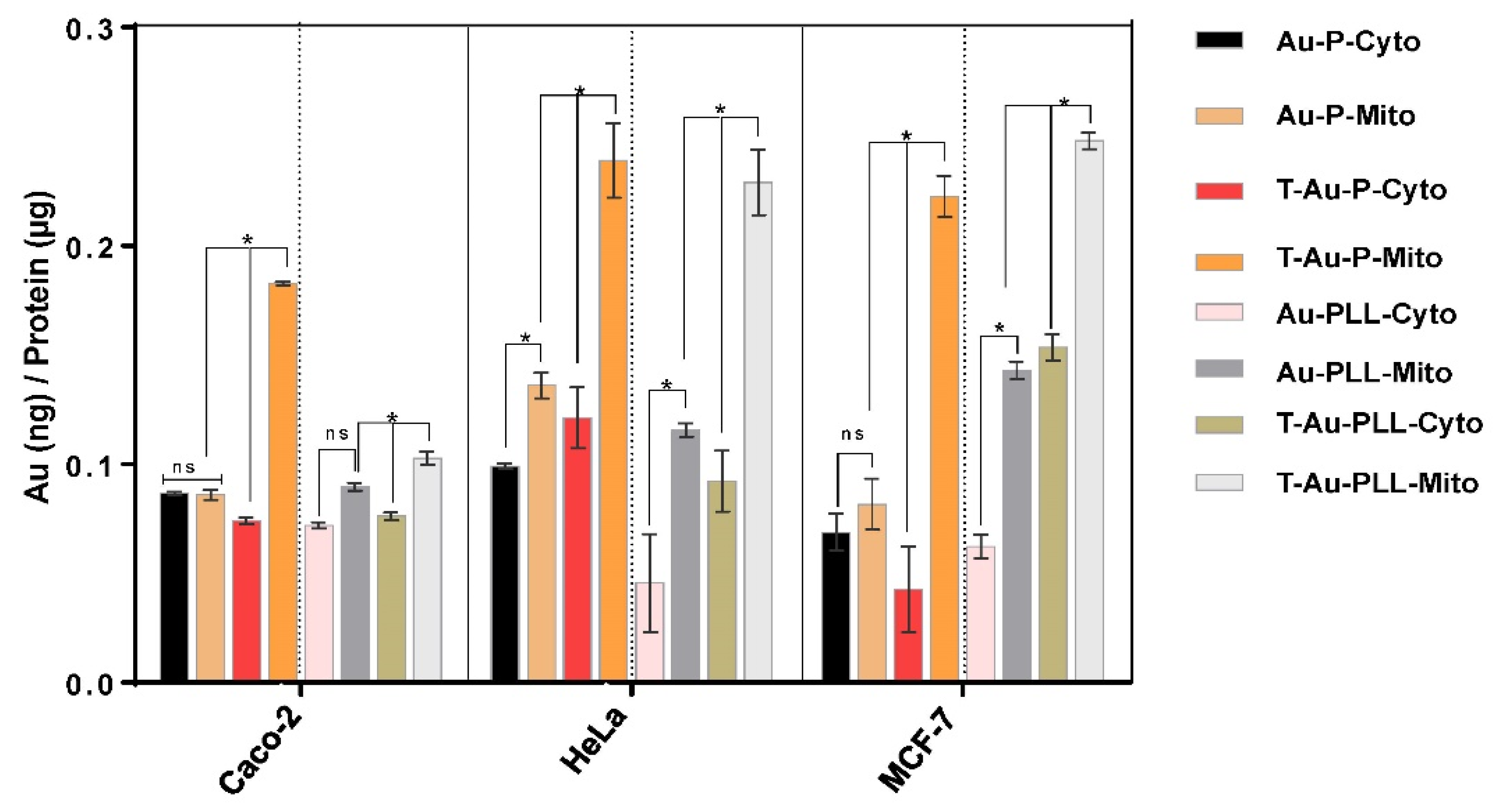
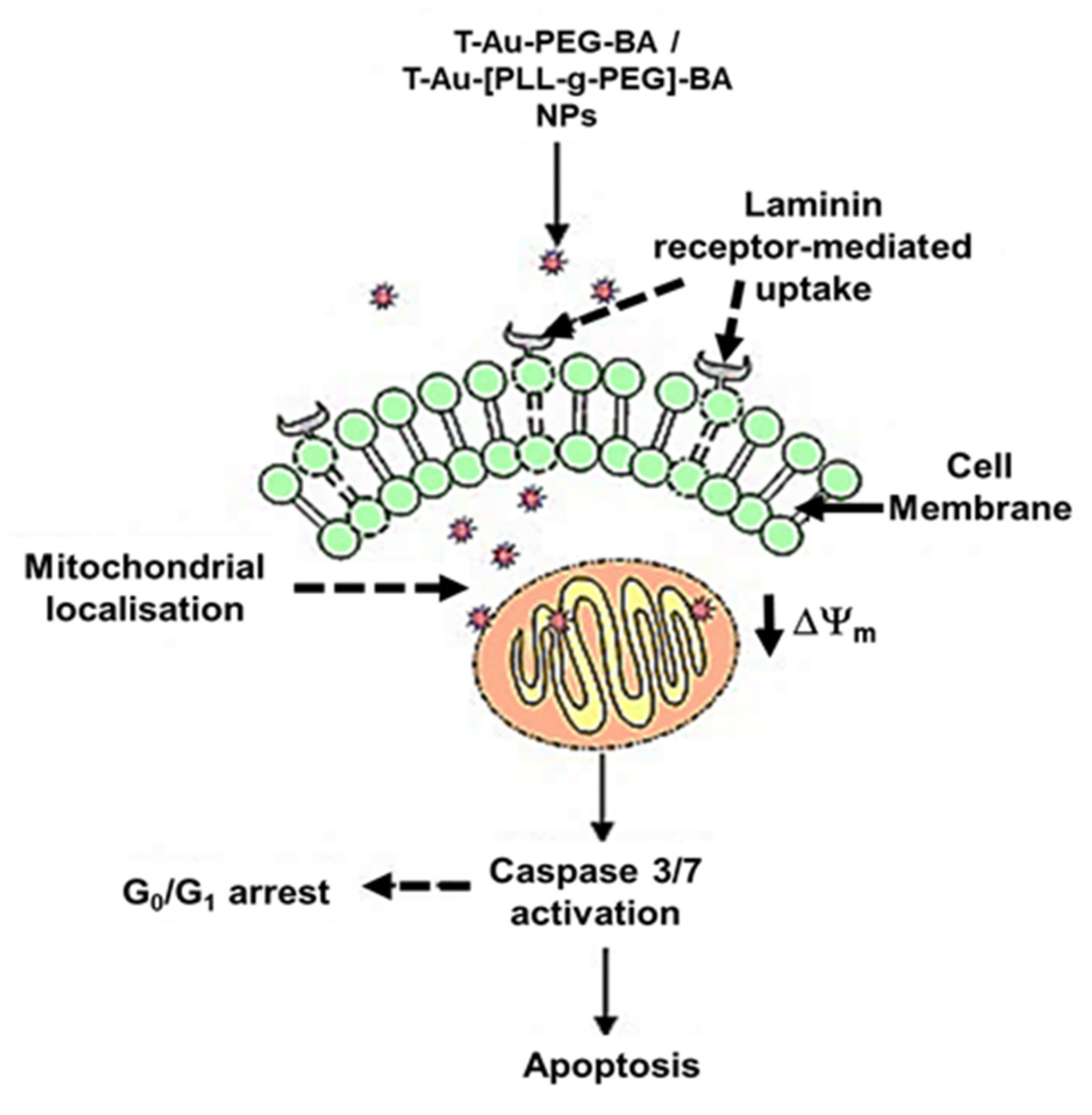
2.5. Cellular Uptake and Mitochondrial Targeting
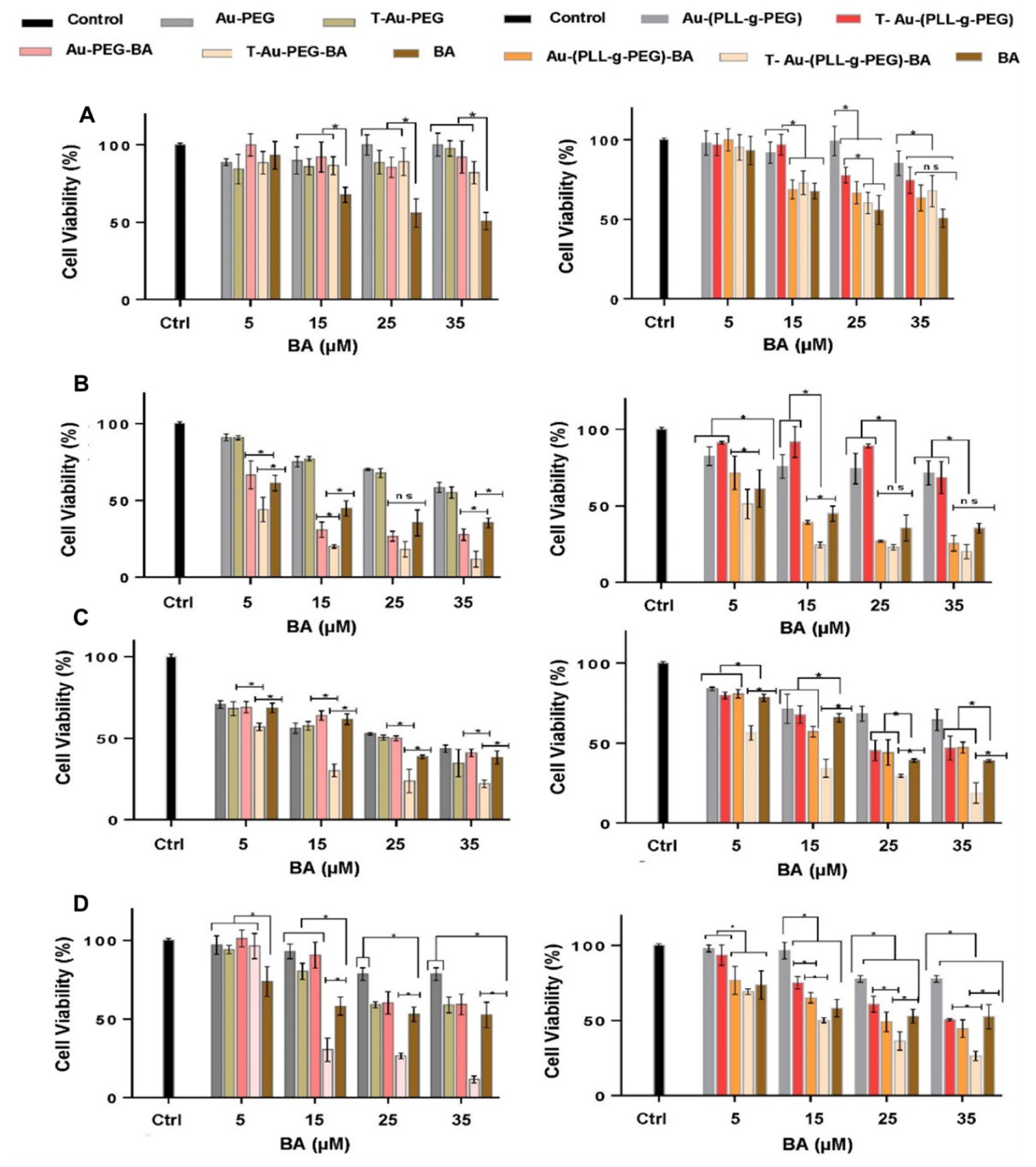
2.6. Cytotoxicity
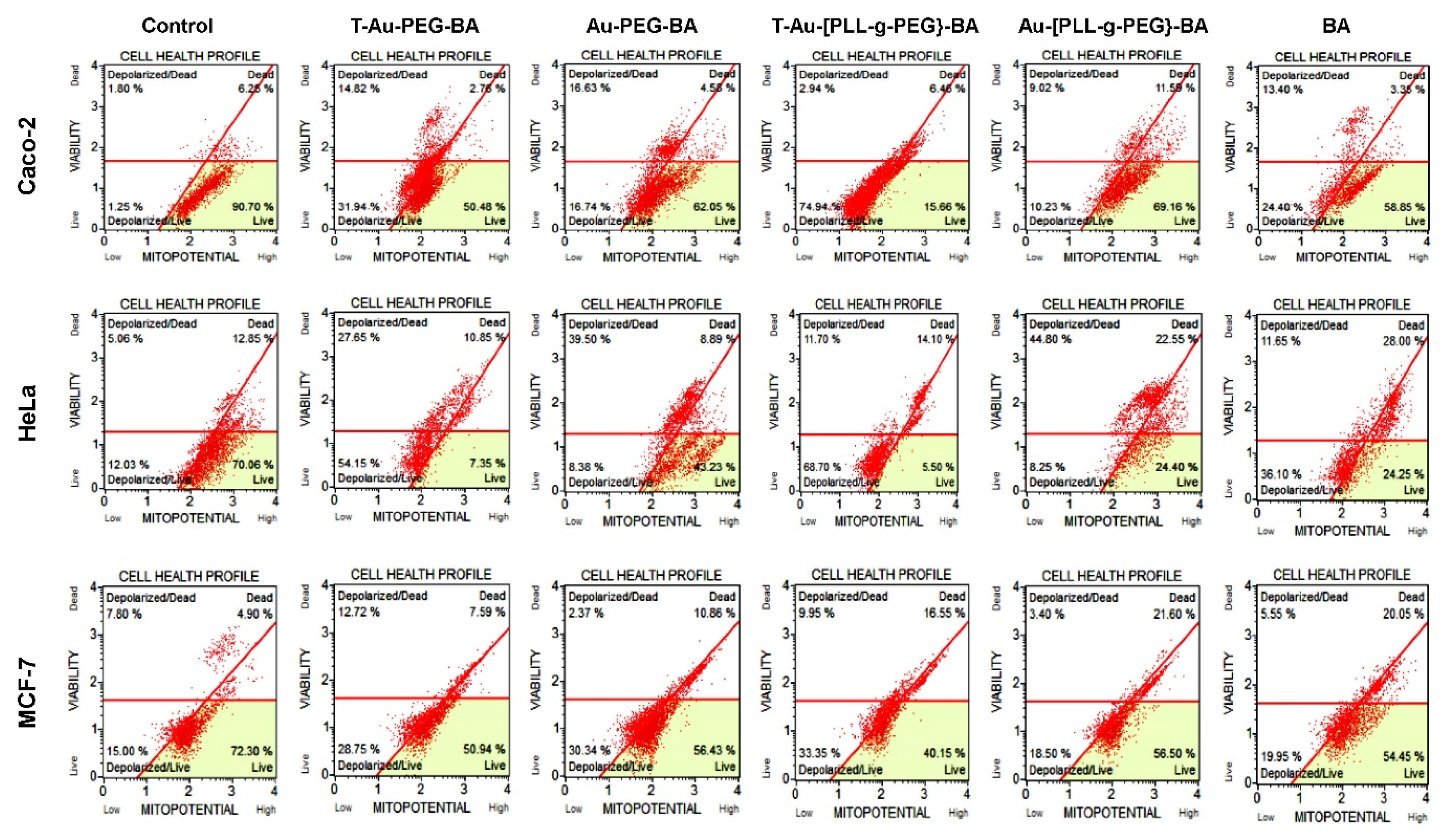
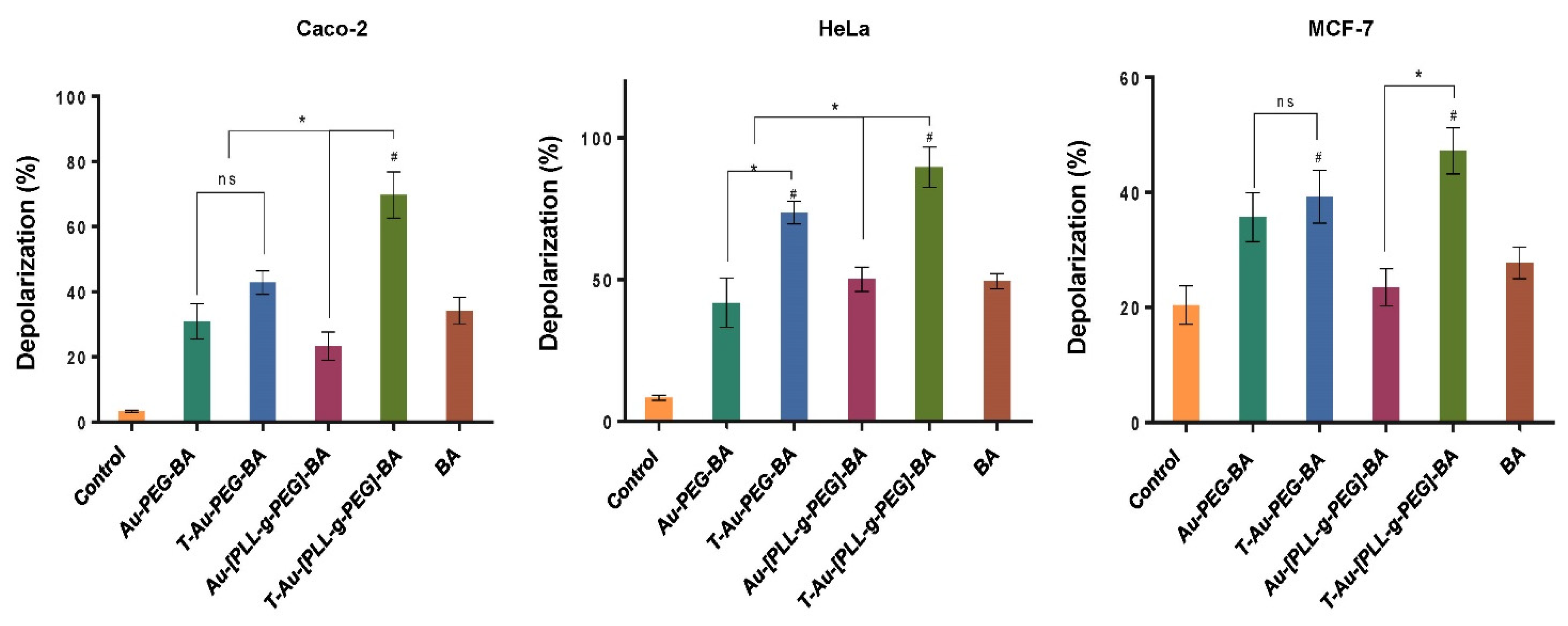
2.7. Effect on Mitochondrial Membrane Potential
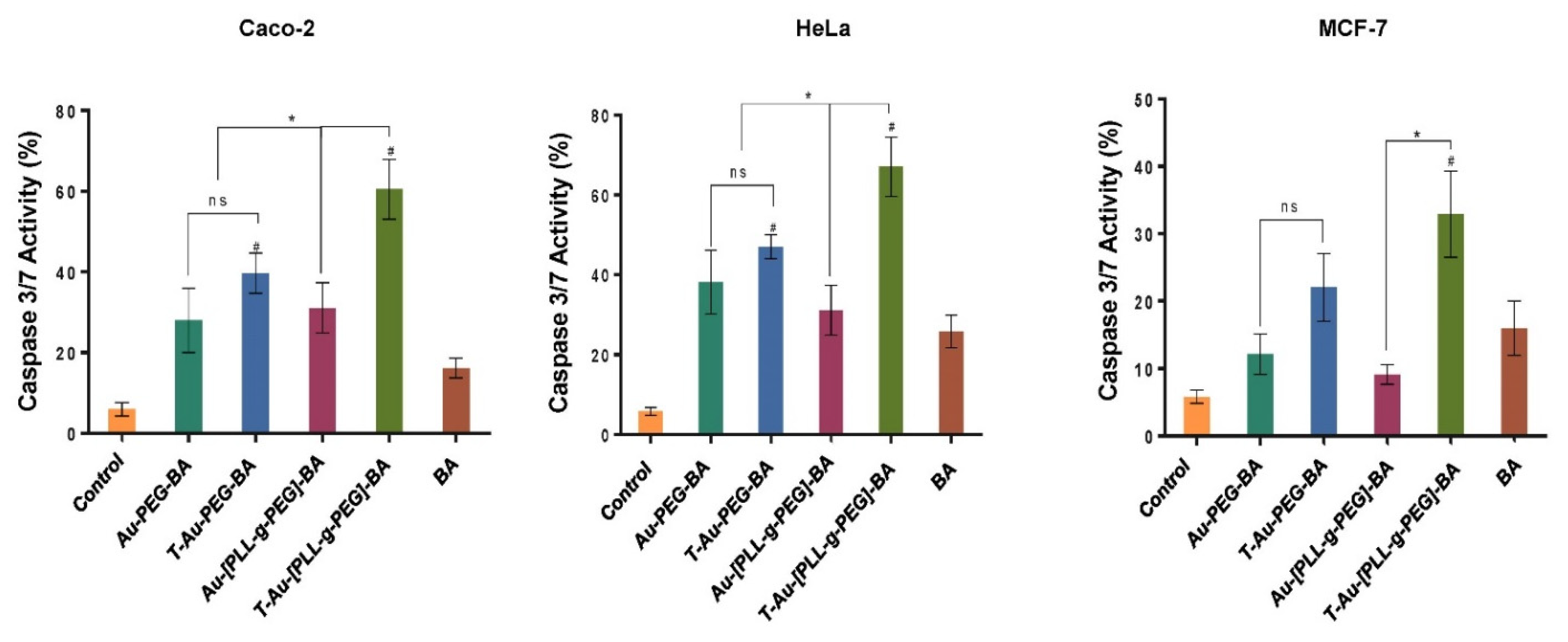
2.8. Impact on Caspases 3 and 7 Activity
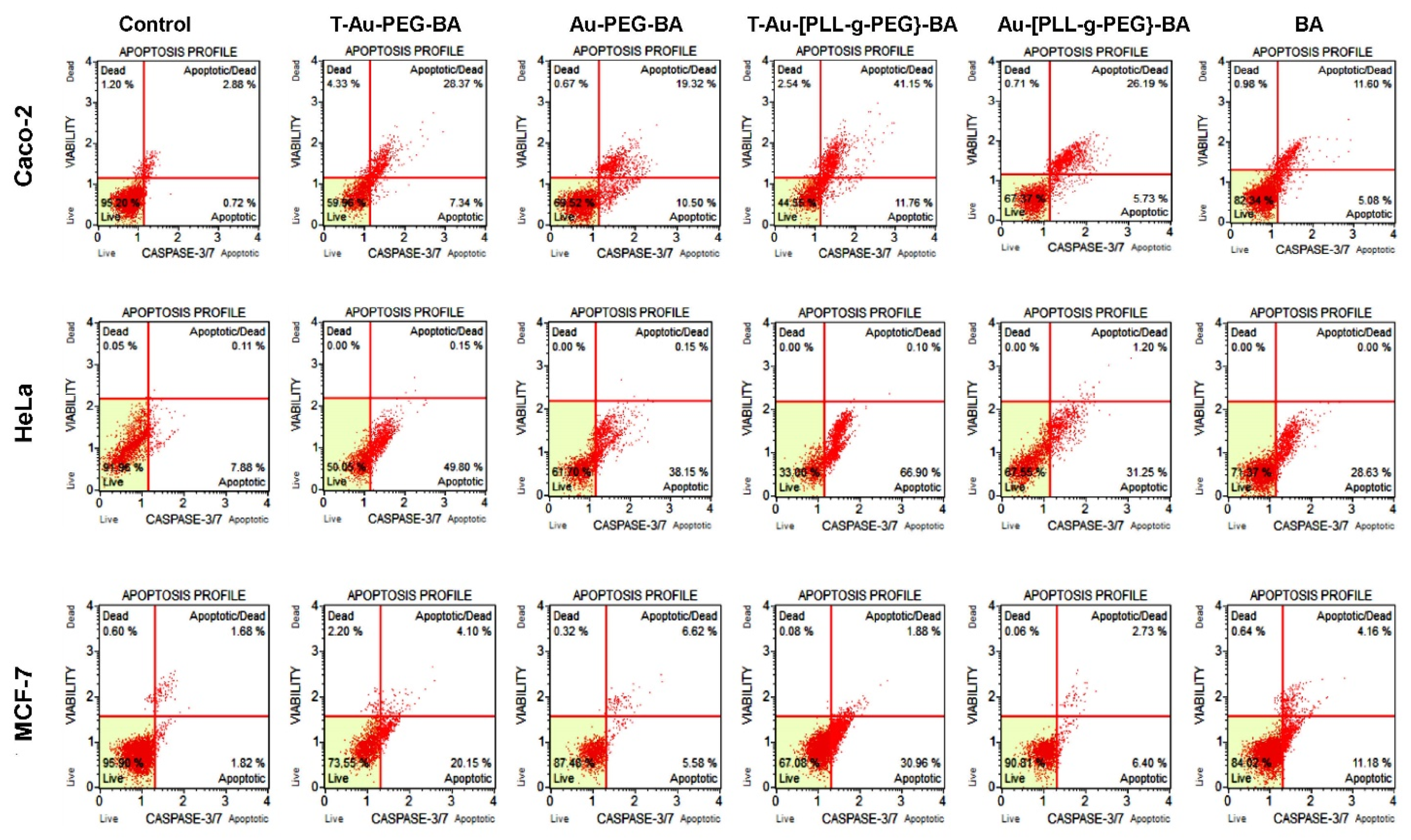
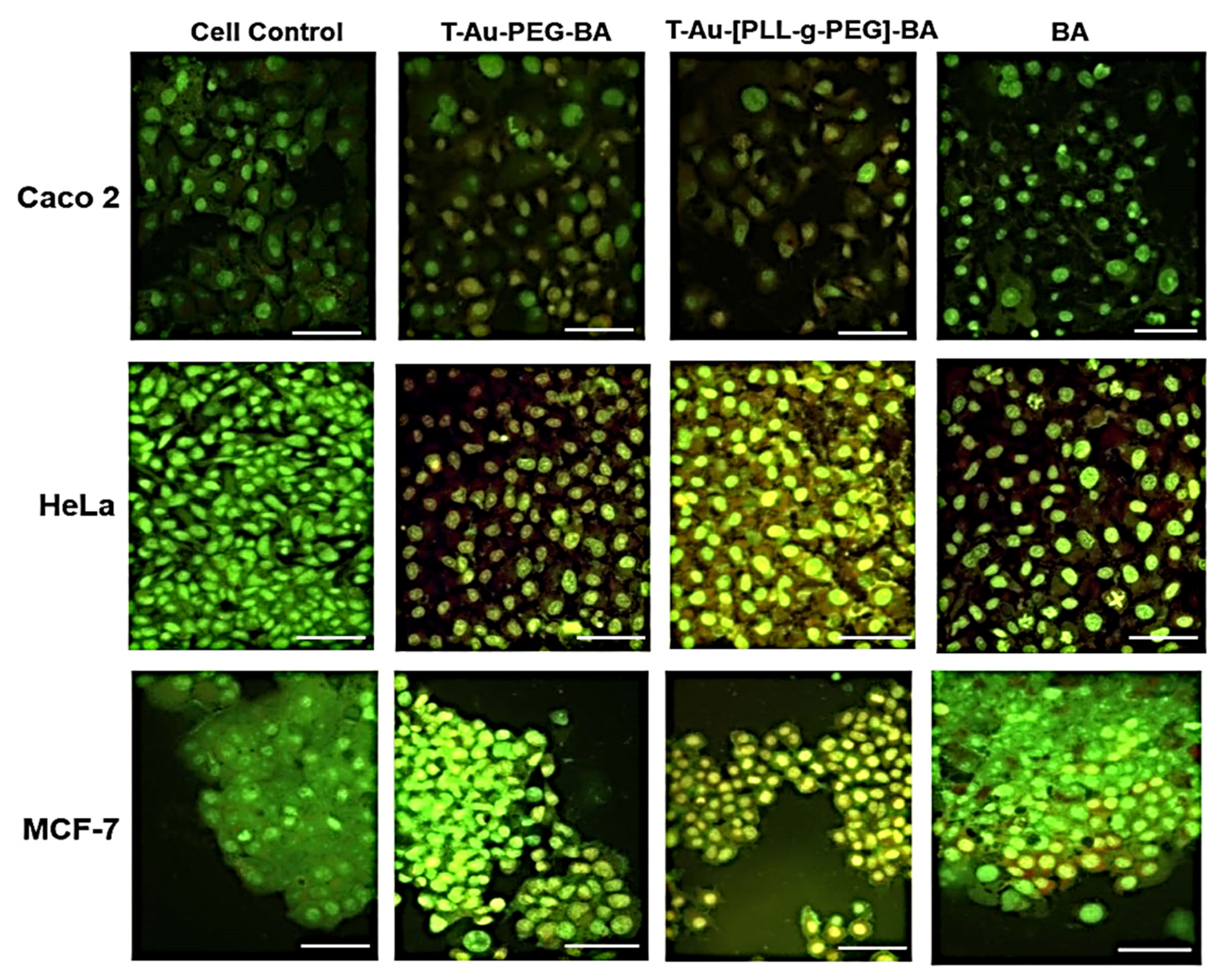
2.9. The Effect of Targeted Nanocomplexes on Apoptosis Induction
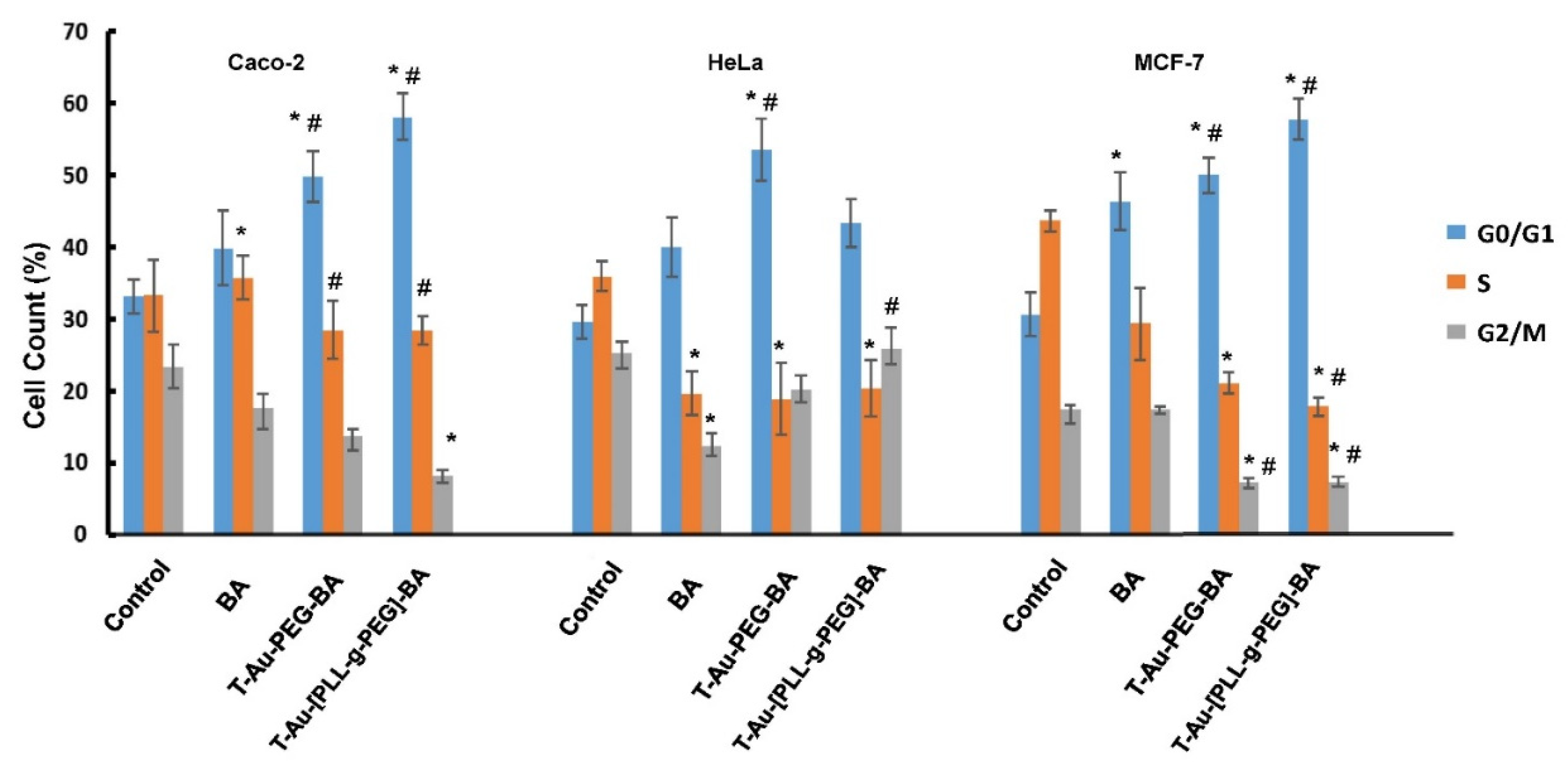
2.10. The Impact of Targeted Nanocomplexes on Cell Cycle Progression
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of EGCG Capped AuNPs
4.3. Synthesis of AuNP-BA
4.4. Synthesis of Poly-L-Lysine-Graft-(g)-Polyethylene Glycol Copolymer (PLL-g-PEG)
4.5. Synthesis of Triphenylphosphine-PLL-g-PEG (T-PLL-g-PEG)
4.6. Synthesis of T-Au-[PLL-g-PEG]-BA), Au-PLL-g-PEG-BA, and T-Au-PEG-BA
4.7. Drug Loading Efficiency
4.8. Nanoparticle Characterization
4.9. Cell Culture
4.10. Cellular Uptake Studies
4.11. Quantitative Determination of Nanoparticle Distribution by ICP-OES Analysis
4.12. MTT Assay
4.13. Cell Cycle Analysis
4.14. Mitopotential Assay
4.15. Caspase 3/7 Analysis
4.16. Apoptosis Assay
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Rais, L.; Masood, A. Sulfur and Nitrogen Co-ordinately Improve Photosynthetic Efficiency, Growth and Proline Accumulation in Two Cultivars of Mustard Under Salt Stress. J. Plant Biochem. Physiol. 2013, 1. [Google Scholar] [CrossRef]
- Maney, V.; Singh, M. The Synergism of Platinum-Gold Bimetallic Nanoconjugates Enhances 5-Fluorouracil Delivery In Vitro. Pharmaceutics 2019, 11, 439. [Google Scholar] [CrossRef]
- Wang, W.; Ning, J.-Y.; Liu, J.-T.; Miao, J.-Y.; Zhao, B.-X. A mitochondria-targeted ratiometric fluorescence sensor for the detection of hypochlorite in living cells. Dye. Pigment. 2019, 171, 107708. [Google Scholar] [CrossRef]
- Tran, S.; DeGiovanni, P.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-G.; Han, Y.-H.; Kankala, R.K.; Wang, S.-B.; Chen, A.-Z. Subcellular Performance of Nanoparticles in Cancer Therapy. Int. J. Nanomed. 2020, 15, 675–704. [Google Scholar] [CrossRef] [PubMed]
- Akinyelu, J.; Singh, M. Nanomedicines for Subcellular Targeting: The Mitochondrial Perspective. Curr. Med. Chem. 2020, 27, 5480–5509. [Google Scholar] [CrossRef]
- Neuzil, J.; Dong, L.-F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef]
- Lee, K.L.; Shukla, S.; Wu, M.; Ayat, N.R.; El Sanadi, C.E.; Wen, A.M.; Edelbrock, J.F.; Pokorski, J.K.; Commandeur, U.; Dubyak, G.R.; et al. Stealth filaments: Polymer chain length and conformation affect the in vivo fate of PEGylated potato virus X. Acta Biomater. 2015, 19, 166–179. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Ostapiuk, A.; Horecka, A.; Kurzepa, J. Betulin and betulinic acid: Triterpenoids derivatives with a powerful biological potential. Phytochem. Rev. 2019, 18, 929–951. [Google Scholar] [CrossRef]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Betulinic Acid for Cancer Treatment and Prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef]
- Chintharlapalli, S.; Papineni, S.; Lei, P.; Pathi, S.; Safe, S. Betulinic acid inhibits colon cancer cell and tumor growth and induces proteasome-dependent and -independent downregulation of specificity proteins (Sp) transcription factors. BMC Cancer 2011, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Ali-Seyed, M.; Jantan, I.; Vijayaraghavan, K.; Bukhari, S.N.A. Betulinic Acid: Recent Advances in Chemical Modifications, Effective Delivery, and Molecular Mechanisms of a Promising Anticancer Therapy. Chem. Biol. Drug Des. 2016, 87, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, J.; Bajzikova, M.; Vondrusová, M.; Stursa, J.; Goodwin, J.; Nguyen, M.; Zobalova, R.; Pesdar, E.A.; Truksa, J.; Tomasetti, M.; et al. Mitochondrial targeting of α-tocopheryl succinate enhances its anti-mesothelioma efficacy. Redox Rep. 2014, 19, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Dahoumane, S.A.; Jeffryes, C.; Mechouet, M.; Agathos, S.N. Biosynthesis of Inorganic Nanoparticles: A Fresh Look at the Control of Shape, Size and Composition. Bioengineers 2017, 4, 14. [Google Scholar] [CrossRef]
- Castronovo, V.; Campo, E.; Brûle, F.A.V.D.; Claysmith, A.P.; Cioce, V.; Liu, F.-T.; Fernandez, P.L.; Sobel, M.E. Inverse Modulation of Steady-State Messenger RNA Levels of Two Non-Integrin Laminin-Binding Proteins in Human Colon Carcinoma. J. Natl. Cancer Inst. 1992, 84, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Basolo, F.; Pollina, L.; Pacini, F.; Fontanini, G.; Ménard, S.; Castronovo, V.; Bevilacqua, G. Expression of the Mr 67,000 laminin receptor is an adverse prognostic indicator in human thyroid cancer: An immunohistochemical study. Clin. Cancer Res. 1996, 2, 1777–1780. [Google Scholar] [PubMed]
- Satoh, K.; Narumi, K.; Abe, T.; Sakai, T.; Kikuchi, T.; Tanaka, M.; Shimo-Oka, T.; Uchida, M.; Tezuka, F.; Isemura, M.; et al. Diminution of 37-kDa laminin binding protein expression reduces tumour formation of murine lung cancer cells. Br. J. Cancer 1999, 80, 1115–1122. [Google Scholar] [CrossRef]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A receptor for green tea polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [CrossRef]
- Shukla, R.; Chanda, N.; Zambre, A.; Upendran, A.; Katti, K.V.; Kulkarni, R.R.; Nune, S.K.; Casteel, S.W.; Smith, C.J.; Vimal, J.; et al. Laminin receptor specific therapeutic gold nanoparticles (198AuNP-EGCg) show efficacy in treating prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 12426–12431. [Google Scholar] [CrossRef] [PubMed]
- Amgoth, C.; Phan, C.; Banavoth, M.; Rompivalasa, S.; Tang, G. Polymer Properties: Functionalization and Surface Modified Nanoparticles. In Role of Novel Drug Delivery Vehicles in Nanobiomedicine; IntechOpen: London, UK, 2020; p. 446. [Google Scholar]
- Yang, D.H.; Kim, H.J.; Park, K.; Kim, J.K.; Chun, H.J. Preparation of poly-l-lysine-based nanoparticles with pH-sensitive release of curcumin for targeted imaging and therapy of liver cancer in vitro and in vivo. Drug Deliv. 2018, 25, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Pan, M.; Zhang, W.; Lin, H.; Wu, S.; Lu, C.; Tang, S.; Liu, D.; Cai, J. Poly(α-l-lysine)-based nanomaterials for versatile biomedical applications: Current advances and perspectives. Bioact. Mater. 2021, 6, 1878–1909. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Singhal, M.; Hirvonen, J. Principles of nanosized drug delivery systems. In Nanoengineered Biomaterials for Advanced Drug Delivery; Elsevier BV: Amsterdam, The Netherlands, 2020; pp. 3–25. [Google Scholar]
- Desai, R.; Mankad, V.; Gupta, S.; Jha, P. Size Distribution of Silver Nanoparticles: UV-Visible Spectroscopic Assessment. Nanosci. Nanotechnol. Lett. 2012, 4, 30–34. [Google Scholar] [CrossRef]
- Mourdikoudis, S.; Pallares, R.M.; Thanh, N.T.K. Characterization techniques for nanoparticles: Comparison and complementarity upon studying nanoparticle properties. Nanoscale 2018, 10, 12871–12934. [Google Scholar] [CrossRef]
- Akinyelu, J.; Singh, M. Folate-tagged chitosan-functionalized gold nanoparticles for enhanced delivery of 5-fluorouracil to cancer cells. Appl. Nanosci. 2018, 9, 7–17. [Google Scholar] [CrossRef]
- Akinyelu, J.; Oladimeji, O.; Singh, M. Lactobionic acid-chitosan functionalised gold-coated poly(lactide-co-glycolide) nanoparticles for hepatocyte targeted gene delivery. Adv. Nat. Sci. Nanosci. Nanotechnol. 2020, 11, 045017. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; John Wiley & Sons Ltd.: Chichester, UK, 2000; pp. 10815–10837. [Google Scholar]
- Cinta-Pinzaru, S.; Dehelean, C.A.; Soica, C.; Culea, M.; Borcan, F. Evaluation and differentiation of the Betulaceae birch bark species and their bioactive triterpene content using analytical FT-vibrational spectroscopy and GC-MS. Chem. Central J. 2012, 6, 67. [Google Scholar] [CrossRef]
- Clayton, K.N.; Salameh, J.W.; Wereley, S.T.; Kinzer-Ursem, T.L. Physical characterization of nanoparticle size and surface modification using particle scattering diffusometry. Biomicrofluidics 2016, 10, 054107. [Google Scholar] [CrossRef]
- Fujimura, Y.; Sumida, M.; Sugihara, K.; Tsukamoto, S.; Yamada, K.; Tachibana, H. Green Tea Polyphenol EGCG Sensing Motif on the 67-kDa Laminin Receptor. PLoS ONE 2012, 7, e37942. [Google Scholar] [CrossRef] [PubMed]
- Moodley, K.; Weiss, S.F.T. Downregulation of the Non-Integrin Laminin Receptor Reduces Cellular Viability by Inducing Apoptosis in Lung and Cervical Cancer Cells. PLoS ONE 2013, 8, e57409. [Google Scholar] [CrossRef]
- Rzeski, W.; Stepulak, A.; Szymański, M.; Juszczak, M.; Grabarska, A.; Sifringer, M.; Kaczor, J.; Kandefer-Szerszeń, M. Betulin Elicits Anti-Cancer Effects in Tumour Primary Cultures and Cell LinesIn Vitro. Basic Clin. Pharmacol. Toxicol. 2009, 105, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jutooru, I.; Lei, P.; Kim, K.; Lee, S.-O.; Brents, L.K.; Prather, P.L.; Safe, S. Betulinic Acid Targets YY1 and ErbB2 through Cannabinoid Receptor-Dependent Disruption of MicroRNA-27a:ZBTB10 in Breast Cancer. Mol. Cancer Ther. 2012, 11, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Boukalova, S.; Rohlenova, K.; Rohlena, J.; Neuzil, J. Mitocans: Mitochondrially Targeted Anti-cancer Drugs. In Mitochondrial Biology and Experimental Therapeutics; Springer: Cham, Swtizerland, 2018; pp. 613–635. [Google Scholar]
- Cai, Y.; Zheng, Y.; Gu, J.; Wang, S.; Wang, N.; Yang, B.; Zhang, F.; Wang, D.; Fu, W.; Wang, Z. Betulinic acid chemosensitizes breast cancer by triggering ER stress-mediated apoptosis by directly targeting GRP78. Cell Death Dis. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, Z.; Yeo, Y. Recent advances in stealth coating of nanoparticle drug delivery systems. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 219–233. [Google Scholar] [CrossRef]
- Oladimeji, O.; Akinyelu, J.; Singh, M. Co-Polymer Functionalised Gold Nanoparticles Show Efficient Mitochondrial Targeted Drug Delivery in Cervical Carcinoma Cells. J. Biomed. Nanotechnol. 2020, 16, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, G.G.; Singh, M. Cationic modified gold nanoparticles show enhanced gene delivery in vitro. Nanotechnol. Rev. 2016, 5, 425–434. [Google Scholar] [CrossRef]
- Barenholz, Y. Liposome application: Problems and prospects. Curr. Opin. Colloid Interface Sci. 2001, 6, 66–77. [Google Scholar] [CrossRef]
- Narainpersad, N.; Singh, M.; Ariatti, M. Novel Neo Glycolipid: Formulation into Pegylated Cationic Liposomes and Targeting of DNA Lipoplexes to the Hepatocyte-Derived Cell Line HepG2. Nucleosides Nucleotides Nucleic Acids 2012, 31, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; El-Sayed, M.A. Gold nanoparticles: Optical properties and implementations in cancer diagnosis and photothermal therapy. J. Adv. Res. 2010, 1, 13–28. [Google Scholar] [CrossRef]
- Kudgus, R.A.; Walden, C.A.; McGovern, R.M.; Reid, J.M.; Robertson, J.D.; Mukherjee, P. Tuning Pharmacokinetics and Biodistribution of a Targeted Drug Delivery System Through Incorporation of a Passive Targeting Component. Sci. Rep. 2014, 4, 5669. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef] [PubMed]
- Marrache, S.; Dhar, S. The energy blocker inside the power house: Mitochondria targeted delivery of 3-bromopyruvate. Chem. Sci. 2014, 6, 1832–1845. [Google Scholar] [CrossRef] [PubMed]
- Greish, K.; Mathur, A.; Bakhiet, M.; Taurin, S. Nanomedicine: Is it lost in translation? Ther. Deliv. 2018, 9, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Marrache, S.; Dhar, S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc. Natl. Acad. Sci. USA 2012, 109, 16288–16293. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Nienhaus, K.; Nienhaus, G.U. Engineered nanoparticles interacting with cells: Size matters. J. Nanobiotechnol. 2014, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pulido, A.; Martín-Molina, A.; Rodríguez-Beas, C.; Llorca, O.; Aicart, E.; Junquera, E.; Sospedra, E.A. A Theoretical and Experimental Approach to the Compaction Process of DNA by Dioctadecyldimethylammonium Bromide/Zwitterionic Mixed Liposomes. J. Phys. Chem. B 2009, 113, 15648–15661. [Google Scholar] [CrossRef] [PubMed]
- Maney, V.; Singh, M. An in vitro assessment of novel chitosan/bimetallic PtAu nanocomposites as delivery vehicles for doxorubicin. Nanomedicine 2017, 12, 2625–2640. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.; Daniels, A.; Ariatti, M.; Singh, M. Anti-c-myc cholesterol based lipoplexes as onco-nanotherapeutic agents in vitro. F1000Research 2020, 9, 770. [Google Scholar] [CrossRef]
- Samimi, S.; Maghsoudnia, N.; Eftekhari, R.B.; Dorkoosh, F. Lipid-Based Nanoparticles for Drug Delivery Systems; Elsevier BV: Amsterdam, The Netherlands, 2019; pp. 47–76. [Google Scholar]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 1–12. [Google Scholar] [CrossRef]
- Boukalova, S.; Stursa, J.; Werner, L.; Ezrova, Z.; Cerny, J.; Bezawork-Geleta, A.; Pecinova, A.; Dong, L.; Drahota, Z.; Neuzil, J. Mitochondrial Targeting of Metformin Enhances Its Activity against Pancreatic Cancer. Mol. Cancer Ther. 2016, 15, 2875–2886. [Google Scholar] [CrossRef] [PubMed]
- Piyaviriyakul, S.; Shimizu, K.; Asakawa, T.; Kan, T.; Siripong, P.; Oku, N. Anti-angiogenic Activity and Intracellular Distribution of Epigallocatechin-3-gallate Analogs. Biol. Pharm. Bull. 2011, 34, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbison, R.A.; Linseman, D.A. Green Tea Epigallocatechin 3-Gallate Accumulates in Mitochondria and Displays a Selective Antiapoptotic Effect Against Inducers of Mitochondrial Oxidative Stress in Neurons. Antioxid. Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Neuzil, J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019, 39, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.E.M.; Mahmoud, N.; Sugimoto, Y.; Efferth, T.; Abdel-Aziz, H. Betulinic Acid Exerts Cytotoxic Activity Against Multidrug-Resistant Tumor Cells via Targeting Autocrine Motility Factor Receptor (AMFR). Front. Pharmacol. 2018, 9, 481. [Google Scholar] [CrossRef] [PubMed]
- Rieber, M.; Rieber, M.S. Induction of p53 Without Increase in p21WAF1 in Betulinic Acid-Mediated Cell Death Is Preferential for Human Metastatic Melanoma. DNA Cell Biol. 1998, 17, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.R.; Mandal, S.; Mittra, B.; Sharma, S.; Mukhopadhyay, S.; Majumder, H.K. Betulinic acid, a potent inhibitor of eukaryotic topoisomerase I: Identification of the inhibitory step, the major functional group responsible and development of more potent derivatives. Med. Sci. Monit. 2002, 8, 254–265. [Google Scholar]
- Kessler, J.H.; Mullauer, F.B.; De Roo, G.M.; Medema, J.P. Broad in vitro efficacy of plant-derived betulinic acid against cell lines derived from the most prevalent human cancer types. Cancer Lett. 2007, 251, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Pucci, B.; Kasten, M.; Giordano, A. Cell Cycle and Apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Alabsi, A.M.; Lim, K.L.; Paterson, I.C.; Ali-Saeed, R.; Muharram, B.A. Cell Cycle Arrest and Apoptosis Induction via Modulation of Mitochondrial Integrity by Bcl-2 Family Members and Caspase Dependence in Dracaena cinnabari-Treated H400 Human Oral Squamous Cell Carcinoma. BioMed Res. Int. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, T.-X.; Ma, Y.; Zhang, Y.; Li, D.-Y.; Xu, H.-R. Bursopentin (BP5) induces G1 phase cell cycle arrest and endoplasmic reticulum stress/mitochondria-mediated caspase-dependent apoptosis in human colon cancer HCT116 cells. Cancer Cell Int. 2019, 19, 130. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.B.; Yazan, L.S.; Tor, Y.S.; Wibowo, A.; Ismail, N.; How, C.W.; Armania, N.; Loh, S.P.; Ismail, I.S.; Cheah, Y.K.; et al. Induction of cell cycle arrest and apoptosis by betulinic acid-rich fraction from Dillenia suffruticosa root in MCF-7 cells involved p53/p21 and mitochondrial signalling pathway. J. Ethnopharmacol. 2015, 166, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, L.; Yang, Y.; Xun, W.; Wei, K.; Zeng, G. Betulinic Acid Inhibits Cell Proliferation in Human Oral Squamous Cell Carcinoma via Modulating ROS-Regulated p53 Signaling. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2017, 25, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPs | Hydrodynamic Size (nm) | PDI | ζ Potential (mV) | BA Loading Efficiency (%) |
|---|---|---|---|---|
| EGCG-AuNP | 127.3 ± 3.1 | 0.10 | −28.3 ± 1.6 | - |
| Au-PEG-BA | 110.1 ± 5.3 | 0.22 | −25.0 ± 0.9 | 25.4 ± 0.04 |
| T-Au-PEG-BA | 97.1 ± 2.5 | 0.035 | −23.1 ± 1.1 | 25.4 ± 0.12 |
| Au-[PLL-g-PEG]-BA | 147.2 ± 4.7 | 0.13 | +11.8 ± 1.6 | 21.0 ± 1.2 |
| T-Au-[PLL-g-PEG]-BA | 119.2 ± 3.5 | 0.11 | +23.4 ± 0.5 | 21.0 ± 0.7 |
| Au-PEG-BA | T-Au-PEG-BA | Au-[PLL-g-PEG]-BA | T-Au-[PLL-g-PEG]-BA | BA | |
|---|---|---|---|---|---|
| Estimated IC50 Values (µM) | |||||
| Caco-2 | 8.20 | 3.13 | 5.72 | 3.12 | 9.74 |
| HeLa | 25.37 | 6.51 | 23.64 | 3.26 | 17.73 |
| MCF-7 | 53.74 | 13.2 | 22.25 | 13.13 | 36.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oladimeji, O.; Akinyelu, J.; Daniels, A.; Singh, M. Modified Gold Nanoparticles for Efficient Delivery of Betulinic Acid to Cancer Cell Mitochondria. Int. J. Mol. Sci. 2021, 22, 5072. https://doi.org/10.3390/ijms22105072
Oladimeji O, Akinyelu J, Daniels A, Singh M. Modified Gold Nanoparticles for Efficient Delivery of Betulinic Acid to Cancer Cell Mitochondria. International Journal of Molecular Sciences. 2021; 22(10):5072. https://doi.org/10.3390/ijms22105072
Chicago/Turabian StyleOladimeji, Olakunle, Jude Akinyelu, Aliscia Daniels, and Moganavelli Singh. 2021. "Modified Gold Nanoparticles for Efficient Delivery of Betulinic Acid to Cancer Cell Mitochondria" International Journal of Molecular Sciences 22, no. 10: 5072. https://doi.org/10.3390/ijms22105072
APA StyleOladimeji, O., Akinyelu, J., Daniels, A., & Singh, M. (2021). Modified Gold Nanoparticles for Efficient Delivery of Betulinic Acid to Cancer Cell Mitochondria. International Journal of Molecular Sciences, 22(10), 5072. https://doi.org/10.3390/ijms22105072