Glutamatergic Dysfunction and Synaptic Ultrastructural Alterations in Schizophrenia and Autism Spectrum Disorder: Evidence from Human and Rodent Studies



Abstract
1. The Glutamatergic System
2. The Glutamatergic System in Neuropsychiatric Disorders
2.1. Schizophrenia (SZ)
2.2. Autism Spectrum Disorder (ASD)
3. Activity-Induced Modulation of Synaptic Ultrastructure
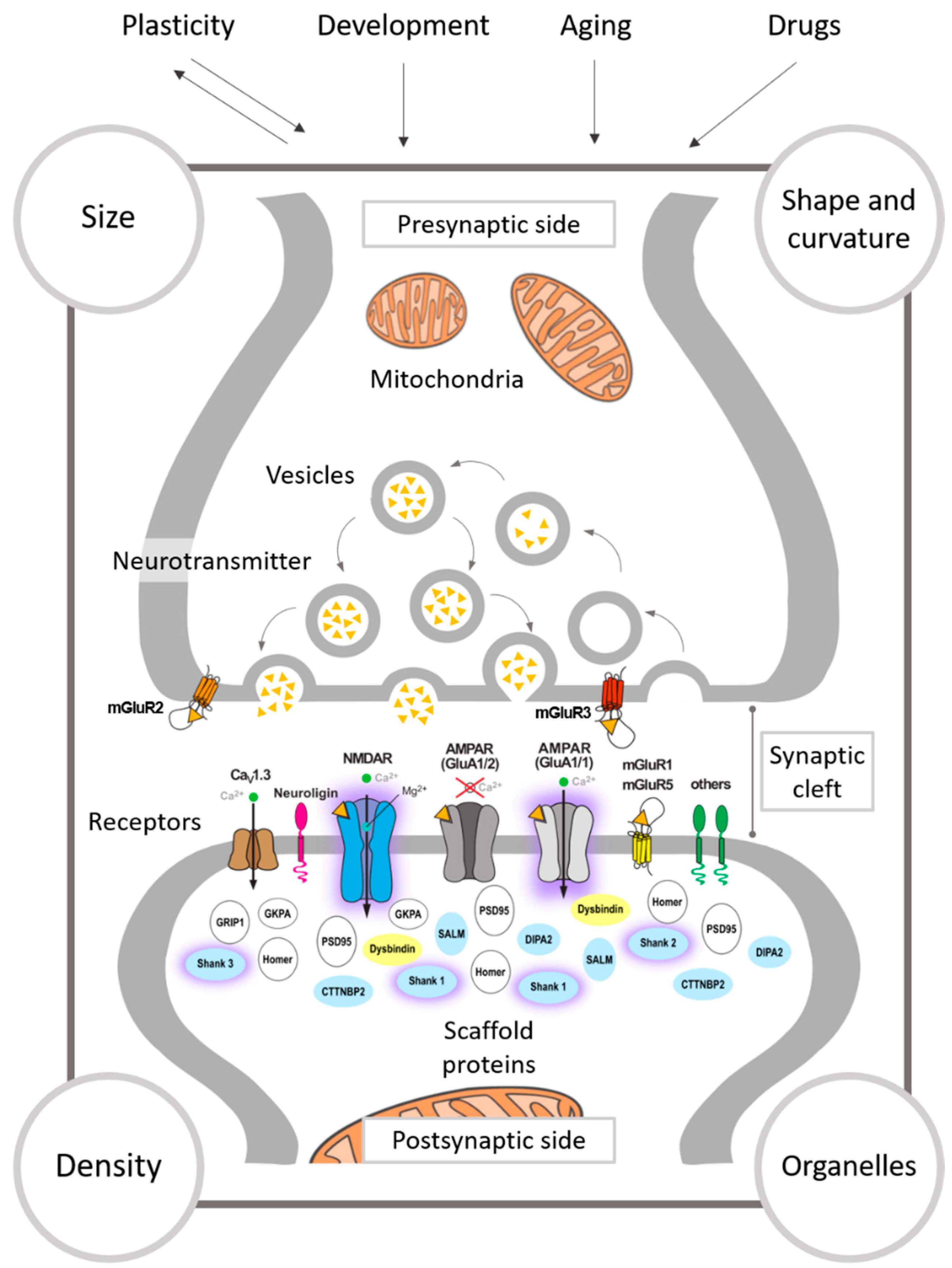
3.1. The Presynapse
3.2. The Synaptic Cleft
3.3. The Postsynapse
3.3.1. Synaptic Size
3.3.2. Shape of Synapses
3.3.3. Curvature of the Synaptic Apposition Surfaces
3.4. Synaptic Density
3.5. Mitochondria
4. Excitatory Synaptic Ultrastructure Alterations in Neuropsychiatric Disorders
4.1. Schizophrenia
4.2. Autism Spectrum Disorder
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Mouse lines
References
- Rothman, D.L.; Behar, K.L.; Hyder, F.; Shulman, R.G. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: Implications for brain function. Annu. Rev. Physiol. 2003, 65, 401–427. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.M. Glutamatergic antipsychotic drugs: A new dawn in the treatment of schizophrenia? Ther. Adv. Psychopharmacol. 2011, 1, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Park, P.; Kang, H.; Sanderson, T.M.; Bortolotto, Z.A.; Georgiou, J.; Zhuo, M.; Kaang, B.-K.; Collingridge, G.L. On the Role of Calcium-Permeable AMPARs in Long-Term Potentiation and Synaptic Tagging in the Rodent Hippocampus. Front. Synaptic Neurosci. 2019, 11. [Google Scholar] [CrossRef]
- Nicoll, R.A.; Malenka, R.C. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 1995, 377, 115–118. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Kew, J.N.; Kemp, J.A. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 2005, 179, 4–29. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Jensen, V.; Kaiser, K.M.; Borchardt, T.; Adelmann, G.; Rozov, A.; Burnashev, N.; Brix, C.; Frotscher, M.; Andersen, P.; Hvalby, O.; et al. A juvenile form of postsynaptic hippocampal long-term potentiation in mice deficient for the AMPA receptor subunit GluR-A. J. Physiol. 2003, 553, 843–856. [Google Scholar] [CrossRef]
- Zamanillo, D.; Sprengel, R.; Hvalby, O.; Jensen, V.; Burnashev, N.; Rozov, A.; Kaiser, K.M.; Köster, H.J.; Borchardt, T.; Worley, P.; et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 1999, 284, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Barch, D.M.; Ceaser, A. Cognition in schizophrenia: Core psychological and neural mechanisms. Trends Cogn. Sci. 2012, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Insel, T.; Cuthbert, B.; Garvey, M.; Heinssen, R.; Pine, D.S.; Quinn, K.; Sanislow, C.; Wang, P. Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. Am. J. Psychiatry 2010, 167, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Scoriels, L.; Barnett, J.H.; Soma, P.K.; Sahakian, B.J.; Jones, P.B. Effects of modafinil on cognitive functions in first episode psychosis. Psychopharmacology 2012, 220, 249–258. [Google Scholar] [CrossRef]
- Uno, Y.; Coyle, J.T. Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 2019, 73, 204–215. [Google Scholar] [CrossRef]
- Catts, V.S.; Lai, Y.L.; Weickert, C.S.; Weickert, T.W.; Catts, S.V. A quantitative review of the postmortem evidence for decreased cortical N-methyl-d-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biol. Psychol. 2016, 116, 57–67. [Google Scholar] [CrossRef]
- Kristiansen, L.V.; Huerta, I.; Beneyto, M.; Meador-Woodruff, J.H. NMDA receptors and schizophrenia. Curr. Opin. Pharmacol. 2007, 7, 48–55. [Google Scholar] [CrossRef]
- Kang, W.S.; Park, J.K.; Kim, S.K.; Park, H.J.; Lee, S.M.; Song, J.Y.; Chung, J.H.; Kim, J.W. Genetic variants of GRIA1 are associated with susceptibility to schizophrenia in Korean population. Mol. Biol. Rep. 2012, 39, 10697–10703. [Google Scholar] [CrossRef]
- Jadi, M.P.; Behrens, M.M.; Sejnowski, T.J. Abnormal Gamma Oscillations in N-methyl-d-aspartate Receptor Hypofunction Models of Schizophrenia. Biol. Psychiatry 2016, 79, 716–726. [Google Scholar] [CrossRef]
- Lisman, J.E.; Coyle, J.T.; Green, R.W.; Javitt, D.C.; Benes, F.M.; Heckers, S.; Grace, A.A. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008, 31, 234–242. [Google Scholar] [CrossRef]
- Uhlhaas, P.J.; Singer, W. High-frequency oscillations and the neurobiology of schizophrenia. Dialogues Clin. Neurosci. 2013, 15, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Mohn, A.R.; Gainetdinov, R.R.; Caron, M.G.; Koller, B.H. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef]
- Bygrave, A.M.; Kilonzo, K.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Can N-methyl-d-aspartate Receptor Hypofunction in Schizophrenia Be Localized to an Individual Cell Type? Front. Psychiatry 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Bygrave, A.M.; Masiulis, S.; Nicholson, E.; Berkemann, M.; Barkus, C.; Sprengel, R.; Harrison, P.J.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Knockout of NMDA-receptors from parvalbumin interneurons sensitizes to schizophrenia-related deficits induced by MK-801. Transl. Psychiatry 2016, 6, e778. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.; Barkus, C.; Feyder, M.; Wiedholz, L.; Chen, Y.-C.; Karlsson, R.-M.; Machado-Vieira, R.; Graybeal, C.; Sharp, T.; Zarate, C.; et al. Does gene deletion of AMPA GluA1 phenocopy features of schizoaffective disorder? Neurobiol. Dis. 2010, 40, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Barkus, C.; Feyder, M.; Graybeal, C.; Wright, T.; Wiedholz, L.; Izquierdo, A.; Kiselycznyk, C.; Schmitt, W.; Sanderson, D.J.; Rawlins, J.N.; et al. Do GluA1 knockout mice exhibit behavioral abnormalities relevant to the negative or cognitive symptoms of schizophrenia and schizoaffective disorder? Neuropharmacology 2012, 62, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, D.J.; Lee, A.; Sprengel, R.; Seeburg, P.H.; Harrison, P.J.; Bannerman, D.M. Altered balance of excitatory and inhibitory learning in a genetically modified mouse model of glutamatergic dysfunction relevant to schizophrenia. Sci. Rep. 2017, 7, 1765. [Google Scholar] [CrossRef]
- Wiedholz, L.M.; Owens, W.A.; Horton, R.E.; Feyder, M.; Karlsson, R.M.; Hefner, K.; Sprengel, R.; Celikel, T.; Daws, L.C.; Holmes, A. Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ‘schizophrenia-related’ behaviors. Mol. Psychiatry 2008, 13, 631–640. [Google Scholar] [CrossRef]
- Inta, D.; Monyer, H.; Sprengel, R.; Meyer-Lindenberg, A.; Gass, P. Mice with genetically altered glutamate receptors as models of schizophrenia: A comprehensive review. Neurosci. Biobehav. Rev. 2009, 34, 285–294. [Google Scholar] [CrossRef]
- Lehman, J.F. The Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar]
- Fernell, E. Further studies of GABA and Glutamate imbalances in autism are important challenges for future research. Acta Paediatr. 2019, 108, 200–201. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill. Summ. 2014, 63, 1–21.
- Lyall, K.; Croen, L.; Daniels, J.; Fallin, M.D.; Ladd-Acosta, C.; Lee, B.K.; Park, B.Y.; Snyder, N.W.; Schendel, D.; Volk, H.; et al. The Changing Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2017, 38, 81–102. [Google Scholar] [CrossRef]
- Horder, J.; Petrinovic, M.M.; Mendez, M.A.; Bruns, A.; Takumi, T.; Spooren, W.; Barker, G.J.; Künnecke, B.; Murphy, D.G. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl. Psychiatry. 2018, 25, 106. [Google Scholar] [CrossRef]
- Carlsson, M.L. Hypothesis: Is infantile autism a hypoglutamatergic disorder? Relevance of glutamate—Serotonin interactions for pharmacotherapy. J. Neural. Transm. 1998, 105, 525–535. [Google Scholar] [CrossRef]
- Fatemi, S.H. The hyperglutamatergic hypothesis of autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 911. [Google Scholar] [CrossRef]
- Uzunova, G.; Hollander, E.; Shepherd, J. The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: Autism spectrum disorders and fragile x syndrome. Curr. Neuropharmacol. 2014, 12, 71–98. [Google Scholar] [CrossRef]
- Moreno, H.; Borjas, L.; Arrieta, A.; Saez, L.; Prassad, A.; Estevez, J.; Bonilla, E. Clinical heterogeneity of the autistic syndrome: A study of 60 families. Investig. Clin. 1992, 33, 13–31. [Google Scholar]
- Moreno-Fuenmayor, H.; Borjas, L.; Arrieta, A.; Valera, V.; Socorro-Candanoza, L. Plasma excitatory amino acids in autism. Investig. Clin. 1996, 37, 113–128. [Google Scholar]
- Aldred, S.; Moore, K.M.; Fitzgerald, M.; Waring, R.H. Plasma amino acid levels in children with autism and their families. J. Autism Dev. Disord. 2003, 33, 93–97. [Google Scholar] [CrossRef]
- Shinohe, A.; Hashimoto, K.; Nakamura, K.; Tsujii, M.; Iwata, Y.; Tsuchiya, K.J.; Sekine, Y.; Suda, S.; Suzuki, K.; Sugihara, G.; et al. Increased serum levels of glutamate in adult patients with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Tirouvanziam, R.; Obukhanych, T.V.; Laval, J.; Aronov, P.A.; Libove, R.; Banerjee, A.G.; Parker, K.J.; O’Hara, R.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Distinct plasma profile of polar neutral amino acids, leucine, and glutamate in children with Autism Spectrum Disorders. J. Autism Dev. Disord. 2012, 42, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Shimmura, C.; Suda, S.; Tsuchiya, K.J.; Hashimoto, K.; Ohno, K.; Matsuzaki, H.; Iwata, K.; Matsumoto, K.; Wakuda, T.; Kameno, Y.; et al. Alteration of plasma glutamate and glutamine levels in children with high-functioning autism. PLoS ONE 2011, 6, e25340. [Google Scholar] [CrossRef] [PubMed]
- Hassan, T.H.; Abdelrahman, H.M.; Abdel Fattah, N.R.; El-Masry, N.M.; Hashim, H.M.; El-Gerby, K.M.; Abdel Fattah, N.R. Blood and brain glutamate levels in children with autistic disorder. Res. Autism Spectr. Disord. 2013, 7, 541–548. [Google Scholar] [CrossRef]
- Khalifa, D.; Shahin, O.; Salem, D.; Raafat, O. Serum glutamate was elevated in children aged 3–10 years with autism spectrum disorders when they were compared with controls. Acta Paediatr. 2019, 108, 295–299. [Google Scholar] [CrossRef]
- Rojas, D.C. The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J. Neural. Transm. 2014, 121, 891–905. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhu, T.; Qu, Y.; Mu, D. Blood Glutamate Levels in Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158688. [Google Scholar] [CrossRef]
- Rinaldi, T.; Kulangara, K.; Antoniello, K.; Markram, H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. USA 2007, 104, 13501–13506. [Google Scholar] [CrossRef]
- Kim, J.W.; Park, K.; Kang, R.J.; Gonzales, E.L.T.; Kim, D.G.; Oh, H.A.; Seung, H.; Ko, M.J.; Kwon, K.J.; Kim, K.C.; et al. Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacology 2019, 44, 314–323. [Google Scholar] [CrossRef]
- Rezaei, V.; Mohammadi, M.R.; Ghanizadeh, A.; Sahraian, A.; Tabrizi, M.; Rezazadeh, S.A.; Akhondzadeh, S. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1269–1272. [Google Scholar] [CrossRef]
- Doyle, C.A.; McDougle, C.J. Pharmacologic treatments for the behavioral symptoms associated with autism spectrum disorders across the lifespan. Dialogues Clin. Neurosci. 2012, 14, 263–279. [Google Scholar]
- Erickson, C.A.; Posey, D.J.; Stigler, K.A.; Mullett, J.; Katschke, A.R.; McDougle, C.J. A retrospective study of memantine in children and adolescents with pervasive developmental disorders. Psychopharmacology 2007, 191, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Owley, T.; Salt, J.; Guter, S.; Grieve, A.; Walton, L.; Ayuyao, N.; Leventhal, B.L.; Cook, E.H., Jr. A prospective, open-label trial of memantine in the treatment of cognitive, behavioral, and memory dysfunction in pervasive developmental disorders. J. Child. Adolesc. Psychopharmacol. 2006, 16, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Chez, M.G.; Burton, Q.; Dowling, T.; Chang, M.; Khanna, P.; Kramer, C. Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: An observation of initial clinical response and maintenance tolerability. J. Child. Neurol. 2007, 22, 574–579. [Google Scholar] [CrossRef] [PubMed]
- King, B.H.; Wright, D.M.; Handen, B.L.; Sikich, L.; Zimmerman, A.W.; McMahon, W.; Cantwell, E.; Davanzo, P.A.; Dourish, C.T.; Dykens, E.M.; et al. Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child. Adolesc. Psychiatry 2001, 40, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Early, M.; Stigler, K.A.; Wink, L.K.; Mullett, J.E.; McDougle, C.J. An open-label naturalistic pilot study of acamprosate in youth with autistic disorder. J. Child. Adolesc. Psychopharmacol. 2011, 21, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Wink, L.K.; Ray, B.; Early, M.C.; Stiegelmeyer, E.; Mathieu-Frasier, L.; Patrick, V.; Lahiri, D.K.; McDougle, C.J. Impact of acamprosate on behavior and brain-derived neurotrophic factor: An open-label study in youth with fragile X syndrome. Psychopharmacology 2013, 228, 75–84. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Tajdar, H.; Mohammadi, M.R.; Mohammadi, M.; Nouroozinejad, G.H.; Shabstari, O.L.; Ghelichnia, H.A. A double-blind placebo controlled trial of piracetam added to risperidone in patients with autistic disorder. Child. Psychiatry Hum. Dev. 2008, 39, 237–245. [Google Scholar] [CrossRef]
- Henter, I.D.; de Sousa, R.T.; Zarate, C.A., Jr. Glutamatergic Modulators in Depression. Harv Rev. Psychiatry 2018, 26, 307–319. [Google Scholar] [CrossRef]
- Posey, D.J.; Kem, D.L.; Swiezy, N.B.; Sweeten, T.L.; Wiegand, R.E.; McDougle, C.J. A Pilot Study of d-Cycloserine in Subjects With Autistic Disorder. Am. J. Psychiatry 2004, 161, 2115–2117. [Google Scholar] [CrossRef]
- Urbano, M.; Okwara, L.; Manser, P.; Hartmann, K.; Deutsch, S.I. A trial of d-cycloserine to treat the social deficit in older adolescents and young adults with autism spectrum disorders. J. Neuropsychiatry Clin. Neurosci. 2015, 27, 133–138. [Google Scholar] [CrossRef]
- Wink, L.K.; Minshawi, N.F.; Shaffer, R.C.; Plawecki, M.H.; Posey, D.J.; Horn, P.S.; Adams, R.; Pedapati, E.V.; Schaefer, T.L.; McDougle, C.J.; et al. d-Cycloserine enhances durability of social skills training in autism spectrum disorder. Mol. Autism. 2017, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Eltokhi, A.; Rappold, G.; Sprengel, R. Distinct Phenotypes of Shank2 Mouse Models Reflect Neuropsychiatric Spectrum Disorders of Human Patients With SHANK2 Variants. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Lee, H.R.; Gee, H.Y.; Mah, W.; Kim, J.I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef]
- Burket, J.A.; Benson, A.D.; Tang, A.H.; Deutsch, S.I. D-Cycloserine improves sociability in the BTBR T+ Itpr3tf/J mouse model of autism spectrum disorders with altered Ras/Raf/ERK1/2 signaling. Brain Res. Bull. 2013, 96, 62–70. [Google Scholar] [CrossRef][Green Version]
- Um, S.M.; Ha, S.; Lee, H.; Kim, J.; Kim, K.; Shin, W.; Cho, Y.S.; Roh, J.D.; Kang, J.; Yoo, T.; et al. NGL-2 Deletion Leads to Autistic-like Behaviors Responsive to NMDAR Modulation. Cell Rep. 2018, 23, 3839–3851. [Google Scholar] [CrossRef]
- Schoch, H.; Kreibich, A.S.; Ferri, S.L.; White, R.S.; Bohorquez, D.; Banerjee, A.; Port, R.G.; Dow, H.C.; Cordero, L.; Pallathra, A.A.; et al. Sociability Deficits and Altered Amygdala Circuits in Mice Lacking Pcdh10, an Autism Associated Gene. Biol. Psychiatry 2017, 81, 193–202. [Google Scholar] [CrossRef]
- Urbano, M.; Okwara, L.; Manser, P.; Hartmann, K.; Herndon, A.; Deutsch, S.I. A trial of D-cycloserine to treat stereotypies in older adolescents and young adults with autism spectrum disorder. Clin. Neuropharmacol. 2014, 37, 69–72. [Google Scholar] [CrossRef]
- Sidorov, M.S.; Auerbach, B.D.; Bear, M.F. Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 2013, 6, 15. [Google Scholar] [CrossRef]
- Cheng, G.R.; Li, X.Y.; Xiang, Y.D.; Liu, D.; McClintock, S.M.; Zeng, Y. The implication of AMPA receptor in synaptic plasticity impairment and intellectual disability in fragile X syndrome. Physiol. Res. 2017, 66, 715–727. [Google Scholar] [CrossRef]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D.; Kneeland, R.E.; Liesch, S.B. Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both Fragile X mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat. Rec. 2011, 294, 1635–1645. [Google Scholar] [CrossRef]
- Lohith, T.G.; Osterweil, E.K.; Fujita, M.; Jenko, K.J.; Bear, M.F.; Innis, R.B. Is metabotropic glutamate receptor 5 upregulated in prefrontal cortex in fragile X syndrome? Mol. Autism 2013, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Blatt, G.J.; Fitzgerald, C.M.; Guptill, J.T.; Booker, A.B.; Kemper, T.L.; Bauman, M.L. Density and distribution of hippocampal neurotransmitter receptors in autism: An autoradiographic study. J. Autism Dev. Disord. 2001, 31, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, E.; Millet, B.; S2D team; et al. Rare mutations in N-methyl-d-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 2011, 1, e55. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Cho, I.H.; Park, M.; Yang, S.Y.; Kim, S.A. Family based association of GRIN2A and GRIN2B with Korean autism spectrum disorders. Neurosci. Lett. 2012, 512, 89–93. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef]
- Barnby, G.; Abbott, A.; Sykes, N.; Morris, A.; Weeks, D.E.; Mott, R.; Lamb, J.; Bailey, A.J.; Monaco, A.P. Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: Evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 2005, 76, 950–966. [Google Scholar] [CrossRef]
- Jamain, S.; Betancur, C.; Quach, H.; Philippe, A.; Fellous, M.; Giros, B.; Gillberg, C.; Leboyer, M.; Bourgeron, T.; Paris Autism Research International Sibpair, S. Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry 2002, 7, 302–310. [Google Scholar] [CrossRef]
- Shuang, M.; Liu, J.; Jia, M.X.; Yang, J.Z.; Wu, S.P.; Gong, X.H.; Ling, Y.S.; Ruan, Y.; Yang, X.L.; Zhang, D. Family-based association study between autism and glutamate receptor 6 gene in Chinese Han trios. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 131, 48–50. [Google Scholar] [CrossRef]
- Strutz-Seebohm, N.; Korniychuk, G.; Schwarz, R.; Baltaev, R.; Ursu, O.; Mack, A.; Ma-Högemeier, Z.; Hollmann, M.; Lang, F.; Seebohm, G. Functional Significance of the Kainate Receptor GluR6(M836I) Mutation that is Linked to Autism. Cell Physiol. Biochem. 2006, 18, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Yonan, A.L.; Alarcon, M.; Cheng, R.; Magnusson, P.K.; Spence, S.J.; Palmer, A.A.; Grunn, A.; Juo, S.H.; Terwilliger, J.D.; Liu, J.; et al. A genomewide screen of 345 families for autism-susceptibility loci. Am. J. Hum. Genet. 2003, 73, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Woodroffe, A.; Flodman, P.L.; Mays, L.Z.; Hanouni, M.; Modahl, C.B.; Steinberg-Epstein, R.; Bocian, M.E.; Spence, M.A.; Smith, M. A case of autism with an interstitial deletion on 4q leading to hemizygosity for genes encoding for glutamine and glycine neurotransmitter receptor sub-units (AMPA 2, GLRA3, GLRB) and neuropeptide receptors NPY1R, NPY5R. BMC Med. Genet. 2004, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Serajee, F.J.; Zhong, H.; Nabi, R.; Huq, A.H. The metabotropic glutamate receptor 8 gene at 7q31: Partial duplication and possible association with autism. J. Med. Genet. 2003, 40, e42. [Google Scholar] [CrossRef] [PubMed]
- Autism Genome Project, C.; Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [CrossRef]
- Bremer, A.; Giacobini, M.; Eriksson, M.; Gustavsson, P.; Nordin, V.; Fernell, E.; Gillberg, C.; Nordgren, A.; Uppstromer, A.; Anderlid, B.M.; et al. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 115–124. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef]
- Gauthier, J.; Siddiqui, T.J.; Huashan, P.; Yokomaku, D.; Hamdan, F.F.; Champagne, N.; Lapointe, M.; Spiegelman, D.; Noreau, A.; Lafreniere, R.G.; et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 2011, 130, 563–573. [Google Scholar] [CrossRef]
- Talebizadeh, Z.; Bittel, D.C.; Veatch, O.J.; Butler, M.G.; Takahashi, T.N.; Miles, J.H. Do known mutations in neuroligin genes (NLGN3 and NLGN4) cause autism? J. Autism Dev. Disord. 2004, 34, 735–736. [Google Scholar] [CrossRef]
- Foldy, C.; Malenka, R.C.; Sudhof, T.C. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 2013, 78, 498–509. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Bonnet-Brilhault, F.; Gomot, M.; Blanc, R.; David, A.; Moizard, M.P.; Raynaud, M.; Ronce, N.; Lemonnier, E.; Calvas, P.; et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004, 74, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, M.; Abrahams, B.S.; Stone, J.L.; Duvall, J.A.; Perederiy, J.V.; Bomar, J.M.; Sebat, J.; Wigler, M.; Martin, C.L.; Ledbetter, D.H.; et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Sampath, S.; Bhat, S.; Gupta, S.; O’Connor, A.; West, A.B.; Arking, D.E.; Chakravarti, A. Defining the contribution of CNTNAP2 to autism susceptibility. PLoS ONE 2013, 8, e77906. [Google Scholar] [CrossRef]
- Gauthier, J.; Spiegelman, D.; Piton, A.; Lafreniere, R.G.; Laurent, S.; St-Onge, J.; Lapointe, L.; Hamdan, F.F.; Cossette, P.; Mottron, L.; et al. Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009, 150B, 421–424. [Google Scholar] [CrossRef]
- Gauthier, J.; Champagne, N.; Lafreniere, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Cote, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsater, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef]
- Sato, D.; Lionel, A.C.; Leblond, C.S.; Prasad, A.; Pinto, D.; Walker, S.; O’Connor, I.; Russell, C.; Drmic, I.E.; Hamdan, F.F.; et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am. J. Hum. Genet. 2012, 90, 879–887. [Google Scholar] [CrossRef]
- Guilmatre, A.; Huguet, G.; Delorme, R.; Bourgeron, T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol. 2014, 74, 113–122. [Google Scholar] [CrossRef]
- Fung, L.K.; Hardan, A.Y. Developing Medications Targeting Glutamatergic Dysfunction in Autism: Progress to Date. CNS Drugs 2015, 29, 453–463. [Google Scholar] [CrossRef]
- Bejjani, A.; O’Neill, J.; Kim, J.A.; Frew, A.J.; Yee, V.W.; Ly, R.; Kitchen, C.; Salamon, N.; McCracken, J.T.; Toga, A.W.; et al. Elevated glutamatergic compounds in pregenual anterior cingulate in pediatric autism spectrum disorder demonstrated by 1H MRS and 1H MRSI. PLoS ONE 2012, 7, e38786. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Anagnostou, E.; Shen, J.; Kolevzon, A.; Buxbaum, J.D.; Hollander, E.; Hof, P.R.; Fan, J. In vivo 1H-magnetic resonance spectroscopy study of the attentional networks in autism. Brain Res. 2011, 1380, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Singel, D.; Hepburn, S.; Rojas, D.C. Increased glutamate concentration in the auditory cortex of persons with autism and first-degree relatives: A (1)H-MRS study. Autism Res. 2013, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Horder, J.; Lavender, T.; Mendez, M.A.; O’Gorman, R.; Daly, E.; Craig, M.C.; Lythgoe, D.J.; Barker, G.J.; Murphy, D.G. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: A [(1)H]MRS study. Transl. Psychiatry 2013, 3, e279. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Biederman, J.; Wozniak, J.; Goldin, R.L.; Crowley, D.; Furtak, S.; Lukas, S.E.; Gonenc, A. Magnetic resonance spectroscopy study of the glutamatergic system in adolescent males with high-functioning autistic disorder: A pilot study at 4T. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 379–384. [Google Scholar] [CrossRef]
- Kubas, B.; Kulak, W.; Sobaniec, W.; Tarasow, E.; Lebkowska, U.; Walecki, J. Metabolite alterations in autistic children: A 1H MR spectroscopy study. Adv. Med. Sci. 2012, 57, 152–156. [Google Scholar] [CrossRef]
- Naaijen, J.; Forde, N.J.; Lythgoe, D.J.; Akkermans, S.E.; Openneer, T.J.; Dietrich, A.; Zwiers, M.P.; Hoekstra, P.J.; Buitelaar, J.K. Fronto-striatal glutamate in children with Tourette’s disorder and attention-deficit/hyperactivity disorder. Neuroimage Clin. 2017, 13, 16–23. [Google Scholar] [CrossRef]
- Page, L.A.; Daly, E.; Schmitz, N.; Simmons, A.; Toal, F.; Deeley, Q.; Ambery, F.; McAlonan, G.M.; Murphy, K.C.; Murphy, D.G. In vivo 1H-magnetic resonance spectroscopy study of amygdala-hippocampal and parietal regions in autism. Am. J. Psychiatry 2006, 163, 2189–2192. [Google Scholar] [CrossRef]
- Tebartz van Elst, L.; Maier, S.; Fangmeier, T.; Endres, D.; Mueller, G.T.; Nickel, K.; Ebert, D.; Lange, T.; Hennig, J.; Biscaldi, M.; et al. Disturbed cingulate glutamate metabolism in adults with high-functioning autism spectrum disorder: Evidence in support of the excitatory/inhibitory imbalance hypothesis. Mol. Psychiatry 2014, 19, 1314–1325. [Google Scholar] [CrossRef]
- Ford, T.C.; Nibbs, R.; Crewther, D.P. Increased glutamate/GABA+ ratio in a shared autistic and schizotypal trait phenotype termed Social Disorganisation. NeuroImage Clin. 2017, 16, 125–131. [Google Scholar] [CrossRef]
- Carlson, G. Glutamate receptor dysfunction and drug targets across models of autism spectrum disorders. Pharmacol. Biochem. Behav. 2011, 100, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lu, J.; Zuo, Y. Changes of Synaptic Structures Associated with Learning, Memory and Diseases. Brain Sci. Adv. 2018, 4, 99–117. [Google Scholar] [CrossRef]
- Kano, M.; Watanabe, T. Developmental synapse remodeling in the cerebellum and visual thalamus [version 1; peer review: 2 approved]. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Robain, O.; Bideau, I.; Farkas, E. Developmental changes of synapses in the cerebellar cortex of the rat. A quantitative analysis. Brain Res. 1981, 206, 1–8. [Google Scholar] [CrossRef]
- Lohmann, C.; Kessels, H.W. The developmental stages of synaptic plasticity. J. Physiol. 2014, 592, 13–31. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; Allen, N.J. Astrocytes, neurons, synapses: A tripartite view on cortical circuit development. Neural Dev. 2018, 13, 7. [Google Scholar] [CrossRef]
- Prokop, A.; Meinertzhagen, I.A. Development and structure of synaptic contacts in Drosophila. Semin. Cell Dev. Biol. 2006, 17, 20–30. [Google Scholar] [CrossRef]
- Benavides-Piccione, R.; Fernaud-Espinosa, I.; Robles, V.; Yuste, R.; DeFelipe, J. Age-based comparison of human dendritic spine structure using complete three-dimensional reconstructions. Cereb. Cortex 2013, 23, 1798–1810. [Google Scholar] [CrossRef]
- Markus, E.J.; Petit, T.L.; LeBoutillier, J.C. Synaptic structural changes during development and aging. Brain Res. 1987, 432, 239–248. [Google Scholar] [CrossRef]
- Russo, S.J.; Dietz, D.M.; Dumitriu, D.; Morrison, J.H.; Malenka, R.C.; Nestler, E.J. The addicted synapse: Mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010, 33, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.C.; Barksdale, K.A.; Roche, J.K.; Lahti, A.C. Decreased synaptic and mitochondrial density in the postmortem anterior cingulate cortex in schizophrenia. Schizophr. Res. 2015, 168, 543–553. [Google Scholar] [CrossRef]
- Aganova, E.A.; Uranova, N.A. Morphometric analysis of synaptic contacts in the anterior limbic cortex in the endogenous psychoses. Neurosci. Behav. Physiol. 1992, 22, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Kolomeets, N.S.; Orlovskaya, D.D.; Rachmanova, V.I.; Uranova, N.A. Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: A postmortem morphometric study. Synapse 2005, 57, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Kolomeets, N.S.; Orlovskaya, D.D.; Uranova, N.A. Decreased numerical density of CA3 hippocampal mossy fiber synapses in schizophrenia. Synapse 2007, 61, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.C.; Roche, J.K.; Conley, R.R. Synaptic differences in the postmortem striatum of subjects with schizophrenia: A stereological ultrastructural analysis. Synapse 2005, 56, 185–197. [Google Scholar] [CrossRef]
- Roberts, R.C.; Roche, J.K.; Conley, R.R. Synaptic differences in the patch matrix compartments of subjects with schizophrenia: A postmortem ultrastructural study of the striatum. Neurobiol. Dis. 2005, 20, 324–335. [Google Scholar] [CrossRef]
- Roberts, R.C.; Roche, R.J.; Somerville, S.M.; Conley, R.R. Ultrastructural distinctions between treatment responders and non-responders in schizophrenia: Postmortem studies of the striatum. In Mental Illnesses—Evaluation, Treatments, and Implications; InTech: London, UK, 2012; pp. 261–286. [Google Scholar]
- McCollum, L.A.; Walker, C.K.; Roche, J.K.; Roberts, R.C. Elevated Excitatory Input to the Nucleus Accumbens in Schizophrenia: A Postmortem Ultrastructural Study. Schizophr. Bull. 2015, 41, 1123–1132. [Google Scholar] [CrossRef]
- Xu, Y.; Deng, C.; Zheng, Y.; Liu, N.; Fu, B. Applying vinpocetine to reverse synaptic ultrastructure by regulating BDNF-related PSD-95 in alleviating schizophrenia-like deficits in rat. Compr. Psychiatry 2019, 94, 152122. [Google Scholar] [CrossRef]
- Chen, X.W.; Feng, Y.Q.; Hao, C.J.; Guo, X.L.; He, X.; Zhou, Z.Y.; Guo, N.; Huang, H.P.; Xiong, W.; Zheng, H.; et al. DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J. Cell Biol. 2008, 181, 791–801. [Google Scholar] [CrossRef]
- Zikopoulos, B.; Barbas, H. Changes in prefrontal axons may disrupt the network in autism. J. Neurosci. 2010, 30, 14595–14609. [Google Scholar] [CrossRef] [PubMed]
- Gassowska-Dobrowolska, M.; Cieslik, M.; Czapski, G.A.; Jesko, H.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Babiec, L.; et al. Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3576. [Google Scholar] [CrossRef] [PubMed]
- Lobzhanidze, G.; Japaridze, N.; Lordkipanidze, T.; Rzayev, F.; MacFabe, D.; Zhvania, M. Behavioural and brain ultrastructural changes following the systemic administration of propionic acid in adolescent male rats. Further development of a rodent model of autism. Int. J. Dev. Neurosci. 2020, 80, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Okabe, S. Nano-scale analysis of synapse morphology in an autism mouse model with 15q11-13 copy number variation using focused ion beam milling and scanning electron microscopy. Microscopy 2019, 68, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.Y.; Futai, K.; Sala, C.; Valtschanoff, J.G.; Ryu, J.; Woodworth, M.A.; Kidd, F.L.; Sung, C.C.; Miyakawa, T.; Bear, M.F.; et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 2008, 28, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef]
- Morimura, N.; Yasuda, H.; Yamaguchi, K.; Katayama, K.I.; Hatayama, M.; Tomioka, N.H.; Odagawa, M.; Kamiya, A.; Iwayama, Y.; Maekawa, M.; et al. Autism-like behaviours and enhanced memory formation and synaptic plasticity in Lrfn2/SALM1-deficient mice. Nat. Commun. 2017, 8, 15800. [Google Scholar] [CrossRef]
- Shih, P.Y.; Hsieh, B.Y.; Lin, M.H.; Huang, T.N.; Tsai, C.Y.; Pong, W.L.; Lee, S.P.; Hsueh, Y.P. CTTNBP2 Controls Synaptic Expression of Zinc-Related Autism-Associated Proteins and Regulates Synapse Formation and Autism-like Behaviors. Cell Rep. 2020, 31, 107700. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, L.Q.; He, Z.X.; He, X.X.; Wang, Y.J.; Jian, Y.L.; Wang, X.; Zhang, B.B.; Su, C.; Lu, J.; et al. Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol. 2019, 17, e3000461. [Google Scholar] [CrossRef]
- Ranneva, S.V.; Maksimov, V.F.; Korostyshevskaja, I.M.; Lipina, T.V. Lack of synaptic protein, calsyntenin-2, impairs morphology of synaptic complexes in mice. Synapse 2020, 74, e22132. [Google Scholar] [CrossRef]
- Kang, M.S.; Choi, T.Y.; Ryu, H.G.; Lee, D.; Lee, S.H.; Choi, S.Y.; Kim, K.T. Autism-like behavior caused by deletion of vaccinia-related kinase 3 is improved by TrkB stimulation. J. Exp. Med. 2017, 214, 2947–2966. [Google Scholar] [CrossRef] [PubMed]
- He, C.X.; Portera-Cailliau, C. The trouble with spines in fragile X syndrome: Density, maturity and plasticity. Neuroscience 2013, 251, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, S.; Kidd, G.J.; Wang, J.; Swetlik, C.; Dutta, R.; Trapp, B.D. Alterations in CA1 hippocampal synapses in a mouse model of fragile X syndrome. Glia 2018, 66, 789–800. [Google Scholar] [CrossRef]
- Padmashri, R.; Reiner, B.C.; Suresh, A.; Spartz, E.; Dunaevsky, A. Altered Structural and Functional Synaptic Plasticity with Motor Skill Learning in a Mouse Model of Fragile X Syndrome. J. Neurosci. 2013, 33, 19715–19723. [Google Scholar] [CrossRef]
- Bailey, C.H.; Chen, M. Morphological basis of long-term habituation and sensitization in Aplysia. Science 1983, 220, 91–93. [Google Scholar] [CrossRef]
- Muller, M.; Liu, K.S.; Sigrist, S.J.; Davis, G.W. RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci. 2012, 32, 16574–16585. [Google Scholar] [CrossRef]
- Goel, P.; Li, X.; Dickman, D. Estimation of the Readily Releasable Synaptic Vesicle Pool at the Drosophila Larval Neuromuscular Junction. Bio-Protocol 2019, 9. [Google Scholar] [CrossRef]
- Lazarevic, V.; Schone, C.; Heine, M.; Gundelfinger, E.D.; Fejtova, A. Extensive remodeling of the presynaptic cytomatrix upon homeostatic adaptation to network activity silencing. J. Neurosci. 2011, 31, 10189–10200. [Google Scholar] [CrossRef]
- Soares, C.; Lee, K.F.H.; Beique, J.C. Metaplasticity at CA1 Synapses by Homeostatic Control of Presynaptic Release Dynamics. Cell Rep. 2017, 21, 1293–1303. [Google Scholar] [CrossRef]
- Savtchenko, L.P.; Rusakov, D.A. The optimal height of the synaptic cleft. Proc. Natl. Acad. Sci. USA 2007, 104, 1823–1828. [Google Scholar] [CrossRef]
- Glebov, O.O.; Cox, S.; Humphreys, L.; Burrone, J. Neuronal activity controls transsynaptic geometry. Sci. Rep. 2016, 6, 22703. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Ellis-Davies, G.C.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001, 4, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Matsuzaki, M.; Noguchi, J.; Yasumatsu, N.; Nakahara, H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003, 26, 360–368. [Google Scholar] [CrossRef]
- Arellano, J.I.; Benavides-Piccione, R.; Defelipe, J.; Yuste, R. Ultrastructure of dendritic spines: Correlation between synaptic and spine morphologies. Front. Neurosci. 2007, 1, 131–143. [Google Scholar] [CrossRef]
- Holderith, N.; Lorincz, A.; Katona, G.; Rozsa, B.; Kulik, A.; Watanabe, M.; Nusser, Z. Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat. Neurosci. 2012, 15, 988–997. [Google Scholar] [CrossRef]
- Quinn, D.P.; Kolar, A.; Harris, S.A.; Wigerius, M.; Fawcett, J.P.; Krueger, S.R. The Stability of Glutamatergic Synapses Is Independent of Activity Level, but Predicted by Synapse Size. Front. Cell Neurosci. 2019, 13, 291. [Google Scholar] [CrossRef]
- Mayford, M.; Siegelbaum, S.A.; Kandel, E.R. Synapses and memory storage. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Kasai, H.; Fukuda, M.; Watanabe, S.; Hayashi-Takagi, A.; Noguchi, J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010, 33, 121–129. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.; Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004, 429, 761–766. [Google Scholar] [CrossRef]
- Minerbi, A.; Kahana, R.; Goldfeld, L.; Kaufman, M.; Marom, S.; Ziv, N.E. Long-Term Relationships between Synaptic Tenacity, Synaptic Remodeling, and Network Activity. PLoS Biol. 2009, 7, e1000136. [Google Scholar] [CrossRef]
- Mongillo, G.; Rumpel, S.; Loewenstein, Y. Intrinsic volatility of synaptic connections—A challenge to the synaptic trace theory of memory. Curr. Opin Neurobiol. 2017, 46, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Susman, L.; Brenner, N.; Barak, O. Stable memory with unstable synapses. Nat. Commun. 2019, 10, 4441. [Google Scholar] [CrossRef] [PubMed]
- Merchan-Perez, A.; Rodriguez, J.R.; Gonzalez, S.; Robles, V.; Defelipe, J.; Larranaga, P.; Bielza, C. Three-dimensional spatial distribution of synapses in the neocortex: A dual-beam electron microscopy study. Cereb. Cortex 2014, 24, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Santuy, A.; Tomas-Roca, L.; Rodriguez, J.R.; Gonzalez-Soriano, J.; Zhu, F.; Qiu, Z.; Grant, S.G.N.; DeFelipe, J.; Merchan-Perez, A. Estimation of the number of synapses in the hippocampus and brain-wide by volume electron microscopy and genetic labeling. Sci. Rep. 2020, 10, 14014. [Google Scholar] [CrossRef] [PubMed]
- Santuy, A.; Rodriguez, J.R.; DeFelipe, J.; Merchan-Perez, A. Study of the Size and Shape of Synapses in the Juvenile Rat Somatosensory Cortex with 3D Electron Microscopy. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Santuy, A.; Rodriguez, J.R.; DeFelipe, J.; Merchan-Perez, A. Volume electron microscopy of the distribution of synapses in the neuropil of the juvenile rat somatosensory cortex. Brain Struct. Funct. 2018, 223, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Tomm, C.; Floyd Sarria, J.C.; Petersen, C.C. The excitatory neuronal network of the C2 barrel column in mouse primary somatosensory cortex. Neuron 2009, 61, 301–316. [Google Scholar] [CrossRef]
- Song, S.; Sjostrom, P.J.; Reigl, M.; Nelson, S.; Chklovskii, D.B. Highly nonrandom features of synaptic connectivity in local cortical circuits. PLoS Biol. 2005, 3, e68. [Google Scholar] [CrossRef]
- Mizuseki, K.; Buzsaki, G. Preconfigured, skewed distribution of firing rates in the hippocampus and entorhinal cortex. Cell Rep. 2013, 4, 1010–1021. [Google Scholar] [CrossRef]
- Buzsaki, G.; Mizuseki, K. The log-dynamic brain: How skewed distributions affect network operations. Nat. Rev. Neurosci. 2014, 15, 264–278. [Google Scholar] [CrossRef]
- Montes, J.; Pena, J.M.; DeFelipe, J.; Herreras, O.; Merchan-Perez, A. The influence of synaptic size on AMPA receptor activation: A Monte Carlo model. PLoS ONE 2015, 10, e0130924. [Google Scholar] [CrossRef] [PubMed]
- Franks, K.M.; Bartol, T.M., Jr.; Sejnowski, T.J. A Monte Carlo model reveals independent signaling at central glutamatergic synapses. Biophys. J. 2002, 83, 2333–2348. [Google Scholar] [CrossRef]
- Gulledge, A.T.; Carnevale, N.T.; Stuart, G.J. Electrical advantages of dendritic spines. PLoS ONE 2012, 7, e36007. [Google Scholar] [CrossRef] [PubMed]
- Araya, R. Input transformation by dendritic spines of pyramidal neurons. Front. Neuroanat. 2014, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Major, G.; Larkum, M.E.; Schiller, J. Active properties of neocortical pyramidal neuron dendrites. Annu. Rev. Neurosci. 2013, 36, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Eyal, G.; Mansvelder, H.D.; de Kock, C.P.; Segev, I. Dendrites impact the encoding capabilities of the axon. J. Neurosci. 2014, 34, 8063–8071. [Google Scholar] [CrossRef]
- Kharazia, V.N.; Weinberg, R.J. Immunogold localization of AMPA and NMDA receptors in somatic sensory cortex of albino rat. J. Comp. Neurol. 1999, 412, 292–302. [Google Scholar] [CrossRef]
- Nusser, Z.; Lujan, R.; Laube, G.; Roberts, J.D.; Molnar, E.; Somogyi, P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 1998, 21, 545–559. [Google Scholar] [CrossRef]
- Racca, C.; Stephenson, F.A.; Streit, P.; Roberts, J.D.; Somogyi, P. NMDA receptor content of synapses in stratum radiatum of the hippocampal CA1 area. J. Neurosci. 2000, 20, 2512–2522. [Google Scholar] [CrossRef]
- Calverley, R.K.; Jones, D.G. Determination of the numerical density of perforated synapses in rat neocortex. Cell Tissue Res. 1987, 248, 399–407. [Google Scholar] [CrossRef]
- Geinisman, Y.; Morrell, F.; de Toledo-Morrell, L. Axospinous synapses with segmented postsynaptic densities: A morphologically distinct synaptic subtype contributing to the number of profiles of ‘perforated’ synapses visualized in random sections. Brain Res. 1987, 423, 179–188. [Google Scholar] [CrossRef]
- Harris, K.M.; Jensen, F.E.; Tsao, B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: Implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci. 1992, 12, 2685–2705. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.G.; Calverley, R.K. Perforated and non-perforated synapses in rat neocortex: Three-dimensional reconstructions. Brain Res. 1991, 556, 247–258. [Google Scholar] [CrossRef]
- Geinisman, Y.; deToledo-Morrell, L.; Morrell, F.; Heller, R.E.; Rossi, M.; Parshall, R.F. Structural synaptic correlate of long-term potentiation: Formation of axospinous synapses with multiple, completely partitioned transmission zones. Hippocampus 1993, 3, 435–445. [Google Scholar] [CrossRef]
- Toni, N.; Buchs, P.A.; Nikonenko, I.; Povilaitite, P.; Parisi, L.; Muller, D. Remodeling of synaptic membranes after induction of long-term potentiation. J. Neurosci. 2001, 21, 6245–6251. [Google Scholar] [CrossRef]
- Ganeshina, O.; Berry, R.W.; Petralia, R.S.; Nicholson, D.A.; Geinisman, Y. Synapses with a segmented, completely partitioned postsynaptic density express more AMPA receptors than other axospinous synaptic junctions. Neuroscience 2004, 125, 615–623. [Google Scholar] [CrossRef]
- Dyson, S.E.; Jones, D.G. Quantitation of terminal parameters and their inter-relationships in maturing central synapses: A perspective for experimental studies. Brain Res. 1980, 183, 43–59. [Google Scholar] [CrossRef]
- Jones, D.G.; Devon, R.M. An ultrastructural study into the effects of pentobarbitone on synaptic organization. Brain Res. 1978, 147, 47–63. [Google Scholar] [CrossRef]
- Oorschot, D.E.; Jones, D.G. A quantitative ultrastructural study of the effects of phenylacetate on synaptic organization in the developing rat cerebral cortex. Dev. Neurosci. 1983, 6, 45–57. [Google Scholar] [CrossRef]
- Calverley, R.K.; Jones, D.G. A serial-section study of perforated synapses in rat neocortex. Cell Tissue Res. 1987, 247, 565–572. [Google Scholar] [CrossRef]
- Jones, D.G.; Dyson, S.E. The influence of protein restriction, rehabilitation and changing nutritional status on synaptic development: A quantitative study in rat brain. Brain Res. 1981, 208, 97–111. [Google Scholar] [CrossRef]
- Medvedev, N.I.; Popov, V.I.; Dallerac, G.; Davies, H.A.; Laroche, S.; Kraev, I.V.; Rodriguez Arellano, J.J.; Doyere, V.; Stewart, M.G. Alterations in synaptic curvature in the dentate gyrus following induction of long-term potentiation, long-term depression, and treatment with the N-methyl-d-aspartate receptor antagonist CPP. Neuroscience 2010, 171, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Marrone, D.F.; Petit, T.L. The role of synaptic morphology in neural plasticity: Structural interactions underlying synaptic power. Brain Res. Brain Res. Rev. 2002, 38, 291–308. [Google Scholar] [CrossRef]
- Tao-Cheng, J.H. Stimulation induces gradual increases in the thickness and curvature of postsynaptic density of hippocampal CA1 neurons in slice cultures. Mol. Brain 2019, 12, 44. [Google Scholar] [CrossRef]
- Alonso-Nanclares, L.; Gonzalez-Soriano, J.; Rodriguez, J.R.; DeFelipe, J. Gender differences in human cortical synaptic density. Proc. Natl. Acad. Sci. USA 2008, 105, 14615–14619. [Google Scholar] [CrossRef]
- Beaulieu, C.; Colonnier, M. A laminar analysis of the number of round-asymmetrical and flat-symmetrical synapses on spines, dendritic trunks, and cell bodies in area 17 of the cat. J. Comp. Neurol. 1985, 231, 180–189. [Google Scholar] [CrossRef]
- Micheva, K.D.; Beaulieu, C. Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J. Comp. Neurol. 1996, 373, 340–354. [Google Scholar] [CrossRef]
- Bosch, C.; Martinez, A.; Masachs, N.; Teixeira, C.M.; Fernaud, I.; Ulloa, F.; Perez-Martinez, E.; Lois, C.; Comella, J.X.; DeFelipe, J.; et al. FIB/SEM technology and high-throughput 3D reconstruction of dendritic spines and synapses in GFP-labeled adult-generated neurons. Front. Neuroanat. 2015, 9, 60. [Google Scholar] [CrossRef]
- Jones, E.G.; Powell, T.P. Morphological variations in the dendritic spines of the neocortex. J. Cell Sci. 1969, 5, 509–529. [Google Scholar]
- Petrak, L.J.; Harris, K.M.; Kirov, S.A. Synaptogenesis on mature hippocampal dendrites occurs via filopodia and immature spines during blocked synaptic transmission. J. Comp. Neurol. 2005, 484, 183–190. [Google Scholar] [CrossRef]
- Popov, V.I.; Deev, A.A.; Klimenko, O.A.; Kraev l, V.; Kuz’minykh, S.B.; Medvedev, N.I.; Patrushev, I.V.; Popov, R.V.; Rogachevskii, V.V.; Khutsiyan, S.S.; et al. Three-dimensional reconstruction of synapses and dendritic spines in the rat and ground squirrel hippocampus: New structural-functional paradigms for synaptic function. Neurosci. Behav. Physiol. 2005, 35, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.W.; Holtmaat, A.; Wilbrecht, L.; Welker, E.; Svoboda, K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat. Neurosci. 2006, 9, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Radwanska, K.; Medvedev, N.I.; Pereira, G.S.; Engmann, O.; Thiede, N.; Moraes, M.F.; Villers, A.; Irvine, E.E.; Maunganidze, N.S.; Pyza, E.M.; et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proc. Natl. Acad. Sci. USA 2011, 108, 18471–18475. [Google Scholar] [CrossRef]
- Giese, K.P.; Aziz, W.; Kraev, I.; Stewart, M.G. Generation of multi-innervated dendritic spines as a novel mechanism of long-term memory formation. Neurobiol. Learn. Mem. 2015, 124, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Toni, N.; Buchs, P.A.; Nikonenko, I.; Bron, C.R.; Muller, D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 1999, 402, 421–425. [Google Scholar] [CrossRef]
- Kozorovitskiy, Y.; Gross, C.G.; Kopil, C.; Battaglia, L.; McBreen, M.; Stranahan, A.M.; Gould, E. Experience induces structural and biochemical changes in the adult primate brain. Proc. Natl. Acad. Sci. USA 2005, 102, 17478–17482. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.B.; Trommald, M.; Andersen, P. An increase in dendritic spine density on hippocampal CA1 pyramidal cells following spatial learning in adult rats suggests the formation of new synapses. Proc. Natl. Acad. Sci. USA 1994, 91, 12673–12675. [Google Scholar] [CrossRef]
- Brockett, A.T.; LaMarca, E.A.; Gould, E. Physical exercise enhances cognitive flexibility as well as astrocytic and synaptic markers in the medial prefrontal cortex. PLoS ONE 2015, 10, e0124859. [Google Scholar] [CrossRef]
- Leuner, B.; Falduto, J.; Shors, T.J. Associative memory formation increases the observation of dendritic spines in the hippocampus. J. Neurosci. 2003, 23, 659–665. [Google Scholar] [CrossRef]
- Patel, S.N.; Stewart, M.G. Changes in the number and structure of dendritic spines 25 hours after passive avoidance training in the domestic chick, Gallus domesticus. Brain Res. 1988, 449, 34–46. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef]
- MacAskill, A.F.; Atkin, T.A.; Kittler, J.T. Mitochondrial trafficking and the provision of energy and calcium buffering at excitatory synapses. Eur. J. Neurosci. 2010, 32, 231–240. [Google Scholar] [CrossRef]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef]
- Rowland, K.C.; Irby, N.K.; Spirou, G.A. Specialized synapse-associated structures within the calyx of Held. J. Neurosci. 2000, 20, 9135–9144. [Google Scholar] [CrossRef]
- Cai, Q.; Sheng, Z.H. Mitochondrial transport and docking in axons. Exp. Neurol. 2009, 218, 257–267. [Google Scholar] [CrossRef]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Chang, D.T.; Honick, A.S.; Reynolds, I.J. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 2006, 26, 7035–7045. [Google Scholar] [CrossRef]
- Obashi, K.; Okabe, S. Regulation of mitochondrial dynamics and distribution by synapse position and neuronal activity in the axon. Eur. J. Neurosci. 2013, 38, 2350–2363. [Google Scholar] [CrossRef]
- MacAskill, A.F.; Kittler, J.T. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010, 20, 102–112. [Google Scholar] [CrossRef]
- Takihara, Y.; Inatani, M.; Eto, K.; Inoue, T.; Kreymerman, A.; Miyake, S.; Ueno, S.; Nagaya, M.; Nakanishi, A.; Iwao, K.; et al. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc. Natl. Acad. Sci. USA 2015, 112, 10515–10520. [Google Scholar] [CrossRef]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect Biol. 2013, 5. [Google Scholar] [CrossRef]
- Santuy, A.; Turegano-Lopez, M.; Rodriguez, J.R.; Alonso-Nanclares, L.; DeFelipe, J.; Merchan-Perez, A. A Quantitative Study on the Distribution of Mitochondria in the Neuropil of the Juvenile Rat Somatosensory Cortex. Cereb. Cortex 2018, 28, 3673–3684. [Google Scholar] [CrossRef]
- Rodriguez-Moreno, J.; Rollenhagen, A.; Arlandis, J.; Santuy, A.; Merchan-Perez, A.; DeFelipe, J.; Lubke, J.H.R.; Clasca, F. Quantitative 3D Ultrastructure of Thalamocortical Synapses from the “Lemniscal” Ventral Posteromedial Nucleus in Mouse Barrel Cortex. Cereb. Cortex 2018, 28, 3159–3175. [Google Scholar] [CrossRef]
- Rintoul, G.L.; Filiano, A.J.; Brocard, J.B.; Kress, G.J.; Reynolds, I.J. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J. Neurosci. 2003, 23, 7881–7888. [Google Scholar] [CrossRef]
- David, G.; Barrett, E.F. Stimulation-evoked increases in cytosolic [Ca2+] in mouse motor nerve terminals are limited by mitochondrial uptake and are temperature-dependent. J. Neurosci. 2000, 20, 7290–7296. [Google Scholar] [CrossRef]
- David, G.; Barrett, E.F. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J. Physiol. 2003, 548, 425–438. [Google Scholar] [CrossRef]
- Li, H.; Chen, Y.; Jones, A.F.; Sanger, R.H.; Collis, L.P.; Flannery, R.; McNay, E.C.; Yu, T.; Schwarzenbacher, R.; Bossy, B.; et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 2169–2174. [Google Scholar] [CrossRef]
- Roberts, R.C.; McCollum, L.A.; Schoonover, K.E.; Mabry, S.J.; Roche, J.K.; Lahti, A.C. Ultrastructural evidence for glutamatergic dysregulation in schizophrenia. Schizophr. Res. 2020. [Google Scholar] [CrossRef]
- Koski, L.; Petrides, M. Time-related changes in task performance after lesions restricted to the frontal cortex. Neuropsychologia 2001, 39, 268–281. [Google Scholar] [CrossRef]
- Barch, D.M.; Braver, T.S.; Akbudak, E.; Conturo, T.; Ollinger, J.; Snyder, A. Anterior cingulate cortex and response conflict: Effects of response modality and processing domain. Cereb. Cortex 2001, 11, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Kerns, J.G.; Cohen, J.D.; MacDonald, A.W., 3rd; Cho, R.Y.; Stenger, V.A.; Carter, C.S. Anterior cingulate conflict monitoring and adjustments in control. Science 2004, 303, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Braver, T.S.; Barch, D.M.; Gray, J.R.; Molfese, D.L.; Snyder, A. Anterior cingulate cortex and response conflict: Effects of frequency, inhibition and errors. Cereb. Cortex 2001, 11, 825–836. [Google Scholar] [CrossRef]
- Mathalon, D.H.; Fedor, M.; Faustman, W.O.; Gray, M.; Askari, N.; Ford, J.M. Response-monitoring dysfunction in schizophrenia: An event-related brain potential study. J. Abnorm. Psychol. 2002, 111, 22–41. [Google Scholar] [CrossRef]
- Bush, G.; Luu, P.; Posner, M.I. Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn. Sci. 2000, 4, 215–222. [Google Scholar] [CrossRef]
- Sowell, E.R.; Thompson, P.M.; Toga, A.W. Mapping Changes in the Human Cortex throughout the Span of Life. Neuroscientist 2004, 10, 372–392. [Google Scholar] [CrossRef]
- Eltokhi, A.; Janmaat, I.E.; Genedi, M.; Haarman, B.C.M.; Sommer, I.E.C. Dysregulation of synaptic pruning as a possible link between intestinal microbiota dysbiosis and neuropsychiatric disorders. J. Neurosci. Res. 2020, 98, 1335–1369. [Google Scholar] [CrossRef]
- Graybiel, A.M.; Ragsdale, C.W., Jr. Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc. Natl. Acad. Sci. USA 1978, 75, 5723–5726. [Google Scholar] [CrossRef]
- Flaherty, A.W.; Graybiel, A.M. Two input systems for body representations in the primate striatal matrix: Experimental evidence in the squirrel monkey. J. Neurosci. 1993, 13, 1120. [Google Scholar] [CrossRef]
- Eblen, F.; Graybiel, A.M. Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J. Neurosci. 1995, 15, 5999–6013. [Google Scholar] [CrossRef] [PubMed]
- Goldman-Rakic, P.S. The cortical dopamine system: Role in memory and cognition. Adv. Pharmacol. 1998, 42, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Groenewegen, H.J.; Trimble, M. The Ventral Striatum as an Interface Between the Limbic and Motor Systems. CNS Spectrums 2007, 12, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Rosen, A.M.; Spellman, T.; Gordon, J.A. Electrophysiological endophenotypes in rodent models of schizophrenia and psychosis. Biol. Psychiatry 2015, 77, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Peykov, S.; Berkel, S.; Schoen, M.; Weiss, K.; Degenhardt, F.; Strohmaier, J.; Weiss, B.; Proepper, C.; Schratt, G.; Nöthen, M.M.; et al. Identification and functional characterization of rare SHANK2 variants in schizophrenia. Mol. Psychiatry 2015, 20, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Uppal, N.; Puri, R.; Yuk, F.; Janssen, W.G.; Bozdagi-Gunal, O.; Harony-Nicolas, H.; Dickstein, D.L.; Buxbaum, J.D.; Hof, P.R. Ultrastructural analyses in the hippocampus CA1 field in Shank3-deficient mice. Mol. Autism 2015, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Lie, E.; Li, Y.; Kim, R.; Kim, E. SALM/Lrfn Family Synaptic Adhesion Molecules. Front. Mol. Neurosci. 2018, 11, 105. [Google Scholar] [CrossRef]
- Shih, P.Y.; Hsieh, B.Y.; Tsai, C.Y.; Lo, C.A.; Chen, B.E.; Hsueh, Y.P. Autism-linked mutations of CTTNBP2 reduce social interaction and impair dendritic spine formation via diverse mechanisms. Acta Neuropathol. Commun. 2020, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- Lipina, T.V.; Prasad, T.; Yokomaku, D.; Luo, L.; Connor, S.A.; Kawabe, H.; Wang, Y.T.; Brose, N.; Roder, J.C.; Craig, A.M. Cognitive Deficits in Calsyntenin-2-deficient Mice Associated with Reduced GABAergic Transmission. Neuropsychopharmacology 2016, 41, 802–810. [Google Scholar] [CrossRef]
- Giza, J.; Urbanski, M.J.; Prestori, F.; Bandyopadhyay, B.; Yam, A.; Friedrich, V.; Kelley, K.; D’Angelo, E.; Goldfarb, M. Behavioral and cerebellar transmission deficits in mice lacking the autism-linked gene islet brain-2. J. Neurosci. 2010, 30, 14805–14816. [Google Scholar] [CrossRef]
- Soda, T.; Mapelli, L.; Locatelli, F.; Botta, L.; Goldfarb, M.; Prestori, F.; D’Angelo, E. Hyperexcitability and Hyperplasticity Disrupt Cerebellar Signal Transfer in the IB2 KO Mouse Model of Autism. J. Neurosci. 2019, 39, 2383–2397. [Google Scholar] [CrossRef] [PubMed]
- Li, V.; Wang, Y.T. Molecular mechanisms of NMDA receptor-mediated excitotoxicity: Implications for neuroprotective therapeutics for stroke. Neural. Regen. Res. 2016, 11, 1752–1753. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Monteiro, P.; Zhou, Y.; Kim, J.A.; Gao, X.; Fu, Z.; Feng, G. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 2016, 530, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Reisel, D.; Bannerman, D.M.; Schmitt, W.B.; Deacon, R.M.; Flint, J.; Borchardt, T.; Seeburg, P.H.; Rawlins, J.N. Spatial memory dissociations in mice lacking GluR1. Nat. Neurosci. 2002, 5, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Bannerman, D.M.; Sprengel, R.; Sanderson, D.J.; McHugh, S.B.; Rawlins, J.N.; Monyer, H.; Seeburg, P.H. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 2014, 15, 181–192. [Google Scholar] [CrossRef]


{kind=link}
| Schizophrenia | |
|---|---|
| Humans | |
| Brain Region | Ultrastructure Modifications |
| Anterior cingulate cortex | ↓ density of axospinous synapses and axonal mitochondria [123]. |
| Anterior limbic cortex | ↑ density of axospinous and convex synapses; ↓ density of synapses on shafts, flat and concave synapses [124]. |
| Hippocampus CA3 | ↓ density of axospinous synapses [125,126]. |
| Caudate and putamen | ↑ density of in axospinous synapses the caudate matrix and putamen patches [127,128,129]. |
| Nucleus accumbens | ↑ density and ↓ size of axospinous synapses in the core [130]. |
| Rodents | |
| Rodent Models | Ultrastructure Modifications |
| Ketamine | In posterior cingulate cortex, ↑ thickness and curvature of the synaptic interface; ↑ synaptic cleft width [131]. |
| Dtnbp1−/− | In CA1, ↑ vesicle size and thickness of PSD; ↓ vesicles of reserve pool and width of synaptic cleft [132]. |
| Autism Spectrum Disorder | |
|---|---|
| Humans | |
| Brain Region | Ultrastructure Modifications |
| Anterior cingulate cortex | ↑ large axons in deep white matter, ↑ small axons in superficial white matter [133]. |
| Rodents | |
| Rodent Model | Ultrastructure Modifications |
| VPA | In Cx and HC, blurred and thickened synaptic cleft; ↓ synaptic vesicles; altered mitochondrial morphology [134]. |
| Propionic acid | In CA1, few atypically enlarged presynaptic terminals; ↓ density of synaptic vesicles and short active zone [135]. |
| 15q11-13 duplication | In somatosensory Cx, ↓size of PSD, spine head volume ↓width, spine neck width ↓density of shaft synapses and mushroom spines; ↑ density of axospinous synapses and filopodial spines [136]. |
| Shank1−/− | In CA1, ↓smaller spines and thinner PSD [137]. |
| Shank3B−/− | In striatum, ↓ thickness and length of PSD and spine density [138]. |
| Lrfn2−/− | In CA1, ↑perforated synapses and synaptic cleft width; ↓PSD length; oddly shaped and spinule-like spines [139]. |
| Cttnbp2−/− | In dorsal HC, ↓PSD length and thickness and synaptic vesicle count [140]. |
| Dip2a−/− | In Cx, a stubby postsynaptic structure and flattened PSD; defect in spine morphology [141]. |
| Clstn2−/− | In MPFC, ↑ density of inhibitory synapses, ↑ negative curved PSD. In ADD, ↑ size of perforated PSD; ↑ density of synaptic vesicles. In HC, ↓ density of synaptic vesicles [142]. |
| Vrk3−/− | In CA1, ↓ PSD length and thickness [143]. |
| Fmr1−/− | For a review reporting the spine phenotypes in different brain regions, see [144]. In CA1, ↑ diameter of secondary dendrites and dendritic spine density; ↑ mature dendritic spines and ↑ mature postsynaptic densities [145]. In the primary motor Cx, normal density but ↑ turnover rate of dendritic spines [146]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eltokhi, A.; Santuy, A.; Merchan-Perez, A.; Sprengel, R. Glutamatergic Dysfunction and Synaptic Ultrastructural Alterations in Schizophrenia and Autism Spectrum Disorder: Evidence from Human and Rodent Studies. Int. J. Mol. Sci. 2021, 22, 59. https://doi.org/10.3390/ijms22010059
Eltokhi A, Santuy A, Merchan-Perez A, Sprengel R. Glutamatergic Dysfunction and Synaptic Ultrastructural Alterations in Schizophrenia and Autism Spectrum Disorder: Evidence from Human and Rodent Studies. International Journal of Molecular Sciences. 2021; 22(1):59. https://doi.org/10.3390/ijms22010059
Chicago/Turabian StyleEltokhi, Ahmed, Andrea Santuy, Angel Merchan-Perez, and Rolf Sprengel. 2021. "Glutamatergic Dysfunction and Synaptic Ultrastructural Alterations in Schizophrenia and Autism Spectrum Disorder: Evidence from Human and Rodent Studies" International Journal of Molecular Sciences 22, no. 1: 59. https://doi.org/10.3390/ijms22010059
APA StyleEltokhi, A., Santuy, A., Merchan-Perez, A., & Sprengel, R. (2021). Glutamatergic Dysfunction and Synaptic Ultrastructural Alterations in Schizophrenia and Autism Spectrum Disorder: Evidence from Human and Rodent Studies. International Journal of Molecular Sciences, 22(1), 59. https://doi.org/10.3390/ijms22010059