The Aryl Hydrocarbon Receptor in Energy Balance: The Road from Dioxin-Induced Wasting Syndrome to Combating Obesity with Ahr Ligands


{kind=link}
Abstract
1. Introduction
2. Part I: TCDD, AHR, and the Wasting Syndrome
3. Part II: AHR Transgenic Mouse Studies and the Age of Endogenous AHR Biology
4. Part III: Antagonist Theory: AHR Inhibition as a Means for Treating Obesity
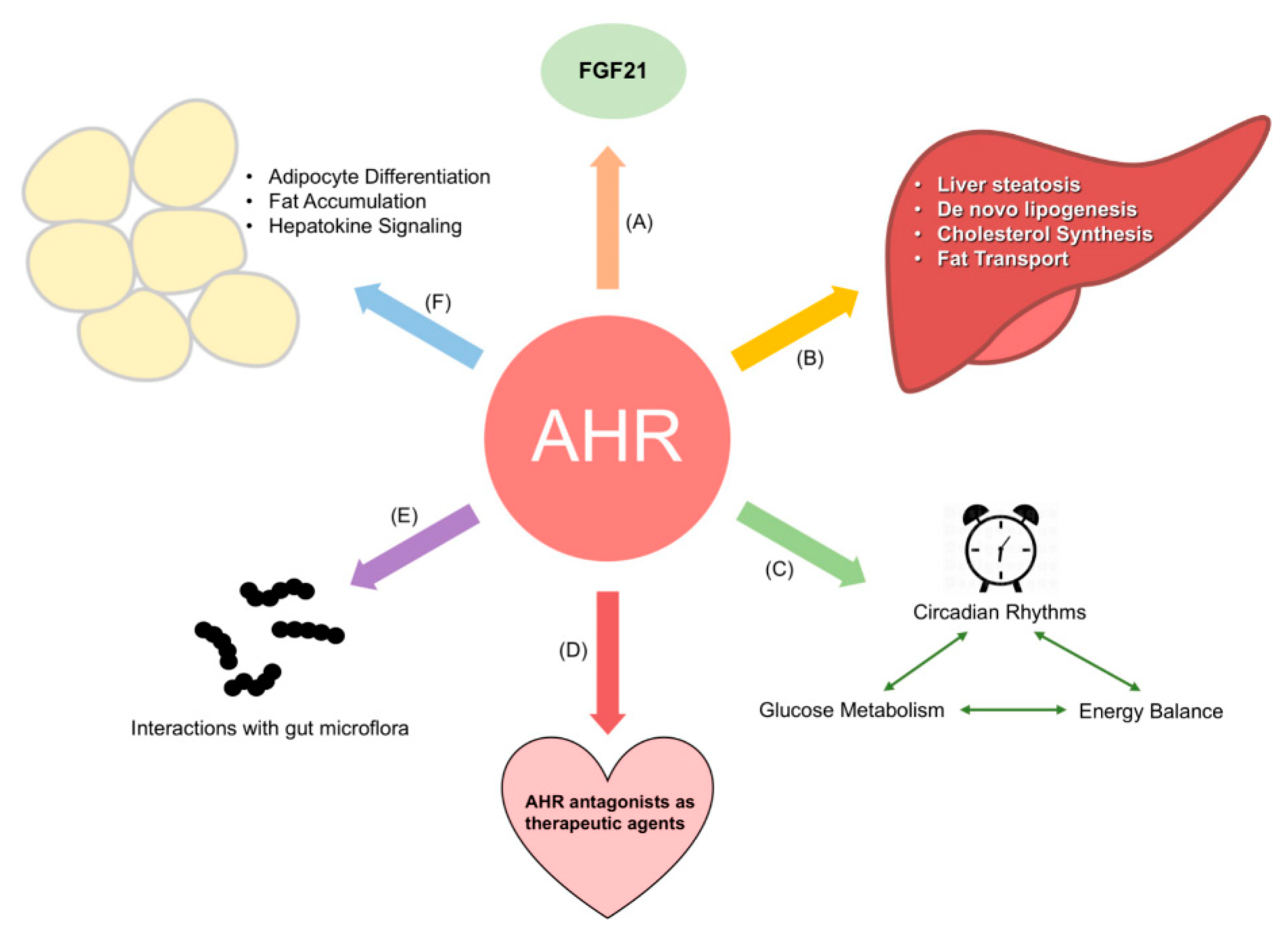
5. Part IV: AHR Regulation of Energy Metabolism: Current and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2,4,5-T | 2,4,5-trichlorophenol |
| ACACA | Acetyl-CoA carboxylase alpha |
| AHR | Aryl hydrocarbon receptor |
| αNF | Alpha-naphthoflavone |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| CA | Constitutively-active |
| CYP1A1 | Cytochrome P450 family 1 subfamily A member 1 |
| FAS | Fatty acid synthase |
| FGF21 | Fibroblast growth factor 21 |
| I3C | Indole-3-carbinol |
| IDO1 | Indoleamine-2,3-deoxygenase 1 |
| KLF6 | Kruppel-like Factor 6 |
| Kyn | Kynurenine |
| PPAR | Peroxisome proliferator-activated receptor |
| SCD1 | Stearoyl-CoA decarboxylase 1 |
| SPP1 | Secreted Phosphoprotein 1 |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
References
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, High Affinity Binding of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin by Hepatic Cytosol: Evidence That the Binding Species Is Receptor for Induction of Aryl Hydrocarbon Hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar] [PubMed]
- Beischlag, T.V.; Morales, J.L.; Hollingshead, B.D.; Perdew, G.H. The Aryl Hydrocarbon Receptor Complex and the Control of Gene Expression. Crit. Rev. Eukaryot. Gene Exp. 2008, 18, 207–250. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The Tumor Suppressor Kruppel-like Factor 6 Is a Novel Aryl Hydrocarbon Receptor DNA Binding Partner. J. Pharm. Exp. 2013, 345, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Fujii-Kuriyama, Y.; Kato, S. AhR Acts as an E3 Ubiquitin Ligase to Modulate Steroid Receptor Functions. Biochem. Pharmacol. 2009, 77, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.P.; Smith, D.; Metter, G.; Possick, P. A Health Survey of Workers in a 2,4-D and 2,4,5-T Plant with Special Attention to Chloracne, Porphyria Cutanea Tarda, and Psychologic Parameters. J. Occup. Environ. Med. 1971, 13, 499–500. [Google Scholar]
- Schulz, K.H. Clinical & experimental studies on the etiology of chloracne. Arch. Klin. Exp. Derm. 1957, 206, 589–596. [Google Scholar]
- Kimmig, J.; Schulz, K.H. Occupational acne (so-called chloracne) due to chlorinated aromatic cyclic ethers. Dermatologica 1957, 115, 540–546. [Google Scholar] [CrossRef]
- Poland, A.; Glover, E. Chlorinated Dibenzo-p-Dioxins: Potent Inducers of Delta-Aminolevulinic Acid Synthetase and Aryl Hydrocarbon Hydroxylase. II. A Study of the Structure-Activity Relationship. Mol. Pharm. 1973, 9, 736–747. [Google Scholar]
- Poland, A.; Glover, E. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: A Potent Inducer of delta-Aminolevulinic Acid Synthetase. Science 1973, 179, 476–477. [Google Scholar] [CrossRef]
- Cunningham, H.M.; Williams, D.T. Effect of Tetrachlorodibenzo-p-Dioxin on Growth Rate and the Synthesis of Lipids and Proteins in Rats. Bull. Environ. Contam. Toxicol. 1972, 7, 45–51. [Google Scholar] [CrossRef]
- Harris, M.W.; Moore, J.A.; Vos, J.G.; Gupta, B.N. General Biological Effects of TCDD in Laboratory Animals. Environ. Health Perspect. 1973, 5, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Putnam, J.P.; Andrews, J.E. Metabolic Studies with TCDD (Dioxin) Treated Rats. Arch. Environ. Contam. Toxicol. 1978, 7, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.N.; Vos, J.G.; Moore, J.A.; Zinkl, J.G.; Bullock, B.C. Pathologic Effects of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Laboratory Animals. Environ. Health Perspect. 1973, 5, 125–140. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.E.; Moore, J.A.; Dalgard, D.W. Toxicity of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Rhesus Monkeys (Macaca Mulatta) Following a Single Oral Dose. Toxicol. Appl. Pharmacol. 1978, 43, 175–187. [Google Scholar] [CrossRef]
- Albro, P.W.; Corbett, J.T.; Harris, M.; Lawson, L.D. Effects of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on Lipid Profiles in Tissue of the Fischer Rat. Chem.-Biol. Interact. 1978, 23, 315–330. [Google Scholar] [CrossRef]
- Seefeld, M.D.; Keesey, R.E.; Peterson, R.E. Body Weight Regulation in Rats Treated with 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Toxicol. Appl. Pharmacol. 1984, 76, 526–536. [Google Scholar] [CrossRef]
- Seefeld, M.D.; Corbett, S.W.; Keesey, R.E.; Peterson, R.E. Characterization of the Wasting Syndrome in Rats Treated with 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Toxicol. Appl. Pharmacol. 1984, 73, 311–322. [Google Scholar] [CrossRef]
- Lakshman, M.R.; Chirtel, S.J.; Chambers, L.L.; Coutlakis, P.J. Effects of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on Lipid Synthesis and Lipogenic Enzymes in the Rat. J. Pharm. Exp. 1989, 248, 62–66. [Google Scholar] [CrossRef]
- Lakshman, M.R.; Campbell, B.S.; Chirtel, S.J.; Ekarohita, N. Effects of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) on de Novo Fatty Acid and Cholesterol Synthesis in the Rat. Lipids 1988, 23, 904–906. [Google Scholar] [CrossRef]
- Weber, L.W.D.; Lebofsky, M.; Stahl, B.U.; Gorski, J.R.; Muzi, G.; Rozman, K. Reduced Activities of Key Enzymes of Gluconeogenesis as Possible Cause of Acute Toxicity of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) in Rats. Toxicology 1991, 66, 133–144. [Google Scholar] [CrossRef]
- Alexander, D.L.; Ganem, L.G.; Fernandez-Salguero, P.; Gonzalez, F.; Jefcoate, C.R. Aryl-Hydrocarbon Receptor Is an Inhibitory Regulator of Lipid Synthesis and of Commitment to Adipogenesis. J. Cell. Sci. 1998, 111 Pt 22, 3311–3322. [Google Scholar]
- Brodie, A.E.; Azarenko, V.A.; Hu, C.Y. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Inhibition of Fat Cell Differentiation. Toxicol. Lett. 1996, 84, 55–59. [Google Scholar] [CrossRef]
- Potter, C.L.; Menahan, L.A.; Peterson, R.E. Relationship of Alterations in Energy Metabolism to Hypophagia in Rats Treated with 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Fundam. Appl. Toxicol. 1986, 6, 89–97. [Google Scholar] [CrossRef]
- Cho, T.E.; Bott, D.; Ahmed, S.; Hutin, D.; Gomez, A.; Tamblyn, L.; Zhou, A.C.; Watts, T.H.; Grant, D.M.; Matthews, J. 3-Methylcholanthrene Induces Chylous Ascites in TCDD-Inducible Poly-ADP-Ribose Polymerase (Tiparp) Knockout Mice. Int. J. Mol. Sci. 2019, 20, 2312. [Google Scholar] [CrossRef] [PubMed]
- Hutin, D.; Tamblyn, L.; Gomez, A.; Grimaldi, G.; Soedling, H.; Cho, T.; Ahmed, S.; Lucas, C.; Kanduri, C.; Grant, D.M.; et al. Hepatocyte-Specific Deletion of TIPARP, a Negative Regulator of the Aryl Hydrocarbon Receptor, Is Sufficient to Increase Sensitivity to Dioxin-Induced Wasting Syndrome. Toxicol. Sci. 2018, 165, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Fingerhut, M.A.; Halperin, W.E.; Marlow, D.A.; Piacitelli, L.A.; Honchar, P.A.; Sweeney, M.H.; Greife, A.L.; Dill, P.A.; Steenland, K.; Suruda, A.J. Cancer Mortality in Workers Exposed to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. N. Engl. J. Med. 1991, 324, 212–218. [Google Scholar] [CrossRef]
- Ott, M.G.; Zober, A. Cause Specific Mortality and Cancer Incidence among Employees Exposed to 2,3,7,8-TCDD after a 1953 Reactor Accident. Occup. Environ. Med. 1996, 53, 606–612. [Google Scholar] [CrossRef]
- Flesch-Janys, D.; Berger, J.; Gurn, P.; Manz, A.; Nagel, S.; Waltsgott, H.; Dwyer, J.H. Exposure to Polychlorinated Dioxins and Furans (PCDD/F) and Mortality in a Cohort of Workers from a Herbicide-Producing Plant in Hamburg, Federal Republic of Germany. Am. J. Epidemiol. 1995, 142, 1165–1175. [Google Scholar] [CrossRef]
- Tritscher, A.M.; Seacat, A.M.; Yager, J.D.; Groopman, J.D.; Miller, B.D.; Bell, D.; Sutter, T.R.; Lucier, G.W. Increased Oxidative DNA Damage in Livers of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Treated Intact but Not Ovariectomized Rats. Cancer Lett. 1996, 98, 219–225. [Google Scholar] [CrossRef]
- Henriksen, G.L.; Ketchum, N.S.; Michalek, J.E.; Swaby, J.A. Serum Dioxin and Diabetes Mellitus in Veterans of Operation Ranch Hand. Epidemiology 1997, 8, 252–258. [Google Scholar] [CrossRef]
- Calvert, G.M.; Sweeney, M.H.; Deddens, J.; Wall, D.K. Evaluation of Diabetes Mellitus, Serum Glucose, and Thyroid Function among United States Workers Exposed to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Occup. Environ. Med. 1999, 56, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Cranmer, M.; Louie, S.; Kennedy, R.H.; Kern, P.A.; Fonseca, V.A. Exposure to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Is Associated with Hyperinsulinemia and Insulin Resistance. Toxicol. Sci. 2000, 56, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Steenland, K.; Piacitelli, L.; Deddens, J.; Fingerhut, M.; Chang, L.I. Cancer, Heart Disease, and Diabetes in Workers Exposed to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. J. Natl. Cancer Inst. 1999, 91, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Pesatori, A.C.; Zocchetti, C.; Guercilena, S.; Consonni, D.; Turrini, D.; Bertazzi, P.A. Dioxin Exposure and Non-Malignant Health Effects: A Mortality Study. Occup. Environ. Med. 1998, 55, 126–131. [Google Scholar] [CrossRef]
- Vena, J.; Boffetta, P.; Becher, H.; Benn, T.; Bueno-de-Mesquita, H.B.; Coggon, D.; Colin, D.; Flesch-Janys, D.; Green, L.; Kauppinen, T.; et al. Exposure to Dioxin and Nonneoplastic Mortality in the Expanded IARC International Cohort Study of Phenoxy Herbicide and Chlorophenol Production Workers and Sprayers. Environ. Health Perspect. 1998, 106, 645–653. [Google Scholar]
- Hooiveld, M.; Heederik, D.J.; Kogevinas, M.; Boffetta, P.; Needham, L.L.; Patterson, D.G.; Bueno-de-Mesquita, H.B. Second Follow-up of a Dutch Cohort Occupationally Exposed to Phenoxy Herbicides, Chlorophenols, and Contaminants. Am. J. Epidemiol. 1998, 147, 891–901. [Google Scholar] [CrossRef]
- Sutter, T.R. Greenlee WF Classification of Members of the Ah Gene Battery. Chemosphere 1992, 25, 223–226. [Google Scholar] [CrossRef]
- Gaido, K.W.; Maness, S.C.; Leonard, L.S.; Greenlee, W.F. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin-Dependent Regulation of Transforming Growth Factors-Alpha and -Beta 2 Expression in a Human Keratinocyte Cell Line Involves Both Transcriptional and Post-Transcriptional Control. J. Biol. Chem. 1992, 267, 24591–24595. [Google Scholar]
- Hoffer, A.; Chang, C.-Y.; Puga, A. Dioxin Induces Transcription of Fos and Jun Genes by Ah Receptor-Dependent and -Independent Pathways. Toxicol. Appl. Pharmacol. 1996, 141, 238–247. [Google Scholar] [CrossRef]
- Puga, A.; Hoffer, A.; Zhou, S.; Bohm, J.M.; Leikauf, G.D.; Shertzer, H.G. Sustained Increase in Intracellular Free Calcium and Activation of Cyclooxygenase-2 Expression in Mouse Hepatoma Cells Treated with Dioxin. Biochem. Pharmacol. 1997, 54, 1287–1296. [Google Scholar] [CrossRef]
- Kraemer, S.A.; Arthur, K.A.; Denison, M.S.; Smith, W.L.; DeWitt, D.L. Regulation of Prostaglandin Endoperoxide H Synthase-2 Expression by 2,3,7,8,-Tetrachlorodibenzo-p-Dioxin. Arch. Biochem. Biophys. 1996, 330, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.; Maier, A.; Medvedovic, M. The Transcriptional Signature of Dioxin in Human Hepatoma HepG2 Cells. Biochem. Pharmacol. 2000, 60, 1129–1142. [Google Scholar] [CrossRef]
- Frueh, F.W.; Hayashibara, K.C.; Brown, P.O.; Whitlock, J.P. Use of CDNA Microarrays to Analyze Dioxin-Induced Changes in Human Liver Gene Expression. Toxicol. Lett. 2001, 122, 189–203. [Google Scholar] [CrossRef]
- Sato, S.; Shirakawa, H.; Tomita, S.; Ohsaki, Y.; Haketa, K.; Tooi, O.; Santo, N.; Tohkin, M.; Furukawa, Y.; Gonzalez, F.J.; et al. Low-Dose Dioxins Alter Gene Expression Related to Cholesterol Biosynthesis, Lipogenesis, and Glucose Metabolism through the Aryl Hydrocarbon Receptor-Mediated Pathway in Mouse Liver. Toxicol. Appl. Pharm. 2008, 229, 10–19. [Google Scholar] [CrossRef]
- Boverhof, D.R.; Burgoon, L.D.; Tashiro, C.; Sharratt, B.; Chittim, B.; Harkema, J.R.; Mendrick, D.L.; Zacharewski, T.R. Comparative Toxicogenomic Analysis of the Hepatotoxic Effects of TCDD in Sprague Dawley Rats and C57BL/6 Mice. Toxicol. Sci. 2006, 94, 398–416. [Google Scholar] [CrossRef]
- Fletcher, N.; Wahlström, D.; Lundberg, R.; Nilsson, C.B.; Nilsson, K.C.; Stockling, K.; Hellmold, H.; Håkansson, H. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Alters the MRNA Expression of Critical Genes Associated with Cholesterol Metabolism, Bile Acid Biosynthesis, and Bile Transport in Rat Liver: A Microarray Study. Toxicol. Appl. Pharmacol. 2005, 207, 1–24. [Google Scholar] [CrossRef]
- Kurachi, M.; Hashimoto, S.; Obata, A.; Nagai, S.; Nagahata, T.; Inadera, H.; Sone, H.; Tohyama, C.; Kaneko, S.; Kobayashi, K.; et al. Identification of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin-Responsive Genes in Mouse Liver by Serial Analysis of Gene Expression. Biochem. Biophys. Res. Commun. 2002, 292, 368–377. [Google Scholar] [CrossRef][Green Version]
- Bjeldanes, L.F.; Kim, J.Y.; Grose, K.R.; Bartholomew, J.C.; Bradfield, C.A. Aromatic Hydrocarbon Responsiveness-Receptor Agonists Generated from Indole-3-Carbinol in Vitro and in Vivo: Comparisons with 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Proc. Natl. Acad. Sci. USA 1991, 88, 9543–9547. [Google Scholar] [CrossRef]
- Loub, W.D.; Wattenberg, L.W.; Davis, D.W. Aryl Hydrocarbon Hydroxylase Induction in Rat Tissues by Naturally Occurring Indoles of Cruciferous Plants. J. Natl. Cancer Inst. 1975, 54, 985–988. [Google Scholar]
- Johansson, G.; Gillner, M.; Högberg, B.; Gustafsson, J.A. The TCDD Receptor in Rat Intestinal Mucosa and Its Possible Dietary Ligands. Nutr. Cancer 1982, 3, 134–144. [Google Scholar] [CrossRef]
- Bradlow, H.L.; Michnovicz, J.; Telang, N.T.; Osborne, M.P. Effects of Dietary Indole-3-Carbinol on Estradiol Metabolism and Spontaneous Mammary Tumors in Mice. Carcinogenesis 1991, 12, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.A.; Peterson, K.S.; Smith, H.J.; Gray, J.C.; Sullivan, D.K.; Mayo, M.S.; Crowell, J.A.; Hurwitz, A. A Phase I Study of Indole-3-Carbinol in Women: Tolerability and Effects. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Fares, F. The Anti-Carcinogenic Effect of Indole-3-Carbinol and 3, 3’-Diindolylmethane and Their Mechanism of Action. Med. Chem. 2014, S1. [Google Scholar] [CrossRef]
- Perdew, G.H.; Babbs, C.F. Production of Ah Receptor Ligands in Rat Fecal Suspensions Containing Tryptophan or Indole-3-Carbinol. Nutr. Cancer 1991, 16, 209–218. [Google Scholar] [CrossRef]
- Heath-Pagliuso, S.; Rogers, W.J.; Tullis, K.; Seidel, S.D.; Cenijn, P.H.; Brouwer, A.; Denison, M.S. Activation of the Ah Receptor by Tryptophan and Tryptophan Metabolites. Biochemistry 1998, 37, 11508–11515. [Google Scholar] [CrossRef]
- Murray, I.A.; Perdew, G.H. Ligand Activation of the Ah Receptor Contributes to Gastrointestinal Homeostasis. Curr. Opin. Toxicol. 2017, 2, 15–23. [Google Scholar] [CrossRef]
- Korecka, A.; Dona, A.; Lahiri, S.; Tett, A.J.; Al-Asmakh, M.; Braniste, V.; D’Arienzo, R.; Abbaspour, A.; Reichardt, N.; Fujii-Kuriyama, Y.; et al. Bidirectional Communication between the Aryl Hydrocarbon Receptor (AhR) and the Microbiome Tunes Host Metabolism. npj Biofilms Microbiomes 2016, 2, 1–10. [Google Scholar] [CrossRef]
- Fernandez-Salguero, P.; Pineau, T.; Hilbert, D.M.; McPhail, T.; Lee, S.S.; Kimura, S.; Nebert, D.W.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Immune System Impairment and Hepatic Fibrosis in Mice Lacking the Dioxin-Binding Ah Receptor. Science 1995, 268, 722–726. [Google Scholar] [CrossRef]
- Schmidt, J.V.; Su, G.H.-T.; Reddy, J.K.; Simon, M.C.; Bradfield, C.A. Characterization of a Murine Ahr Null Allele: Involvement of the Ah Receptor in Hepatic Growth and Development. Proc. Natl. Acad. Sci. USA 1996, 93, 6731–6736. [Google Scholar] [CrossRef]
- Mimura, J.; Yamashita, K.; Nakamura, K.; Morita, M.; Takagi, T.N.; Nakao, K.; Ema, M.; Sogawa, K.; Yasuda, M.; Katsuki, M.; et al. Loss of Teratogenic Response to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) in Mice Lacking the Ah (Dioxin) Receptor. Genes Cells 1997, 2, 645–654. [Google Scholar] [CrossRef]
- Walisser, J.A.; Bunger, M.K.; Glover, E.; Bradfield, C.A. Gestational Exposure of Ahr and Arnt Hypomorphs to Dioxin Rescues Vascular Development. Proc. Natl. Acad. Sci. USA 2004, 101, 16677–16682. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, A.; Atallah-Yunes, N.; Smith, F.C.; You, X.; Chase, S.E.; Silverstone, A.E.; Vikstrom, K.L. A Role for the Aryl Hydrocarbon Receptor in Cardiac Physiology and Function as Demonstrated by AhR Knockout Mice. Cardiovasc. Toxicol. 2003, 3, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Gonzalez, J.M.; Roman, A.C.; Cerezo-Guisado, M.I.; Rico-Leo, E.M.; Martin-Partido, G.; Fernandez-Salguero, P.M. Loss of Dioxin-Receptor Expression Accelerates Wound Healing in Vivo by a Mechanism Involving TGFbeta. J. Cell. Sci. 2009, 122, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Mimura, J.; Nakamura, N.; Harada, N.; Yamamoto, M.; Morohashi, K.-I.; Fujii-Kuriyama, Y. Intrinsic Function of the Aryl Hydrocarbon (Dioxin) Receptor as a Key Factor in Female Reproduction. Mol. Cell. Biol. 2005, 25, 10040–10051. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wada, T.; Febbraio, M.; He, J.; Matsubara, T.; Lee, M.J.; Gonzalez, F.J.; Xie, W. A Novel Role for the Dioxin Receptor in Fatty Acid Metabolism and Hepatic Steatosis. Gastroenterology 2010, 139, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, C.-X.; Krager, S.L.; Bottum, K.M.; Liao, D.-F.; Tischkau, S.A. Aryl Hydrocarbon Receptor Deficiency Enhances Insulin Sensitivity and Reduces PPAR-α Pathway Activity in Mice. Environ. Health Perspect. 2011, 119, 1739–1744. [Google Scholar] [CrossRef]
- Xu, C.-X.; Wang, C.; Zhang, Z.-M.; Jaeger, C.D.; Krager, S.L.; Bottum, K.M.; Liu, J.; Liao, D.-F.; Tischkau, S.A. Aryl Hydrocarbon Receptor Deficiency Protects Mice from Diet-Induced Adiposity and Metabolic Disorders through Increased Energy Expenditure. Int. J. Obes. (Lond.) 2015, 39, 1300–1309. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Ma, J.; Jiao, C.; Qiu, X.; Cui, X.; Wang, D.; Zhang, H. MiR-124a Mediates the Impairment of Intestinal Epithelial Integrity by Targeting Aryl Hydrocarbon Receptor in Crohn’s Disease. Inflammation 2020, 43, 1862–1875. [Google Scholar] [CrossRef]
- Makhloufi, C.; Nicolas, F.; McKay, N.; Fernandez, S.; Hache, G.; Garrigue, P.; Brunet, P.; Guillet, B.; Burtey, S.; Poitevin, S. Female AhR Knockout Mice Develop a Minor Renal Insufficiency in an Adenine-Diet Model of Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 2483. [Google Scholar] [CrossRef]
- Keys, B.; Piskorska-Pliszczynska, J.; Safe, S. Polychlorinated Dibenzofurans as 2,3,7,8-Tcdd Antagonists: In Vitro Inhibition of Monooxygenase Enzyme Induction. Toxicol. Lett. 1986, 31, 151–158. [Google Scholar] [CrossRef]
- Astroff, B.; Safe, S. 6-Substituted-1,3,8-Trichlorodibenzofurans as 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Antagonists in the Rat: Structure Activity Relationships. Toxicology 1989, 59, 285–296. [Google Scholar] [CrossRef]
- Harris, M.; Zacharewski, T.; Astroff, B.; Safe, S. Partial Antagonism of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin-Mediated Induction of Aryl Hydrocarbon Hydroxylase by 6-Methyl-1,3,8-Trichlorodibenzofuran: Mechanistic Studies. Mol. Pharm. 1989, 35, 729–735. [Google Scholar]
- Biegel, L.; Harris, M.; Davis, D.; Rosengren, R.; Safe, L.; Safe, S. 2,2′,4,4′,5,5′-Hexachlorobiphenyl as a 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Antagonist in C57BL 6J Mice. Toxicol. Appl. Pharmacol. 1989, 97, 561–571. [Google Scholar] [CrossRef]
- Chen, G.; Bunce, N.J. Polybrominated Diphenyl Ethers as Ah Receptor Agonists and Antagonists. Toxicol. Sci. 2003, 76, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Casper, R.F.; Quesne, M.; Rogers, I.M.; Shirota, T.; Jolivet, A.; Milgrom, E.; Savouret, J.-F.O. Resveratrol Has Antagonist Activity on the Aryl Hydrocarbon Receptor: Implications for Prevention of Dioxin Toxicity. Mol. Pharm. 1999, 56, 784–790. [Google Scholar]
- Murray, I.A.; Krishnegowda, G.; DiNatale, B.C.; Flaveny, C.; Chiaro, C.; Lin, J.-M.; Sharma, A.K.; Amin, S.; Perdew, G.H. Development of a Selective Modulator of Aryl Hydrocarbon (Ah) Receptor Activity That Exhibits Anti-Inflammatory Properties. Chem. Res. Toxicol. 2010, 23, 955–966. [Google Scholar] [CrossRef]
- Kim, S.-H.; Henry, E.C.; Kim, D.-K.; Kim, Y.-H.; Shin, K.J.; Han, M.S.; Lee, T.G.; Kang, J.-K.; Gasiewicz, T.A.; Ryu, S.H.; et al. Novel Compound 2-Methyl-2H-Pyrazole-3-Carboxylic Acid (2-Methyl-4-o-Tolylazo-Phenyl)-Amide (CH-223191) Prevents 2,3,7,8-TCDD-Induced Toxicity by Antagonizing the Aryl Hydrocarbon Receptor. Mol. Pharm. 2006, 69, 1871–1878. [Google Scholar] [CrossRef]
- Poland, A.P.; Glover, E.; Robinson, J.R.; Nebert, D.W. Genetic Expression of Aryl Hydrocarbon Hydroxylase Activity: Induction of Monooxygenase Activities Andcytochrome P1-450 Formation by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Mice Genetically “Nonresponsive” to Other Aromatic Hydrocarbons. J. Biol. Chem. 1974, 249, 5599–5606. [Google Scholar]
- Kerley-Hamilton, J.S.; Trask, H.W.; Ridley, C.J.A.; DuFour, E.; Ringelberg, C.S.; Nurinova, N.; Wong, D.; Moodie, K.L.; Shipman, S.L.; Moore, J.H.; et al. Obesity Is Mediated by Differential Aryl Hydrocarbon Receptor Signaling in Mice Fed a Western Diet. Environ. Health Perspect. 2012, 120, 1252–1259. [Google Scholar] [CrossRef]
- Blank, J.A.; Tucker, A.N.; Sweatlock, J.; Gasiewicz, T.A.; Luster, M.I. Alpha-Naphthoflavone Antagonism of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin-Induced Murine Lymphocyte Ethoxyresorufin-O-Deethylase Activity and Immunosuppression. Mol. Pharm. 1987, 32, 169–172. [Google Scholar]
- Merchant, M.; Arellano, L.; Safe, S. The Mechanism of Action of α-Naphthoflavone as an Inhibitor of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin-Induced CYP1A1 Gene Expression. Arch. Biochem. Biophys. 1990, 281, 84–89. [Google Scholar] [CrossRef]
- Zhao, B.; DeGroot, D.E.; Hayashi, A.; He, G.; Denison, M.S. CH223191 Is a Ligand-Selective Antagonist of the Ah (Dioxin) Receptor. Toxicol. Sci. 2010, 117, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Hazlett, H.F.; Nemani, K.V.; Trask, H.W.; West, R.J.; Lupien, L.E.; Collins, A.J.; Ringelberg, C.S.; et al. Inhibition of the Aryl Hydrocarbon Receptor Prevents Western Diet-Induced Obesity. Model for AHR Activation by Kynurenine via Oxidized-LDL, TLR2/4, TGFβ, and IDO1. Toxicol. Appl. Pharm. 2016, 300, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Nemani, K.V.; Trask, H.W.; Ringelberg, C.S.; Gimi, B.; Demidenko, E.; Tomlinson, C.R. Obesity and Fatty Liver Are Prevented by Inhibition of the Aryl Hydrocarbon Receptor in Both Female and Male Mice. Nutr. Res. 2017, 44, 38–50. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Nguyen, L.P.; Bradfield, C.A. The Search for Endogenous Activators of the Aryl Hydrocarbon Receptor. Chem. Res. Toxicol. 2008, 21, 102–116. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The Roles of Aryl Hydrocarbon Receptor in Immune Responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hirota, K.; Christensen, J.; O’Garra, A.; Stockinger, B. Natural Agonists for Aryl Hydrocarbon Receptor in Culture Medium Are Essential for Optimal Differentiation of Th17 T Cells. J. Exp. Med. 2009, 206, 43–49. [Google Scholar] [CrossRef]
- Rojas, I.Y.; Moyer, B.J.; Ringelberg, C.S.; Wilkins, O.M.; Pooler, D.B.; Ness, D.B.; Coker, S.; Tosteson, T.D.; Lewis, L.D.; Chamberlin, M.D.; et al. Kynurenine-Induced Aryl Hydrocarbon Receptor Signaling in Mice Causes Body Mass Gain, Liver Steatosis, and Hyperglycemia. Obesity 2020, in press. [Google Scholar]
- Rojas, I.Y.; Moyer, B.J.; Ringelberg, C.S.; Tomlinson, C.R. Reversal of Obesity and Liver Steatosis in Mice via Inhibition of Aryl Hydrocarbon Receptor and Altered Gene Expression of CYP1B1, PPARα, SCD1, and Osteopontin. Int. J. Obes. 2020, 44, 948–963. [Google Scholar] [CrossRef]
- Pohjanvirta, R. AHR in Energy Balance Regulation. Curr. Opin. Toxicol. 2017, 2, 8–14. [Google Scholar] [CrossRef]
- Tischkau, S.A. Mechanisms of Circadian Clock Interactions with Aryl Hydrocarbon Receptor Signalling. Eur. J. Neurosci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Shoemaker, R.; English, V.; Larian, N.; Sunkara, M.; Morris, A.J.; Walker, M.; Yiannikouris, F.; Cassis, L.A. Effects of Adipocyte Aryl Hydrocarbon Receptor Deficiency on PCB-Induced Disruption of Glucose Homeostasis in Lean and Obese Mice. Environ. Health Perspect. 2015, 123, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Fisher ffolliott, M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 Regulates PGC-1α and Browning of White Adipose Tissues in Adaptive Thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef]
- Hondares, E.; Rosell, M.; Gonzalez, F.J.; Giralt, M.; Iglesias, R.; Villarroya, F. Hepatic FGF21 Expression Is Induced at Birth via PPARalpha in Response to Milk Intake and Contributes to Thermogenic Activation of Neonatal Brown Fat. Cell Metab. 2010, 11, 206–212. [Google Scholar] [CrossRef]
- Talukdar, S.; Owen, B.M.; Song, P.; Hernandez, G.; Zhang, Y.; Zhou, Y.; Scott, W.T.; Paratala, B.; Turner, T.; Smith, A.; et al. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab. 2016, 23, 344–349. [Google Scholar] [CrossRef]
- von Holstein-Rathlou, S.; BonDurant, L.D.; Peltekian, L.; Naber, M.C.; Yin, T.C.; Claflin, K.E.; Urizar, A.I.; Madsen, A.N.; Ratner, C.; Holst, B.; et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016, 23, 335–343. [Google Scholar] [CrossRef]
- Bookout, A.L.; de Groot, M.H.M.; Owen, B.M.; Lee, S.; Gautron, L.; Lawrence, H.L.; Ding, X.; Elmquist, J.K.; Takahashi, J.S.; Mangelsdorf, D.J.; et al. FGF21 Regulates Metabolism and Circadian Behavior by Acting on the Nervous System. Nat. Med. 2013, 19, 1147–1152. [Google Scholar] [CrossRef]
- Foltz, I.N.; Hu, S.; King, C.; Wu, X.; Yang, C.; Wang, W.; Weiszmann, J.; Stevens, J.; Chen, J.S.; Nuanmanee, N.; et al. Treating Diabetes and Obesity with an FGF21-Mimetic Antibody Activating the ΒKlotho/FGFR1c Receptor Complex. Sci. Transl. Med. 2012, 4, 162ra153. [Google Scholar] [CrossRef]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The Effects of LY2405319, an FGF21 Analog, in Obese Human Subjects with Type 2 Diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 Gene Therapy as Treatment for Obesity and Insulin Resistance. Embo Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Cheng, X.; Vispute, S.G.; Liu, J.; Cheng, C.; Kharitonenkov, A.; Klaassen, C.D. Fibroblast Growth Factor (Fgf) 21 Is a Novel Target Gene of the Aryl Hydrocarbon Receptor (AhR). Toxicol. Appl. Pharmacol. 2014, 278, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Yan, J.; Liu, K.; Garbacz, W.G.; Wang, P.; Xu, M.; Ma, X.; Xie, W. Activation of Aryl Hydrocarbon Receptor Dissociates Fatty Liver from Insulin Resistance by Inducing FGF21. Hepatology 2015, 61, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Girer, N.G.; Murray, I.A.; Omiecinski, C.J.; Perdew, G.H. Hepatic Aryl Hydrocarbon Receptor Attenuates Fibroblast Growth Factor 21 Expression. J. Biol. Chem. 2016, 291, 15378–15387. [Google Scholar] [CrossRef] [PubMed]
- Girer, N.G.; Carter, D.; Bhattarai, N.; Mustafa, M.; Denner, L.; Porter, C.; Elferink, C.J. Inducible Loss of the Aryl Hydrocarbon Receptor Activates Perigonadal White Fat Respiration and Brown Fat Thermogenesis via Fibroblast Growth Factor 21. Int. J. Mol. Sci. 2019, 20, 950. [Google Scholar] [CrossRef]
- Geng, L.; Lam, K.S.L.; Xu, A. The Therapeutic Potential of FGF21 in Metabolic Diseases: From Bench to Clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef]
- Lu, W.; Li, X.; Luo, Y. FGF21 in Obesity and Cancer: New Insights. Cancer Lett. 2020. [Google Scholar] [CrossRef]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous Stimuli Maintain Intraepithelial Lymphocytes via Aryl Hydrocarbon Receptor Activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan Catabolites from Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Fukumoto, S.; Toshimitsu, T.; Matsuoka, S.; Maruyama, A.; Oh-Oka, K.; Takamura, T.; Nakamura, Y.; Ishimaru, K.; Fujii-Kuriyama, Y.; Ikegami, S.; et al. Identification of a Probiotic Bacteria-Derived Activator of the Aryl Hydrocarbon Receptor That Inhibits Colitis. Immunol. Cell Biol. 2014, 92, 460–465. [Google Scholar] [CrossRef]
- Jin, U.-H.; Lee, S.-O.; Sridharan, G.; Lee, K.; Davidson, L.A.; Jayaraman, A.; Chapkin, R.S.; Alaniz, R.; Safe, S. Microbiome-Derived Tryptophan Metabolites and Their Aryl Hydrocarbon Receptor-Dependent Agonist and Antagonist Activities. Mol. Pharm. 2014, 85, 777–788. [Google Scholar] [CrossRef]
- Lamas, B.; Hernandez-Galan, L.; Galipeau, H.J.; Constante, M.; Clarizio, A.; Jury, J.; Breyner, N.M.; Caminero, A.; Rueda, G.; Hayes, C.L.; et al. Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is Decreased in Celiac Disease Leading to Intestinal Inflammation. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-R.; Miao, H.; Deng, D.-Q.; Vaziri, N.D.; Li, P.; Zhao, Y.-Y. Gut Microbiota-Derived Tryptophan Metabolism Mediates Renal Fibrosis by Aryl Hydrocarbon Receptor Signaling Activation. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ghiboub, M.; Verburgt, C.M.; Sovran, B.; Benninga, M.A.; de Jonge, W.J.; Van Limbergen, J.E. Nutritional Therapy to Modulate Tryptophan Metabolism and Aryl Hydrocarbon-Receptor Signaling Activation in Human Diseases. Nutrients 2020, 12, 2846. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, L.; Ciocan, D.; Hugot, C.; Spatz, M.; Dupeux, M.; Houron, C.; Moal, V.L.-L.; Puchois, V.; Ferrere, G.; Trainel, N.; et al. Microbiota Tryptophan Metabolism Induces Aryl Hydrocarbon Receptor Activation and Improves Alcohol-Induced Liver Injury. Gut 2020. [Google Scholar] [CrossRef]
- Sári, Z.; Mikó, E.; Kovács, T.; Jankó, L.; Csonka, T.; Lente, G.; Sebő, É.; Tóth, J.; Tóth, D.; Árkosy, P.; et al. Indolepropionic Acid, a Metabolite of the Microbiome, Has Cytostatic Properties in Breast Cancer by Activating AHR and PXR Receptors and Inducing Oxidative Stress. Cancers 2020, 12, 2411. [Google Scholar] [CrossRef]
- Sári, Z.; Mikó, E.; Kovács, T.; Boratkó, A.; Ujlaki, G.; Jankó, L.; Kiss, B.; Uray, K.; Bai, P. Indoxylsulfate, a Metabolite of the Microbiome, Has Cytostatic Effects in Breast Cancer via Activation of AHR and PXR Receptors and Induction of Oxidative Stress. Cancers 2020, 12, 2915. [Google Scholar] [CrossRef]
- Murray, I.A.; Perdew, G.H. How Ah Receptor Ligand Specificity Became Important in Understanding Its Physiological Function. Int. J. Mol. Sci. 2020, 21, 9614. [Google Scholar] [CrossRef]
- Muku, G.E.; Lahoti, T.S.; Murray, I.A.; Podolsky, M.A.; Smith, K.J.; Hubbard, T.D.; Kuzu, G.; Gowda, K.; Amin, S.G.; Perdew, G.H. Ligand-Mediated Cytoplasmic Retention of the Ah Receptor Inhibits Macrophage-Mediated Acute Inflammatory Responses. Lab. Investig. 2017, 97, 1471–1487. [Google Scholar] [CrossRef]
- Patel, R.D.; Murray, I.A.; Flaveny, C.A.; Kusnadi, A.; Perdew, G.H. Ah Receptor Represses Acute-Phase Response Gene Expression without Binding to Its Cognate Response Element. Lab. Investig. 2009, 89, 695–707. [Google Scholar] [CrossRef]
- Tanos, R.; Patel, R.D.; Murray, I.A.; Smith, P.B.; Perdew, G.H. Ah Receptor Regulates the Cholesterol Biosynthetic Pathway in a Dioxin Response Element-Independent Manner. Hepatology 2012, 55, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Tanos, R.; Murray, I.A.; Smith, P.B.; Patterson, A.; Perdew, G.H. Role of the Ah Receptor in Homeostatic Control of Fatty Acid Synthesis in the Liver. Toxicol. Sci. 2012, 129, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Muku, G.E.; Kusnadi, A.; Kuzu, G.; Tanos, R.; Murray, I.A.; Gowda, K.; Amin, S.; Perdew, G.H. Selective Ah Receptor Modulators Attenuate NPC1L1-Mediated Cholesterol Uptake through Repression of SREBP-2 Transcriptional Activity. Lab. Investig. 2020, 100, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Mustafa, M.G.; Lichti, C.F.; Elferink, C.J. Homocitrullination Is a Novel Histone H1 Epigenetic Mark Dependent on Aryl Hydrocarbon Receptor Recruitment of Carbamoyl Phosphate Synthase 1. J. Biol. Chem. 2015, 290, 27767–27778. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girer, N.G.; Tomlinson, C.R.; Elferink, C.J. The Aryl Hydrocarbon Receptor in Energy Balance: The Road from Dioxin-Induced Wasting Syndrome to Combating Obesity with Ahr Ligands. Int. J. Mol. Sci. 2021, 22, 49. https://doi.org/10.3390/ijms22010049
Girer NG, Tomlinson CR, Elferink CJ. The Aryl Hydrocarbon Receptor in Energy Balance: The Road from Dioxin-Induced Wasting Syndrome to Combating Obesity with Ahr Ligands. International Journal of Molecular Sciences. 2021; 22(1):49. https://doi.org/10.3390/ijms22010049
Chicago/Turabian StyleGirer, Nathaniel G., Craig R. Tomlinson, and Cornelis J. Elferink. 2021. "The Aryl Hydrocarbon Receptor in Energy Balance: The Road from Dioxin-Induced Wasting Syndrome to Combating Obesity with Ahr Ligands" International Journal of Molecular Sciences 22, no. 1: 49. https://doi.org/10.3390/ijms22010049
APA StyleGirer, N. G., Tomlinson, C. R., & Elferink, C. J. (2021). The Aryl Hydrocarbon Receptor in Energy Balance: The Road from Dioxin-Induced Wasting Syndrome to Combating Obesity with Ahr Ligands. International Journal of Molecular Sciences, 22(1), 49. https://doi.org/10.3390/ijms22010049