Metabolome-Driven Regulation of Adenovirus-Induced Cell Death

 , and
, and
Abstract
1. Introduction
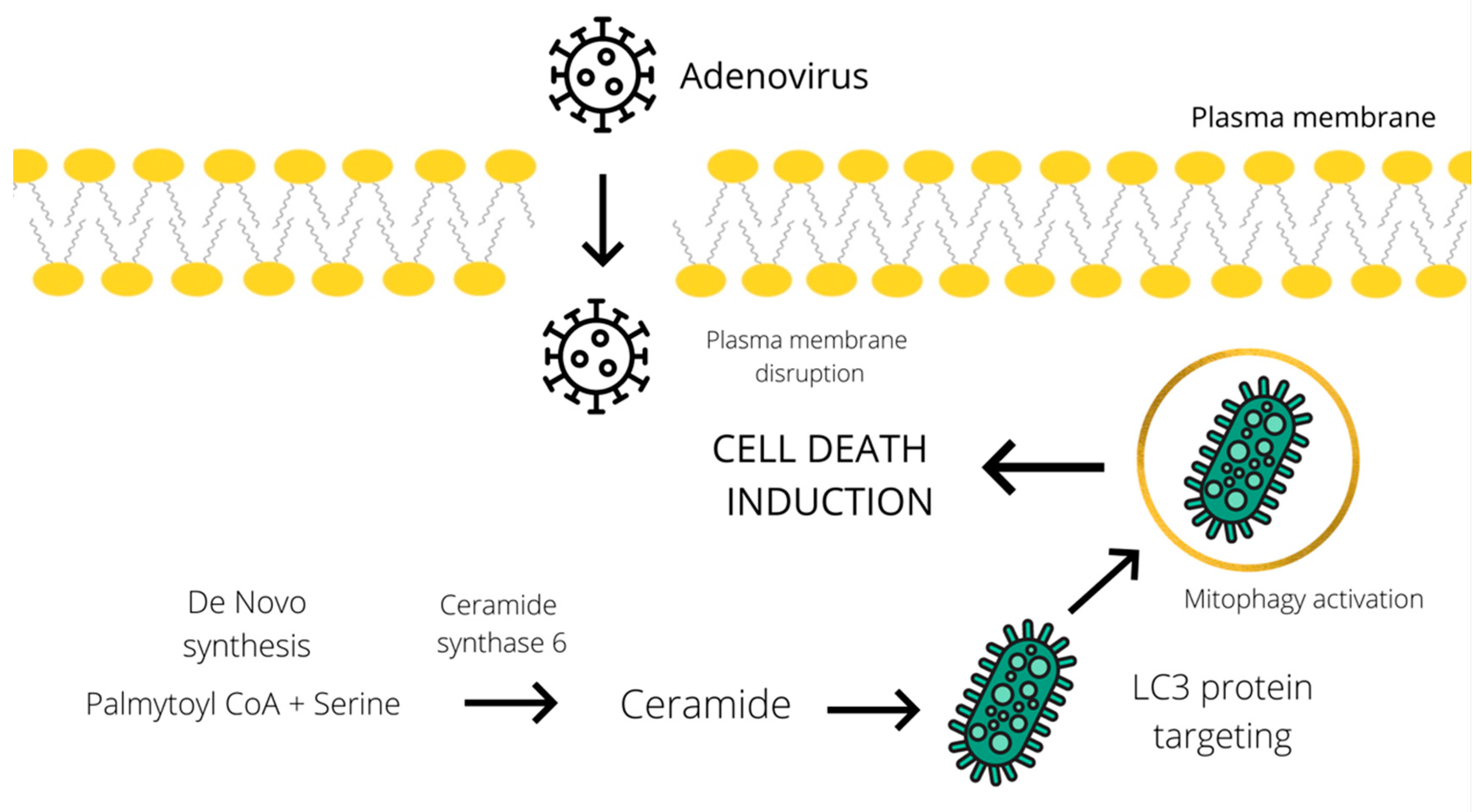
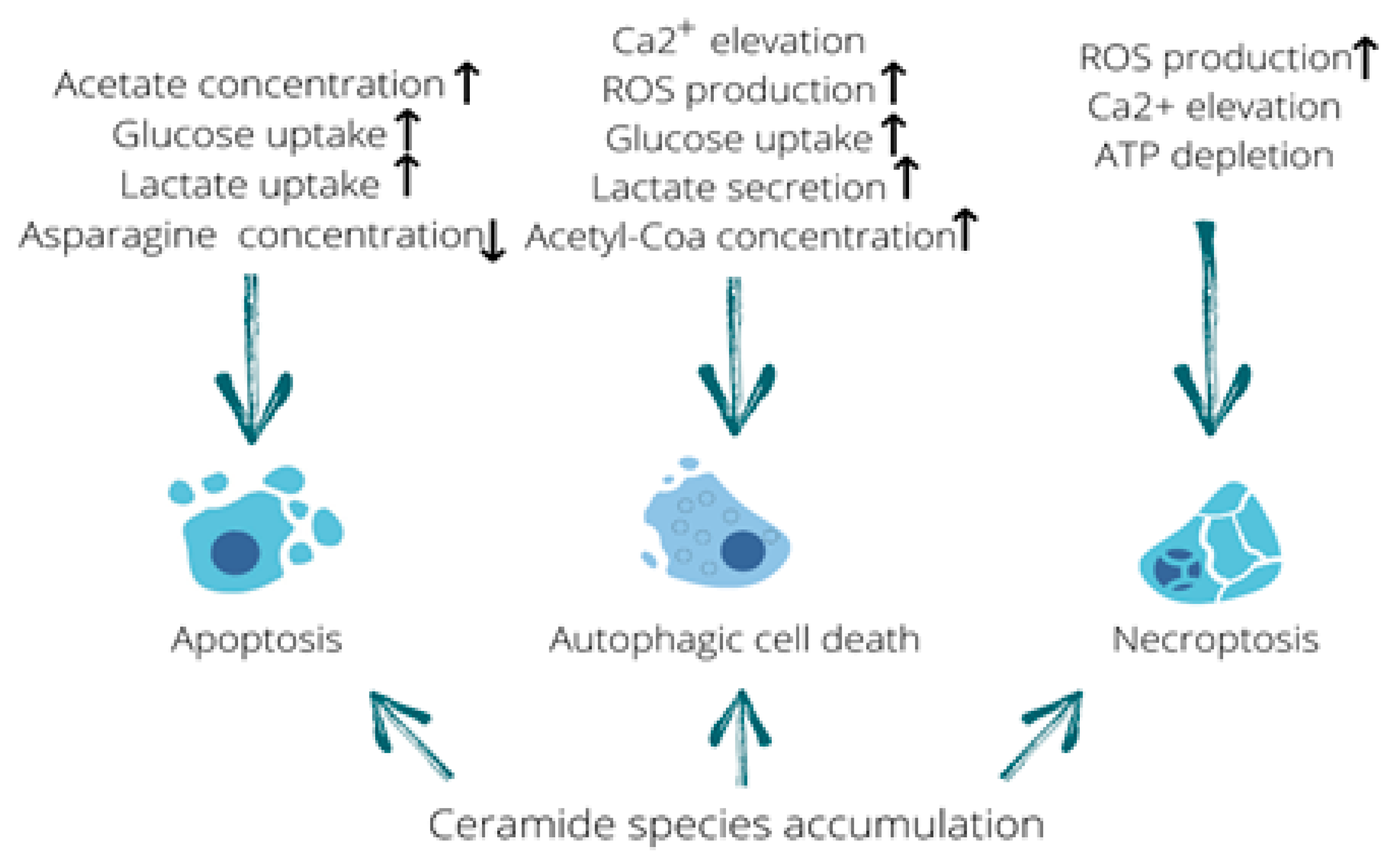
2. Metabolites Associated with the Adenovirus-Induced Autophagic Cell Death
3. Adenovirus Infection-Associated Metabolome in the Regulation of Apoptosis
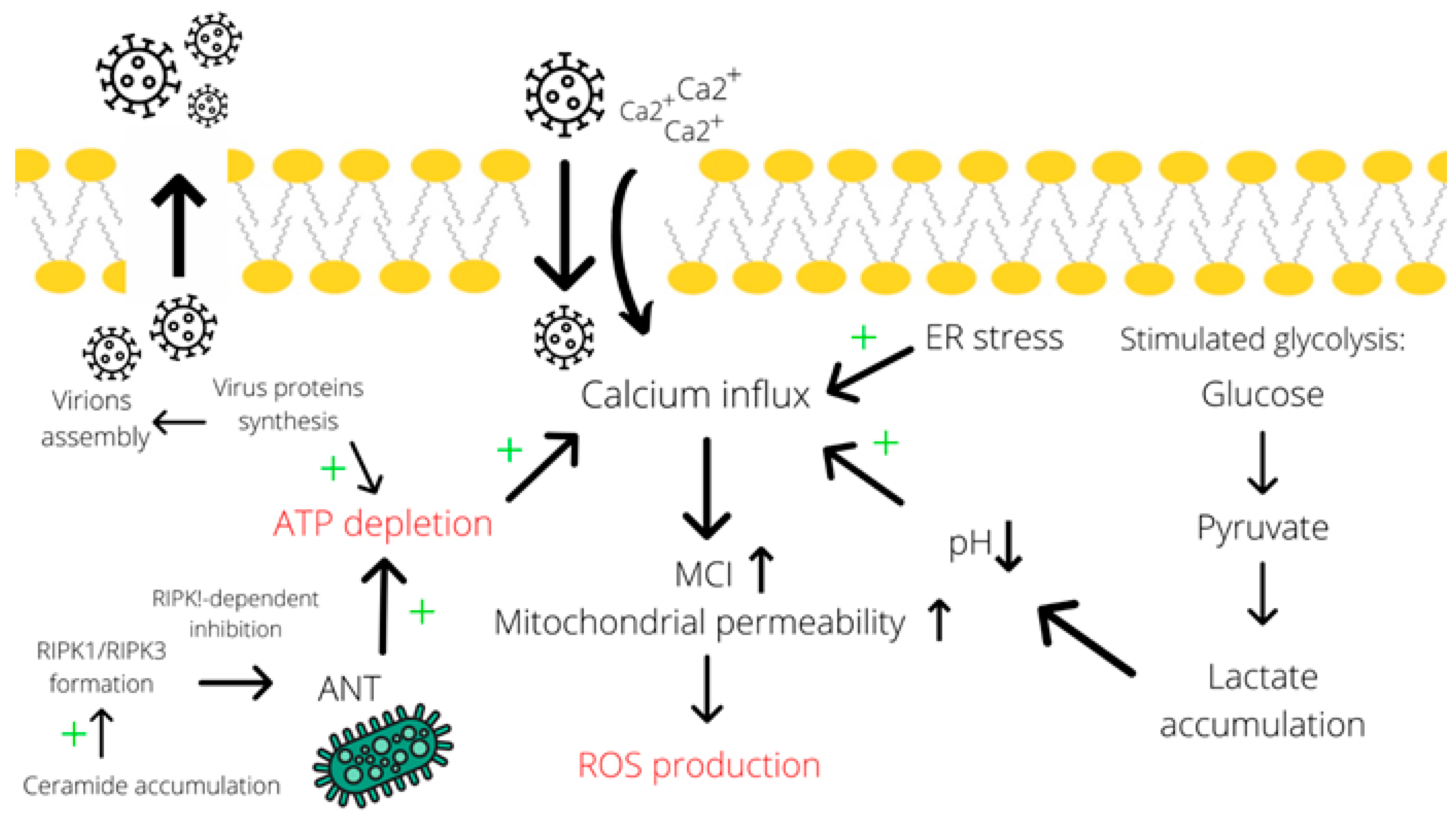
4. Adenovirus Controls Necrosis and Necroptosis with the Help of Cellular Metabolites
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amberg, A.; Riefke, B.; Schlotterbeck, G.; Ross, A.; Senn, H.; Dieterle, F.; Keck, M. NMR and MS Methods for Metabolomics. In Drug Safety Evaluation; Gautier, J.C., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1641, pp. 229–258. [Google Scholar]
- Bizzarri, M.; D’Anselmi, F.; Valerio, M.; Cucina, A.; Proietti, S.; Dinicola, S.; Pasqualato, A.; Manetti, C.; Galli, L.; Giuliani, A. Metabolomic profile and fractal dimensions in breast cancer cells. In Metabolomics Metabolites, Metabonomics, and Analytical Technologies 87–119; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2011. [Google Scholar]
- Endo, Y.; Sakai, R.; Ouchi, M.; Onimatsu, H.; Hioki, M.; Kagawa, S.; Uno, F.; Watanabe, Y.; Urata, Y.; Tanaka, N.; et al. Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene 2008, 27, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Diop, F.; Vial, T.; Ferraris, P.; Wichit, S.; Bengue, M.; Hamel, R.; Talignani, L.; Liegeois, F.; Pompon, J.; Yssel, H.; et al. Zika virus infection modulates the metabolomic profile of microglial cells. PLoS ONE 2018, 13, e0206093. [Google Scholar] [CrossRef] [PubMed]
- Sato-Dahlman, M.; Yamamoto, M. The Development of Oncoltyic Adenovirus Therapy in the Past and Future—For the Case of Pancreatic Cancer. Curr. Cancer Drug Targets 2018, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.-K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Kaverina, N.V.; Kadagidze, Z.G.; Borovjagin, A.V.; Karseladze, A.I.; Kim, C.K.; Lesniak, M.S.; Miska, J.; Zhang, P.; Baryshnikova, M.A.; Xiao, T.; et al. Tamoxifen overrides autophagy inhibition in Beclin-1-deficient glioma cells and their resistance to adenovirus-mediated oncolysis via upregulation of PUMA and BAX. Oncogene 2018, 37, 6069–6082. [Google Scholar] [CrossRef]
- Tazawa, H.; Kuroda, S.; Hasei, J.; Kagawa, S.; Fujiwara, T. Impact of Autophagy in Oncolytic Adenoviral Therapy for Cancer. Int. J. Mol. Sci. 2017, 18, 1479. [Google Scholar] [CrossRef]
- Rodriguez-Rocha, H.; Gomez-Gutierrez, J.G.; Garcia-Garcia, A.; Rao, X.-M.; Chen, L.; McMasters, K.M.; Zhou, H.S. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011, 416, 9–15. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Tyler, M.A.; Zhu, Z.B.; Han, Y.; He, T.-C.; Lesniak, M.S. Oncolytic adenoviral vectors which employ the survivin promoter induce glioma oncolysis via a process of beclin-dependent autophagy. Int. J. Oncol. 2009, 34, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Sonabend, A.M.; Nandi, S.; Khramtsov, A.; Han, Y.; Lesniak, M.S. Combination of adenoviral virotherapy and temozolomide chemotherapy eradicates malignant glioma through autophagic and apoptotic cell death in vivo. Br. J. Cancer 2009, 100, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, R.; Stichling, N.; Koelen, J.; Kuryk, L.; Lipiec, A.; Greber, U.F. Innate Immunity to Adenovirus. Hum. Gene Ther. 2014, 25, 265–284. [Google Scholar] [CrossRef]
- Tazawa, H.; Kagawa, S.; Fujiwara, T. Oncolytic adenovirus-induced autophagy: Tumor-suppressive efect and molecular basis. Acta Med. Okayama 2013, 67, 333–342. [Google Scholar] [CrossRef]
- Yacoub, A.; Hamed, H.A.; Allegood, J.; Mitchell, C.; Spiegel, S.; Lesniak, M.S.; Ogretmen, B.; Dash, R.; Sarkar, D.; Broaddus, W.C.; et al. PERK–Dependent Regulation of Ceramide Synthase 6 and Thioredoxin Play a Key Role in mda-7/IL-24–Induced Killing of Primary Human Glioblastoma Multiforme Cells. Cancer Res. 2010, 70, 1120–1129. [Google Scholar] [CrossRef]
- Luisoni, S.; Suomalainen, M.; Boucke, K.; Tanner, L.B.; Wenk, M.R.; Guan, X.L.; Grzybek, M.; Coskun, Ü.; Greber, U.F. Co-option of Membrane Wounding Enables Virus Penetration into Cells. Cell Host Microbe 2015, 18, 75–85. [Google Scholar] [CrossRef]
- Park, M.A.; Hamed, H.A.; Mitchell, C.; Cruickshanks, N.; Dash, R.; Allegood, J.; Dmitriev, I.P.; Tye, G.; Ogretmen, B.; Spiegel, S.; et al. A Serotype 5/3 Adenovirus Expressing MDA-7/IL-24 Infects Renal Carcinoma Cells and Promotes Toxicity of Agents That Increase Ros and Ceramide Levels. Mol. Pharmacol. 2010, 79, 368–380. [Google Scholar] [CrossRef]
- Kanj, S.S.; Dandashi, N.; El-Hed, A.; Harik, H.; Maalouf, M.; Kozhaya, L.; Mousallem, T.; Tollefson, A.E.; Wold, W.S.; Chalfant, C.E.; et al. Ceramide regulates SR protein phosphorylation during adenoviral infection. Virology 2006, 345, 280–289. [Google Scholar] [CrossRef]
- Cruickshanks, N.; Roberts, J.L.; Bareford, M.D.; Tavallai, M.; Poklepovic, A.; Booth, L.; Spiegel, S.; Dent, P. Differential regulation of autophagy and cell viability by ceramide species. Cancer Biol. Ther. 2015, 16, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.E.; Murphy, J.P.; Clements, D.R.; Konda, P.; Holay, N.; Kim, Y.; Pathak, G.P.; Giacomantonio, M.A.; El Hiani, Y.; Gujar, S. Inhibition of Pyruvate Dehydrogenase Kinase Enhances the Antitumor Efficacy of Oncolytic Reovirus. Cancer Res. 2019, 79, 3824–3836. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ogretmen, B. Ceramide stress in survival versus lethal autophagy paradox. Autophagy 2013, 9, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L. Starvation in the midst of plenty: Making sense of ceramide-induced autophagy by analysing nutrient transporter expression. Biochem. Soc. Trans. 2009, 37, 253–258. [Google Scholar] [CrossRef]
- Prusinkiewicz, M.A.; Mymryk, J.S. Metabolic Reprogramming of the Host Cell by Human Adenovirus Infection. Viruses 2019, 11, 141. [Google Scholar] [CrossRef]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.-J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef]
- Carinhas, N.; Pais, D.A.M.; Koshkin, A.; Fernandes, P.; Coroadinha, A.S.; Carrondo, M.J.T.; Alves, P.M.; Teixeira, A.P. Metabolic flux profiling of MDCK cells during growth and canine adenovirus vector production. Sci. Rep. 2016, 6, 23529. [Google Scholar] [CrossRef]
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of Autophagy by Cytosolic Acetyl-Coenzyme A. Mol. Cell 2014, 53, 710–725. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Liikanen, I.; Ahtiainen, L.; Hirvinen, M.L.M.; Bramante, S.; Cerullo, V.; Nokisalmi, P.; Hemminki, O.; Diaconu, I.; Pesonen, S.; Koski, A.; et al. Oncolytic Adenovirus with Temozolomide Induces Autophagy and Antitumor Immune Responses in Cancer Patients. Mol. Ther. 2013, 21, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, Apoptosis, Mitoptosis and Necrosis: Interdependence between Those Pathways and Effects on Cancer. Arch. Immunol. Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2012, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O’Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef]
- Braithwaite, A.W.; Russell, I.A. Induction of cell death by adenoviruses. Apoptosis 2001, 6, 359–370. [Google Scholar] [CrossRef]
- Wohlfahrt, M.E.; Beard, B.C.; Lieber, A.; Kiem, H.-P. A Capsid-Modified, Conditionally Replicating Oncolytic Adenovirus Vector Expressing TRAIL Leads to Enhanced Cancer Cell Killing in Human Glioblastoma Models. Cancer Res. 2007, 67, 8783–8790. [Google Scholar] [CrossRef]
- Radke, J.R.; Siddiqui, Z.K.; Figueroa, I.; Cook, J.L. E1A enhances cellular sensitivity to DNA-damage-induced apoptosis through PIDD-dependent caspase-2 activation. Cell Death Discov. 2016, 2. [Google Scholar] [CrossRef]
- Silva, A.C.; Teixeira, A.P.; Alves, P.M. Impact of Adenovirus infection in host cell metabolism evaluated by 1 H-NMR spectroscopy. J. Biotechnol. 2016, 231, 16–23. [Google Scholar] [CrossRef]
- Madhu, B.; Narita, M.; Jauhiainen, A.; Menon, S.; Stubbs, M.; Tavaré, S.; Narita, M.; Griffiths, J.R. Metabolomic changes during cellular transformation monitored by metabolite–metabolite correlation analysis and correlated with gene expression. Metabolomics 2015, 11, 1848–1863. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; De Oliveira, C.S.F.; Alves, S.; Chaves, S.R.; Coutinho, O.P.; Côrte-Real, M.; Preto, A. Acetate-induced apoptosis in colorectal carcinoma cells involves lysosomal membrane permeabilization and cathepsin D release. Cell Death Dis. 2013, 4, e507. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.S.F.; Pereira, H.; Alves, S.; Castro, L.; Baltazar, F.; Chaves, S.R.; Preto, A.; Côrte-Real, M. Cathepsin D protects colorectal cancer cells from acetate-induced apoptosis through autophagy-independent degradation of damaged mitochondria. Cell Death Dis. 2015, 6, e1788. [Google Scholar] [CrossRef] [PubMed]
- Jan, G.; Belzacq, A.-S.; Haouzi, D.; Rouault, A.; Métivier, D.; Kroemer, G.; Brenner, C. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ. 2002, 9, 179–188. [Google Scholar] [CrossRef]
- Liu, J.; Wang, J.; Shi, Y.; Su, W.; Chen, J.; Zhang, Z.; Wang, G.; Wang, F. Short Chain Fatty Acid Acetate Protects against Ethanol-Induced Acute Gastric Mucosal Lesion in Mice. Biol. Pharm. Bull. 2017, 40, 1439–1446. [Google Scholar] [CrossRef]
- Cheng, Q.; Li, X.; Wang, Y.; Dong, M.; Zhan, F.-H.; Liu, J. The ceramide pathway is involved in the survival, apoptosis and exosome functions of human multiple myeloma cells in vitro. Acta Pharmacol. Sin. 2018, 39, 561–568. [Google Scholar] [CrossRef]
- Colombini, M. Ceramide channels and mitochondrial outer membrane permeability. J. Bioenerg. Biomembr. 2017, 49, 57–64. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, Q.; Feng, M.; Li, J.; Guan, Z.; An, D.; Dong, M.; Peng, Y.; Kuerban, K.; Ye, L. C2-ceramide enhances sorafenib-induced caspase-dependent apoptosis via PI3K/AKT/mTOR and Erk signaling pathways in HCC cells. Appl. Microbiol. Biotechnol. 2017, 101, 1535–1546. [Google Scholar] [CrossRef]
- Na, H.-N.; Dubuisson, O.; Hegde, V.K.; Nam, J.-H.; Dhurandhar, N.V. Human adenovirus Ad36 and its E4orf1 gene enhance cellular glucose uptake even in the presence of inflammatory cytokines. Biochimie 2016, 124, 3–10. [Google Scholar] [CrossRef]
- Kasinskas, R.W.; Venkatasubramanian, R.; Forbes, N.S. Rapid uptake of glucose and lactate, and not hypoxia, induces apoptosis in three-dimensional tumor tissue culture. Integr. Biol. 2014, 6, 399–410. [Google Scholar] [CrossRef]
- Carinhas, N.; Koshkin, A.; Pais, D.A.M.; Alves, P.M.; Teixeira, A.P. 13C-metabolic flux analysis of human adenovirus infection: Implications for viral vector production. Biotechnol. Bioeng. 2016, 114, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Wu, T.T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine Plays a Critical Role in Regulating Cellular Adaptation to Glutamine Depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Li, Q.-H.; Yang, J.-L.; Liu, F.; Wang, L.; Xu, W.-M.; Zhao, W.-X. The antitumor effects of oncolytic adenovirus H101 against lung cancer. Int. J. Oncol. 2015, 47, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Xiao, T.; He, L.; Ji, H.; Liu, X.-Y. Interferon-β-armed oncolytic adenovirus induces both apoptosis and necroptosis in cancer cells. Acta Biochim. Biophys. Sin. 2012, 44, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.; Kraaij, R.; Leadley, R.; De Ridder, C.; Van Weerden, W.; Van Schie, K.; Van Der Kroeg, M.; Hoeben, R.; Maitland, N.; Lindholm, L. A Transductionally Retargeted Adenoviral Vector for Virotherapy of Her2/neu-Expressing Prostate Cancer. Hum. Gene Ther. 2012, 23, 70–82. [Google Scholar] [CrossRef]
- Weigert, M.; Binks, A.; Dowson, S.; Leung, E.Y.L.; Athineos, D.; Yu, X.; Mullin, M.; Walton, J.B.; Orange, C.; Ennis, D.; et al. RIPK3 promotes adenovirus type 5 activity. Cell Death Dis. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Hemminki, A.; Immonen, R.; Närväinen, J.; Kipar, A.; Paasonen, J.; Jokivarsi, K.T.; Yli-Ollila, H.; Soininen, P.; Partanen, K.; Joensuu, T.; et al. In vivo magnetic resonance imaging and spectroscopy identifies oncolytic adenovirus responders. Int. J. Cancer 2014, 134, 2878–2890. [Google Scholar] [CrossRef]
- Berghe, T.V.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef]
- Wu, W.; Liu, P.; Li, J. Necroptosis: An emerging form of programmed cell death. Crit. Rev. Oncol. Hematol. 2012, 82, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Temkin, V.; Huang, Q.; Liu, H.; Osada, H.; Pope, R.M. Inhibition of ADP/ATP Exchange in Receptor-Interacting Protein-Mediated Necrosis. Mol. Cell. Biol. 2006, 26, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar] [PubMed]
- Dyer, A.; Di, Y.; Calderon, H.; Illingworth, S.; Kueberuwa, G.; Tedcastle, A.; Jakeman, P.; Chia, S.L.; Brown, A.; Silva, M.A.; et al. Oncolytic Group B Adenovirus Enadenotucirev Mediates Non-apoptotic Cell Death with Membrane Disruption and Release of Inflammatory Mediators. Mol. Ther. Oncolytics 2017, 4, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.; Kumar, M.; Taruishi, M.; Javier, R.T. The Human Adenovirus E4-ORF1 Protein Subverts Discs Large 1 to Mediate Membrane Recruitment and Dysregulation of Phosphatidylinositol 3-Kinase. PLoS Pathog. 2014, 10, e1004102. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, B.; Hou, W.; Lin, H.; Rebetz, J.; Hong, S.-S.; Wang, Y.; Ran, L.; Fan, X. Active and separate secretion of fiber and penton base during the early phase of Ad2 or Ad5 infection. Virology 2017, 505, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Chen, F.-H.; Yuan, F.-L.; Zhang, T.-Y.; Wu, F.-R.; Rong, C.; Jiang, S.; Tang, J.; Zhang, C.-C.; Lin, M.-Y. Blockade of acid-sensing ion channels protects articular chondrocytes from acid-induced apoptotic injury. Inflamm. Res. 2012, 61, 327–335. [Google Scholar] [CrossRef]
- Gao, W.-F.; Xu, Y.-Y.; Ge, J.-F.; Chen, F.-H. Inhibition of acid-sensing ion channel 1a attenuates acid-induced activation of autophagy via a calcium signaling pathway in articular chondrocytes. Int. J. Mol. Med. 2019, 43, 1778–1788. [Google Scholar] [CrossRef]
- Festjens, N.; Berghe, T.V.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 2006, 1757, 1371–1387. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Ramalho-Santos, J. Mitochondrial dysfunction in reproductive and developmental toxicity. Reprod. Dev. Toxicol. 2011, 815–824. [Google Scholar] [CrossRef]
- Taverner, W.K.; Jacobus, E.J.; Christianson, J.; Champion, B.; Paton, A.W.; Paton, J.C.; Su, W.; Cawood, R.; Seymour, L.; Lei-Rossmann, J. Calcium Influx Caused by ER Stress Inducers Enhances Oncolytic Adenovirus Enadenotucirev Replication and Killing through PKCα Activation. Mol. Ther. Oncolytics 2019, 15, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.; Hewett, T.E.; Robbins, J.; et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007, 117, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Thon, L.; Möhlig, H.; Mathieu, S.; Lange, A.; Bulanova, E.; Winoto-Morbach, S.; Schütze, S.; Bulfone-Paus, S.; Adam, D. Ceramide mediates caspase-independent programmed cell death. FASEB J. 2005, 19, 1945–1956. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, X.; Zhou, Y.; Wang, H. C2-Ceramide Induces Cell Death and Protective Autophagy in Head and Neck Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2014, 15, 3336–3355. [Google Scholar] [CrossRef]
- Yokoyama, T.; Iwado, E.; Kondo, Y.; Aoki, H.; Hayashi, Y.; Georgescu, M.; Sawaya, R.; Hess, K.R.; Mills, G.B.; Kawamura, H.; et al. Autophagy-inducing agents augment the antitumor effect of telerase-selve oncolytic adenovirus OBP-405 on glioblastoma cells. Gene Ther. 2008, 15, 1233–1239. [Google Scholar] [CrossRef]
- Botta, G.; Passaro, C.; Libertini, S.; Abagnale, A.; Barbato, S.; Maione, A.S.; Hallden, G.; Beguinot, F.; Formisano, P.; Portella, G. Inhibition of Autophagy Enhances the Effects of E1A-Defective Oncolytic Adenovirus dl922–947 Against Glioma Cells In Vitro and In Vivo. Hum. Gene Ther. 2012, 23, 623–634. [Google Scholar] [CrossRef]
- Alonso, M.M.; Jiang, H.; Yokoyama, T.; Xu, J.; Bekele, N.B.; Lang, F.F.; Kondo, S.; Gomez-Manzano, C.; Fueyo, J. Delta-24-RGD in Combination with RAD001 Induces Enhanced Anti-glioma Effect via Autophagic Cell Death. Mol. Ther. 2008, 16, 487–493. [Google Scholar] [CrossRef]
- Jiang, H.; White, E.J.; Ríos-Vicil, C.I.; Xu, J.; Gomez-Manzano, C.; Fueyo, J. Human Adenovirus Type 5 Induces Cell Lysis through Autophagy and Autophagy-Triggered Caspase Activity. J. Virol. 2011, 85, 4720–4729. [Google Scholar] [CrossRef]
- Klein, S.R.; Piya, S.; Lu, Z.; Xia, Y.; Alonso, M.; White, E.J.; Wei, J.; Gomezmanzano, C.; Jiang, H.; Fueyo, J. C-Jun N-terminal kinases are required for oncolytic adenovirus-mediated autophagy. Oncogene 2015, 34, 5295–5301. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Shah, N.; Kaverina, N.V.; Lee, H.; Lin, B.; Lieber, A.; Kadagidze, Z.G.; Yoon, J.-G.; Schroeder, B.; Hothi, P.; et al. Tamoxifen improves cytopathic effect of oncolytic adenovirus in primary glioblastoma cells mediated through autophagy. Oncotarget 2015, 6, 3977–3987. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the Therapeutic Potential of Delta-24-RGD in Brain Tumor Stem Cells: Role of Autophagic Cell Death. J. Natl. Cancer Inst. 2007, 99, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Mao, Q.; Wang, D.; Zhang, W.; Xia, H. A Fiber Chimeric CRAd Vector Ad5/11-D24 Double-Armed with TRAIL and Arresten for Enhanced Glioblastoma Therapy. Hum. Gene Ther. 2012, 23, 589–596. [Google Scholar] [CrossRef]
- Tsamis, K.I.; Alexiou, G.A.; Vartholomatos, E.; Kyritsis, A.P. Combination Treatment for Glioblastoma Cells with Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand and Oncolytic Adenovirus Delta-24. Cancer Investig. 2013, 31, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Hong, J.; Kwon, O.-J.; Yun, C.-O. A hypoxia- and telomerase-responsive oncolytic adenovirus expressing secretable trimeric TRAIL triggers tumour-specific apoptosis and promotes viral dispersion in TRAIL-resistant glioblastoma. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Bil-Lula, I.; Krzywonos-Zawadzka, A.; Sawicki, G.; Woźniak, M. An infection of human adenovirus 31 affects the differentiation of preadipocytes into fat cells, its metabolic profile and fat accumulation. J. Med. Virol. 2016, 88, 400–407. [Google Scholar] [CrossRef]
- Miyake-Stoner, S.J.; O’Shea, C.C. Metabolism goes viral. Cell Metab. 2014, 19, 549–550. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell Death Type | Autophagic Cell Death | Apoptosis | Necrotic Cell Death |
|---|---|---|---|
| General description | Catabolic process that presents degradation of intracellular components in autophagosomes. | Form of programmed cell death that proceeded through caspase activation [32,33] | Necrosis: “accidental” cell death caused be excessive damage. Necroptosis: programmed necrotic cell death, controlled by MLKL and RIPK1/3 |
| Role | Pro-survival role to maintain cell’s homeostasis under harsh conditions [6,7,8,9]. While coping with excessive stress switches to mechanism inducing cell death resembling self-consumption [10,11,12,13,14]. Can be triggered when apoptosis is inhibited. | Elimination of damaged or infected cells without immune response activation | Controlled and uncontrolled death mechanism in the context of heavy damage. Necroptosis can be triggered when apoptosis is inhibited. |
| Morphology | Formation of double-membraned autophagosomes surrounding cell’s organelles. | Cell shrinkage, DNA fragmentation and formation of apoptotic bodies [9,34]. | Cell swelling, nuclear degradation, plasma membrane destruction and cell contents’ spilling into surrounding [32,33] |
| Markers | p62, LC3B expression | Caspase activation, DNA fragmentation | MLKL, RIPK1/3 expression |
| Immune response | Activated due to DAMP release | Immunologically asymptomatic | Activated due to DAMP release |
| Metabolic triggers | Ceramide accumulation, increased glucose consumption and lactate secretion, acetyl-CoA accumulation. Accompanied by ATP release. | Ceramide accumulation, acetate accumulation, rapid glucose uptake, glutamine and asparagine depletion | Ceramide accumulation, ATP depletion and ROS production |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laevskaya, A.; Borovjagin, A.; Timashev, P.S.; Lesniak, M.S.; Ulasov, I. Metabolome-Driven Regulation of Adenovirus-Induced Cell Death. Int. J. Mol. Sci. 2021, 22, 464. https://doi.org/10.3390/ijms22010464
Laevskaya A, Borovjagin A, Timashev PS, Lesniak MS, Ulasov I. Metabolome-Driven Regulation of Adenovirus-Induced Cell Death. International Journal of Molecular Sciences. 2021; 22(1):464. https://doi.org/10.3390/ijms22010464
Chicago/Turabian StyleLaevskaya, Anastasia, Anton Borovjagin, Peter S. Timashev, Maciej S. Lesniak, and Ilya Ulasov. 2021. "Metabolome-Driven Regulation of Adenovirus-Induced Cell Death" International Journal of Molecular Sciences 22, no. 1: 464. https://doi.org/10.3390/ijms22010464
APA StyleLaevskaya, A., Borovjagin, A., Timashev, P. S., Lesniak, M. S., & Ulasov, I. (2021). Metabolome-Driven Regulation of Adenovirus-Induced Cell Death. International Journal of Molecular Sciences, 22(1), 464. https://doi.org/10.3390/ijms22010464