Energy Metabolism and Ketogenic Diets: What about the Skeletal Health? A Narrative Review and a Prospective Vision for Planning Clinical Trials on this Issue


 ,
,  ,
,  ,
, 

Abstract
1. Introduction
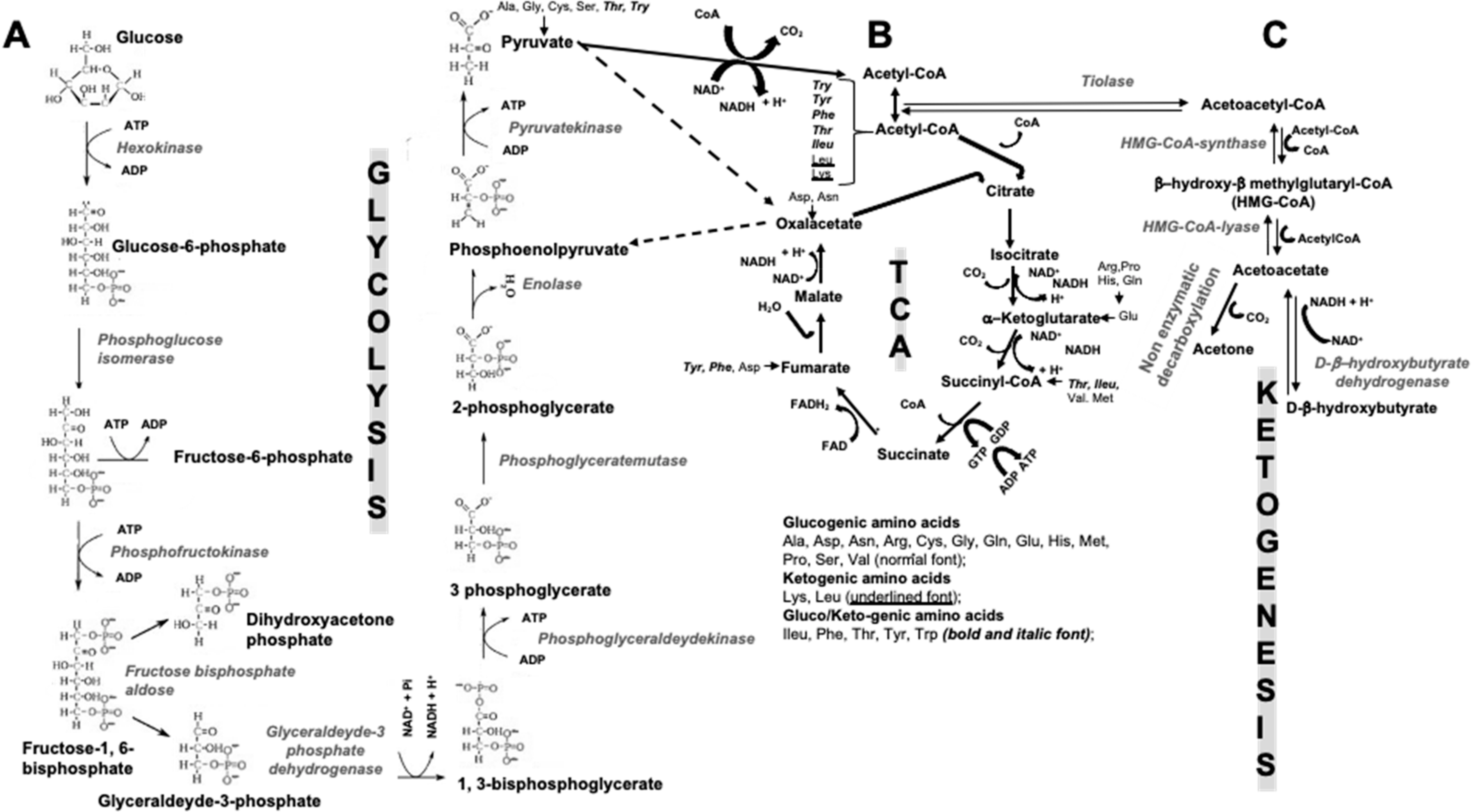
2. Ketogenic Diets
2.1. Forms of KDs
2.2. Are KBs the Only Main Source of Energy in Fast and/or Low Carbohydrate Periods?
3. Skeleton and Energy Metabolism: A Complex “Multiplayers Ping Pong Match Model”
3.1. Bone Remodeling Needs of Boosting Energy
3.2. Insulin, Osteocalcin, Osteoprotegerin, Receptor Activator of Nuclear Factor Kappa-B Ligand: A Complex, Interconnected, Network
Genetic Data on UcOC in Glucose Metabolism Regulation
4. The Role of Wingless-Type Mouse Mammary Virus Integration Site (Wnt) Pathway in Bone Pathophysiology and Energy Metabolism
4.1. Bone Cells-Specific Bioenergetic Program
4.1.1. “Fuel” Selection according to Bone Cells Differentiation Steps
4.1.2. Glutamine and Wnt-Dependent Bone Metabolism
4.1.3. Inhibiting the Inhibitor of the Wnt-Dependent Differentiation and Maturation of OBLs: Improvements of the Insulin Sensitivity in an Animal Model
4.2. Osteoanabolic Signals Stimulate Glycolysis
Is the Existence of an Insulin/PTH Axis Conceivable?
4.3. What Do We Know about the Role of IGF-1 in Bone Health?
4.4. Other Energetic Substrates in OBLs
5. How Could KDs and KBs Affect Bone Metabolism?
5.1. Animal Studies on KDs and Bone Metabolism
5.2. Clinical Studies on KDs and Human Bone Health
5.2.1. KDs, Growth Delay, and the Developing Skeleton
5.2.2. KDs and Calcium Metabolism
5.3. Duration of the KD Regimen and Study of Bone Health Parameters
5.4. VLCKD and Acid-Ash Proteins Diet
5.5. KDs and the Role of Mineral Supplements
6. Some Specific Key Points to Be Considered to Adequately Perform Clinical Study Having Bone Health as Primary Endpoint in Subjects Treated by KDs, VLCKD
6.1. The Importance of Least Significant Change Concept
6.1.1. DXA Measurements
6.1.2. BTMs Measurements
6.2. Other Suggestions
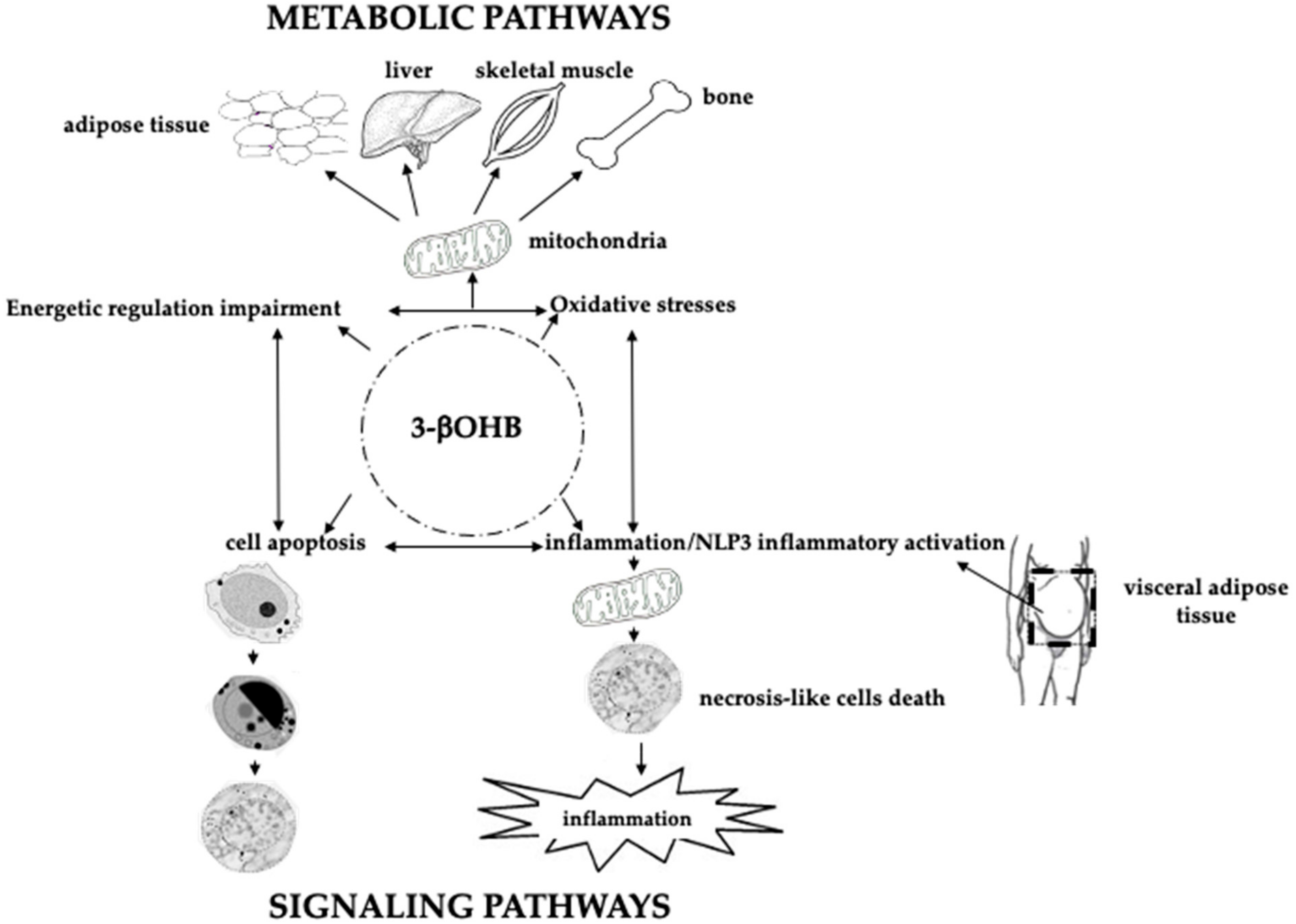
7. Inflammation, Diet, and Bone Health
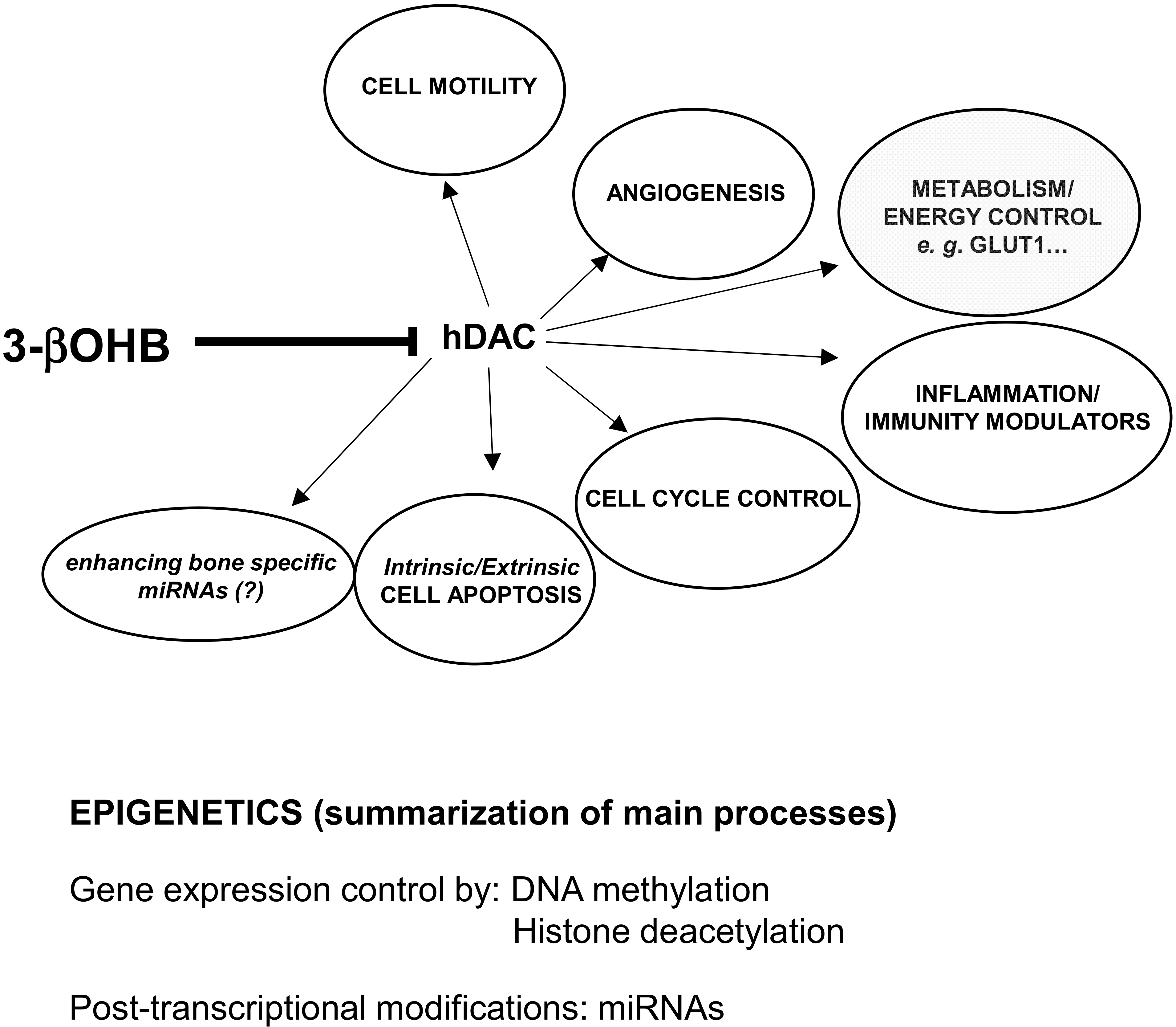
8. Future Perspectives on KDs, Microbiota, microRNAs, Bone Marrow Lipids Composition, and Bone Health
8.1. Microbiota, Short-Chain Fatty Acids, Butyrate, and Bone Health
8.2. KDs, miRNAs, and Bone Health
8.3. Bone Marrow Lipids Composition and Bone Health
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Perrot, P. A to Z of Thermodynamics; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Groesbeck, D.K.; Bluml, R.M.; Kossoff, E.H. Long-term use of the ketogenic diet in the treatment of epilepsy. Dev. Med. Child Neurol. 2007, 48, 978–981. [Google Scholar] [CrossRef]
- Kang, H.C.; Chung, D.E.; Kim, D.W.; Kim, H.D. Early- and Late-onset Complications of the Ketogenic Diet for Intractable Epilepsy. Epilepsia 2004, 45, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, A.C.; Schall, J.I.; Stallings, V.A. Vitamin D Status in Children with Intractable Epilepsy, and Impact of the Ketogenic Diet. Epilepsia 2007, 48, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, A.C.; Schall, J.I.; Stallings, V.A.; Zemel, B.S. Progressive bone mineral content loss in children with intractable epilepsy treated with the ketogenic diet. Am. J. Clin. Nutr. 2008, 88, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Bergqvist, A.C.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; the Cardiovascular Endocrinology Club of the Italian Society of Endocrinology; Infante, M.; Moriconi, E.; Armani, A.; Fabbri, A.; Mantovani, G.; Mariani, S.; Lubrano, C.; Poggiogalle, E.; et al. Very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: Systematic review and consensus statement from the Italian Society of Endocrinology (SIE). J. Endocrinol. Investig. 2019, 42, 1365–1386. [Google Scholar] [CrossRef]
- Daniel, S.; Soleymani, T.; Garvey, W.T. A complications-based clinical staging of obesity to guide treatment modality and intensity. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 377–388. [Google Scholar] [CrossRef]
- Stryer, L. Biochemistry, 4th ed.; W.H. Freeman and Company: New York, NY, USA, 1995; pp. 510–515, 581–613, 775–778. [Google Scholar]
- Koeslag, J.H.; Noakes, T.D.; Sloan, A.W. Post-exercise ketosis. J. Physiol. 1980, 301, 79–90. [Google Scholar] [CrossRef]
- Møller, N. Ketone Body, 3-Hydroxybutyrate: Minor Metabolite—Major Medical Manifestations. J. Clin. Endocrinol. Metab. 2020, 105, 2884–2892. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by -Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Devlin, M.J.; Rosen, C.J. The bone–fat interface: Basic and clinical implications of marrow adiposity. Lancet Diabetes Endocrinol. 2015, 3, 141–147. [Google Scholar] [CrossRef]
- Kanazawa, I. Osteocalcin as a hormone regulating glucose metabolism. World J. Diabetes 2015, 6, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Lacombe, J. Regulation of energy metabolism by the skeleton: Osteocalcin and beyond. Arch. Biochem. Biophys. 2014, 561, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Duffhues, G.; Hiepen, C.; Knaus, P.; Dijke, P.T. Bone morphogenetic protein signaling in bone homeostasis. Bone 2015, 80, 43–59. [Google Scholar] [CrossRef]
- Nakahama, K.-I. Cellular communications in bone homeostasis and repair. Cell. Mol. Life Sci. 2010, 67, 4001–4009. [Google Scholar] [CrossRef]
- Tsukamoto, I.; Hie, M.; Iitsuka, N.; Otsuka, T. Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activation. Int. J. Mol. Med. 2011, 28, 455–462. [Google Scholar] [CrossRef]
- Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 2007, 13, 791–801. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-β1–induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef]
- Rey, C.; Combes, C.; Drouet, C.; Glimcher, M.J. Bone mineral: Update on chemical composition and structure. Osteoporos. Int. 2009, 20, 1013–1021. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Kneissel, M.; Johnson, M. Preface: The Osteocyte. Bone 2013, 54, 181. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin Binds to LRP5/6 and Antagonizes Canonical Wnt Signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.; Guntur, A.R.; Long, F.; Rosen, C.J. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr. Rev. 2017, 38, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef]
- Lucero, C.M.; Vega, O.A.; Osorio, M.M.; Tapia, J.C.; Antonelli, M.; Stein, G.S.; Van Wijnen, A.J.; Galindo, M. The cancer-related transcription factor Runx2 modulates cell proliferation in human osteosarcoma cell lines. J. Cell. Physiol. 2013, 228, 714–723. [Google Scholar] [CrossRef]
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a Novel Regulator of Osteoblast Differentiation and Skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Kitamura, T.; Kitamura, Y.; Biggs, W.H.; Arden, K.C.; Accili, D. The Forkhead Transcription Factor Foxo1 Regulates Adipocyte Differentiation. Dev. Cell 2003, 4, 119–129. [Google Scholar] [CrossRef]
- Pi, M.; Wu, Y.; Quarles, L.D. GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J. Bone Miner. Res. 2011, 26, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Oury, F.; Ferron, M.; Huizhen, W.; Confavreux, C.; Xu, L.; Lacombe, J.; Srinivas, P.; Chamouni, A.; Lugani, F.; Lejeune, H.; et al. Osteocalcin regulates murine and human fertility through a pancreas-bone-testis axis. J. Clin. Investig. 2013, 123, 2421–2433. [Google Scholar] [CrossRef]
- Korostishevsky, M.; Malkin, I.; Trofimov, S.; Pei, Y.; Deng, H.-W.; Livshits, G. Significant association between body composition phenotypes and the osteocalcin genomic region in normative human population. Bone 2012, 51, 688–694. [Google Scholar] [CrossRef]
- Das, S.K.; Sharma, N.K.; Elbein, S.C. Analysis of Osteocalcin as a Candidate Gene for Type 2 Diabetes (T2D) and Intermediate Traits in Caucasians and African Americans. Dis. Markers 2010, 28, 281–286. [Google Scholar] [CrossRef]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; Depinho, R.A.; Teti, A.; Ducy, P.; Karsenty, G. Insulin Signaling in Osteoblasts Integrates Bone Remodeling and Energy Metabolism. Cell 2010, 142, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Midura, R.J.; Vasanji, A.; Wang, A.J.; Hascall, V.C. Hyperglycemia Diverts Dividing Osteoblastic Precursor Cells to an Adipogenic Pathway and Induces Synthesis of a Hyaluronan Matrix That Is Adhesive for Monocytes. J. Biol. Chem. 2014, 289, 11410–11420. [Google Scholar] [CrossRef] [PubMed]
- García-Hernández, A.; Arzate, H.; Gil-Chavarría, I.; Rojo, R.; Moreno-Fierros, L. High glucose concentrations alter the biomineralization process in human osteoblastic cells. Bone 2012, 50, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Ehnert, S.; Thomas, F.; Ihle, C.; Mayer, L.; Braun, B.; Graeser, J.; Flesch, I.; Stöckle, U.; Nussler, A.K.; Pscherer, S. Factors circulating in the blood of type 2 diabetes mellitus patients affect osteoblast maturation —Description of a novel in vitro model. Exp. Cell Res. 2015, 332, 247–258. [Google Scholar] [CrossRef]
- Kim, H.K.; Oxendine, I.; Kamiya, N. High-concentration of BMP2 reduces cell proliferation and increases apoptosis via DKK1 and SOST in human primary periosteal cells. Bone 2013, 54, 141–150. [Google Scholar] [CrossRef]
- Ke, H.Z.; Richards, W.G.; Li, X.; Ominsky, M.S. Sclerostin and Dickkopf-1 as Therapeutic Targets in Bone Diseases. Endocr. Rev. 2012, 33, 747–783. [Google Scholar] [CrossRef]
- Gennari, L.; Merlotti, D.; Falchetti, A.; Vainicher, C.E.; Cosso, R.; Chiodini, I. Emerging therapeutic targets for osteoporosis. Expert Opin. Ther. Targets 2020, 24, 115–130. [Google Scholar] [CrossRef]
- Fu, C.; Zhang, X.; Ye, F.; Yang, J. High Insulin Levels in KK-Ay Diabetic Mice Cause Increased Cortical Bone Mass and Impaired Trabecular Micro-Structure. Int. J. Mol. Sci. 2015, 16, 8213–8226. [Google Scholar] [CrossRef]
- Hie, M.; Shimono, M.; Fujii, K.; Tsukamoto, I. Increased cathepsin K and tartrate-resistant acid phosphatase expression in bone of streptozotocin-induced diabetic rats. Bone 2007, 41, 1045–1050. [Google Scholar] [CrossRef]
- Fujii, H.; Hamada, Y.; Fukagawa, M. Bone formation in spontaneously diabetic Torii-newly established model of non-obese type 2 diabetes rats. Bone 2008, 42, 372–379. [Google Scholar] [CrossRef]
- Won, H.Y.; Lee, J.-A.; Park, Z.S.; Song, J.S.; Kim, H.Y.; Jang, S.-M.; Yoo, S.-E.; Rhee, Y.; Hwang, E.S.; Bae, M.-A. Prominent Bone Loss Mediated by RANKL and IL-17 Produced by CD4+ T Cells in TallyHo/JngJ Mice. PLoS ONE 2011, 6, e18168. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ueharu, H.; Mishina, Y. Energy metabolism: A newly emerging target of BMP signaling in bone homeostasis. Bone 2020, 138, 115467. [Google Scholar] [CrossRef] [PubMed]
- Shum, L.C.; White, N.S.; Mills, B.N.; Bentley, K.L.D.M.; Eliseev, R.A. Energy Metabolism in Mesenchymal Stem Cells During Osteogenic Differentiation. Stem Cells Dev. 2016, 25, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Karner, C.; Long, F. Glucose metabolism in bone. Bone 2018, 115, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Fini, M.; Torricelli, P.; Giavaresi, G.; Carpi, A.; Nicolini, A.; Giardino, R. Effect of L-lysine and L-arginine on primary osteoblast cultures from normal and osteopenic rats. Biomed. Pharmacother. 2001, 55, 213–220. [Google Scholar] [CrossRef]
- El Refaey, M.; Watkins, C.P.; Kennedy, E.J.; Chang, A.; Zhong, Q.; Ding, K.-H.; Shi, X.-M.; Xu, J.; Bollag, W.B.; Hill, W.D.; et al. Oxidation of the aromatic amino acids tryptophan and tyrosine disrupts their anabolic effects on bone marrow mesenchymal stem cells. Mol. Cell. Endocrinol. 2015, 410, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-Q.; Cheung, L.S.; Feng, L.; Tanner, W.; Frommer, W.B. Transport of Sugars. Annu. Rev. Biochem. 2015, 84, 865–894. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. Membrane Fatty Acid Transporters as Regulators of Lipid Metabolism: Implications for Metabolic Disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef]
- Kushwaha, P.; Wolfgang, M.J.; Riddle, R.C. Fatty acid metabolism by the osteoblast. Bone 2018, 115, 8–14. [Google Scholar] [CrossRef]
- Schweikhard, E.S.; Ziegler, C.M. Amino Acid Secondary Transporters. Na Channels Phyla Funct. 2012, 70, 1–28. [Google Scholar] [CrossRef]
- Zoch, M.L.; Abou, D.S.; Clemens, T.L.; Thorek, D.L.J.; Riddle, R.C. In vivo radiometric analysis of glucose uptake and distribution in mouse bone. Bone Res. 2016, 4, 16004. [Google Scholar] [CrossRef]
- Wei, J.; Shimazu, J.; Makinistoglu, M.P.; Maurizi, A.; Kajimura, D.; Zong, H.; Takarada, T.; Iezaki, T.; Pessin, J.E.; Hinoi, E.; et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 2015, 161, 1576–1591. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.N.; Lim, J.; Shi, Y.; Joeng, K.S.; Arbeit, J.M.; Shohet, R.V.; Long, F. Up-regulation of glycolytic metabolism is required for HIF1 -driven bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 8673–8678. [Google Scholar] [CrossRef] [PubMed]
- Esen, E.; Lee, S.-Y.; Wice, B.M.; Long, F. PTH Promotes Bone Anabolism by Stimulating Aerobic Glycolysis via IGF Signaling. J. Bone Miner. Res. 2015, 30, 1959–1968. [Google Scholar] [CrossRef]
- Esen, E.; Chen, J.; Karner, C.M.; Okunade, A.L.; Patterson, B.W.; Long, F. WNT-LRP5 Signaling Induces Warburg Effect through mTORC2 Activation during Osteoblast Differentiation. Cell Metab. 2013, 17, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Hannah, S.S.; McFadden, S.; McNeilly, A.; McClean, C. “Take My Bone Away?” Hypoxia and bone: A narrative review. J. Cell. Physiol. 2021, 236, 721–740. [Google Scholar] [CrossRef] [PubMed]
- Riddle, R.C. Parathyroid Hormone Reprograms Osteoblast Metabolism. J. Bone Miner. Res. 2015, 30, 1956–1958. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, B.; Nichols, G. Parathyroid Inhibition of Bone Collagen Synthesis. Endocrinology 1964, 74, 180–186. [Google Scholar] [CrossRef]
- Moorer, M.C.; Riddle, R.C. Regulation of Osteoblast Metabolism by Wnt Signaling. Endocrinol. Metab. 2018, 33, 318–330. [Google Scholar] [CrossRef]
- Thompson, J.; Wu, G. The effect of ketone bodies on nitrogen metabolism in skeletal muscle. Comp. Biochem. Physiol. Part B Comp. Biochem. 1991, 100, 209–216. [Google Scholar] [CrossRef]
- HoleČek, M. Side Effects of Long-Term Glutamine Supplementation. J. Parenter. Enter. Nutr. 2012, 37, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Karner, C.M.; Esen, E.; Okunade, A.L.; Patterson, B.W.; Long, F. Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. J. Clin. Investig. 2015, 125, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Frey, J.L.; Li, Z.; Ellis, J.M.; Zhang, Q.; Farber, C.R.; Aja, S.; Wolfgang, M.J.; Clemens, T.L.; Riddle, R.C. Wnt-Lrp5 Signaling Regulates Fatty Acid Metabolism in the Osteoblast. Mol. Cell. Biol. 2015, 35, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Elefteriou, F.; Benson, M.D.; Sowa, H.; Starbuck, M.; Liu, X.; Ron, D.; Parada, L.F.; Karsenty, G. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 2006, 4, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xia, R.; Yue, C.; Zhai, W.; Du, W.; Yang, Q.; Cao, H.; Chen, X.; Obando, D.; Zhu, Y.; et al. ATF4 Regulates CD4+ T Cell Immune Responses through Metabolic Reprogramming. Cell Rep. 2018, 23, 1754–1766. [Google Scholar] [CrossRef]
- Kim, S.P.; Frey, J.L.; Lim, N.H.; Kushwaha, P.; Zoch, M.L.; Tomlinson, R.E.; Da, H.; Aja, S.; Noh, H.L.; Kim, J.K.; et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E11238–E11247. [Google Scholar] [CrossRef]
- Kim, H.; Lee, Y.D.; Kim, H.J.; Lee, Z.H.; Kim, H.-H. SOD2 and Sirt3 Control Osteoclastogenesis by Regulating Mitochondrial ROS. J. Bone Miner. Res. 2017, 32, 397–406. [Google Scholar] [CrossRef]
- Huh, J.-E.; Shin, J.H.; Jang, E.S.; Park, S.J.; Park, D.R.; Ko, R.; Seo, D.-H.; Kim, H.-S.; Lee, S.H.; Choi, Y.; et al. Sirtuin 3 (SIRT3) maintains bone homeostasis by regulating AMPK-PGC-1β axis in mice. Sci. Rep. 2016, 6, 22511. [Google Scholar] [CrossRef]
- Robling, A.G.; Bonewald, L.F. The Osteocyte: New Insights. Annu. Rev. Physiol. 2020, 82, 485–506. [Google Scholar] [CrossRef]
- Hinoi, E.; Takarada, T.; Tsuchihashi, Y.; Fujimori, S.; Moriguchi, N.; Wang, L.; Uno, K.; Yoneda, Y. A Molecular Mechanism of Pyruvate Protection against Cytotoxicity of Reactive Oxygen Species in Osteoblasts. Mol. Pharmacol. 2006, 70, 925–935. [Google Scholar] [CrossRef]
- Clowes, J.A.; Allen, H.C.; Prentis, D.M.; Eastell, R.; Blumsohn, A. Octreotide Abolishes the Acute Decrease in Bone Turnover in Response to Oral Glucose. J. Clin. Endocrinol. Metab. 2003, 88, 4867–4873. [Google Scholar] [CrossRef]
- D’Amelio, P.; Sassi, F.; Buondonno, I.; Spertino, E.; Tamone, C.; Piano, S.; Zugna, D.; Richiardi, L.; Isaia, G.C. Effect of intermittent PTH treatment on plasma glucose in osteoporosis: A randomized trial. Bone 2015, 76, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Sass, M.R.; Albrechtsen, N.J.W.; Pedersen, J.; Hare, K.J.; Borbye-Lorenzen, N.; Kiss, K.; Vilsbøll, T.; Knop, F.K.; Poulsen, S.S.; Jørgensen, N.R.; et al. Secretion of parathyroid hormone may be coupled to insulin secretion in humans. Endocr. Connect. 2020, 9, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.M.; Lai, T.; Holly, J.M.P.; Wheeler, M.H.; Stewart, C.; Farndon, J.R. Insulin-like Growth Factors (IGF) I and II Utilize Different Calcium Signaling Pathways in a Primary Human Parathyroid Cell Culture Model. World J. Surg. 2006, 30, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Giustina, A.; Mazziotti, G.; Canalis, E. Growth Hormone, Insulin-Like Growth Factors, and the Skeleton. Endocr. Rev. 2008, 29, 535–559. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Werner, H.; Rosen, C.J. 40 YEARS OF IGF1: Insulin-like growth factors: Actions on the skeleton. J. Mol. Endocrinol. 2018, 61, T115–T137. [Google Scholar] [CrossRef]
- Rosen, C.J. Insulin-like growth factor I and bone mineral density: Experience from animal models and human observational studies. Best Pr. Res. Clin. Endocrinol. Metab. 2004, 18, 423–435. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kanatani, M.; Yamauchi, M.; Kaji, H.; Sugishita, T.; Baylink, D.J.; Mohan, S.; Chihara, K.; Sugimoto, T. Serum Levels of Insulin-Like Growth Factor (IGF); IGF-Binding Proteins-3, -4, and -5; and Their Relationships to Bone Mineral Density and the Risk of Vertebral Fractures in Postmenopausal Women. Calcif. Tissue Int. 2006, 78, 18–24. [Google Scholar] [CrossRef]
- Garnero, P.; Sornay-Rendu, E.; Delmas, P. Low serum IGF-1 and occurrence of osteoporotic fractures in postmenopausal women. Lancet 2000, 355, 898–899. [Google Scholar] [CrossRef]
- Yanovski, J.A.; Sovik, K.N.; Nguyen, T.T.; Sebring, N.G. Insulin-like growth factors and bone mineral density in African American and white girls. J. Pediatr. 2000, 137, 826–832. [Google Scholar] [CrossRef]
- Lloyd, M.E.; Hart, D.J.; Nandra, D.; McAlindon, T.E.; Wheeler, M.; Doyle, D.V.; Spector, T.D. Relation between insulin-like growth factor-I concentrations, osteoarthritis, bone density, and fractures in the general population: The Chingford study. Ann. Rheum. Dis. 1996, 55, 870–874. [Google Scholar] [CrossRef]
- Martini, G.; Valenti, R.; Giovani, S.; Franci, B.; Campagna, S.; Nuti, R. Influence of insulin-like growth factor-1 and leptin on bone mass in healthy postmenopausal women. Bone 2001, 28, 113–117. [Google Scholar] [CrossRef]
- Schurch, M.-A.; Rizzoli, R.; Slosman, D.; Vadas, L.; Vergnaud, P.; Bonjour, J.-P. Protein Supplements Increase Serum Insulin-Like Growth Factor-I Levels and Attenuate Proximal Femur Bone Loss in Patients with Recent Hip Fracture. Ann. Intern. Med. 1998, 128, 801–809. [Google Scholar] [CrossRef]
- Friedlander, A.L.; Butterfield, G.E.; Moynihan, S.; Grillo, J.; Pollack, M.; Holloway, L.; Friedman, L.; Yesavage, J.; Matthias, D.; Lee, S.; et al. One Year of Insulin-Like Growth Factor I Treatment Does Not Affect Bone Density, Body Composition, or Psychological Measures in Postmenopausal Women1. J. Clin. Endocrinol. Metab. 2001, 86, 1496–1503. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.; Cheng, C.M.; Kopchick, J.J.; A Bondy, C. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J. Endocrinol. 2004, 180, 247–255. [Google Scholar] [CrossRef]
- A Fraser, D.; Thoen, J.; Bondhus, S.; Haugen, M.; E Reseland, J.; Djøseland, O.; Førre, O.; Kjeldsen-Kragh, J. Reduction in serum leptin and IGF-1 but preserved T-lymphocyte numbers and activation after a ketogenic diet in rheumatoid arthritis patients. Clin. Exp. Rheumatol. 2000, 18, 209–214. [Google Scholar]
- Spulber, G.; Spulber, S.; Hagenäs, L.; Åmark, P.; Dahlin, M. Growth dependence on insulin-like growth factor-1 during the ketogenic diet. Epilepsia 2009, 50, 297–303. [Google Scholar] [CrossRef]
- Groleau, V.; I Schall, J.; A Stallings, V.; A Bergqvist, C. Long-term impact of the ketogenic diet on growth and resting energy expenditure in children with intractable epilepsy. Dev. Med. Child Neurol. 2014, 56, 898–904. [Google Scholar] [CrossRef]
- Rendina-Ruedy, E.; Guntur, A.R.; Rosen, C.J. Intracellular lipid droplets support osteoblast function. Adipocyte 2017, 6, 250–258. [Google Scholar] [CrossRef]
- Maridas, D.E.; Rendina-Ruedy, E.; Helderman, R.C.; DeMambro, V.E.; Brooks, D.; Guntur, A.R.; Lanske, B.; Bouxsein, M.L.; Rosen, C.J. Progenitor recruitment and adipogenic lipolysis contribute to the anabolic actions of parathyroid hormone on the skeleton. FASEB J. 2018, 33, 2885–2898. [Google Scholar] [CrossRef]
- Rendina-Ruedy, E.; Rosen, C.J. Lipids in the Bone Marrow: An Evolving Perspective. Cell Metab. 2020, 31, 219–231. [Google Scholar] [CrossRef]
- Frey, J.L.; Kim, S.P.; Li, Z.; Wolfgang, M.J.; Riddle, R.C. β-Catenin Directs Long-Chain Fatty Acid Catabolism in the Osteoblasts of Male Mice. Endocrinology 2017, 159, 272–284. [Google Scholar] [CrossRef]
- Kevorkova, O.; Martineau, C.; Martin-Falstrault, L.; Sanchez-Dardon, J.; Brissette, L.; Moreau, R. Low-Bone-Mass Phenotype of Deficient Mice for the Cluster of Differentiation 36 (CD36). PLoS ONE 2013, 8, e77701. [Google Scholar] [CrossRef]
- Gao, B.; Huang, Q.; Jie, Q.; Lu, W.-G.; Wang, L.; Li, X.-J.; Sun, Z.; Hu, Y.-Q.; Chen, L.; Liu, B.-H.; et al. GPR120: A bi-potential mediator to modulate the osteogenic and adipogenic differentiation of BMMSCs. Sci. Rep. 2015, 5, 14080. [Google Scholar] [CrossRef]
- Pegorier, J.P.; Garcia-Garcia, M.V.; Prip-Buus, C.; Duée, P.H.; Kohl, C.; Girard, J. Induction of ketogenesis and fatty acid oxidation by glucagon and cyclic AMP in cultured hepatocytes from rabbit fetuses. Evidence for a decreased sensitivity of carnitine palmitoyltransferase I to malonyl-CoA inhibition after glucagon or cyclic AMP treatment. Biochem. J. 1989, 264, 93–100. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Dominy, J.E.; Blättler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.-H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA Pathway Rapidly Activates SIRT1 to Promote Fatty Acid Oxidation Independently of Changes in NAD+. Mol. Cell 2011, 44, 851–863. [Google Scholar] [CrossRef]
- Saito, A.; Yoshimura, K.; Miyamoto, Y.; Kaneko, K.; Chikazu, D.; Yamamoto, M.; Kamijo, R. Enhanced and suppressed mineralization by acetoacetate and β-hydroxybutyrate in osteoblast cultures. Biochem. Biophys. Res. Commun. 2016, 473, 537–544. [Google Scholar] [CrossRef]
- Yamasaki, M.; Hasegawa, S.; Suzuki, H.; Hidai, K.; Saitoh, Y.; Fukui, T. Acetoacetyl-CoA synthetase gene is abundant in rat adipose, and related with fatty acid synthesis in mature adipocytes. Biochem. Biophys. Res. Commun. 2005, 335, 215–219. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef]
- Cao, Q.; Zhang, J.; Liu, H.; Wu, Q.; Chen, J.; Chen, G.-Q. The mechanism of anti-osteoporosis effects of 3-hydroxybutyrate and derivatives under simulated microgravity. Biomaterials 2014, 35, 8273–8283. [Google Scholar] [CrossRef]
- Wu, X.; Huang, Z.; Wang, X.; Fu, Z.; Liu, J.; Huang, Z.; Kong, G.; Xu, X.; Ding, J.; Zhu, Q. Ketogenic Diet Compromises Both Cancellous and Cortical Bone Mass in Mice. Calcif. Tissue Int. 2017, 101, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Rubenstein, J.E.; Kossoff, E.H. Intermittent fasting: A “new” historical strategy for controlling seizures? Epilepsy Res. 2012, 104, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Q.; Zhou, J.; Wu, X.; Zhu, Q. β hydroxybutyrate levels in serum and cerebrospinal fluid under ketone body metabolism in rats. Exp. Anim. 2017, 66, 177–182. [Google Scholar] [CrossRef]
- Xu, X.; Ding, J.; Wu, X.; Huang, Z.; Kong, G.; Liu, Q.; Yang, Z.; Huang, Z.; Zhu, Q. Bone microstructure and metabolism changes under the combined intervention of ketogenic diet with intermittent fasting: An in vivo study of rats. Exp. Anim. 2019, 68, 371–380. [Google Scholar] [CrossRef]
- Ding, J.; Xu, X.; Wu, X.; Huang, Z.; Kong, G.; Liu, J.; Huang, Z.; Liu, Q.; Li, R.; Yang, Z.; et al. Bone loss and biomechanical reduction of appendicular and axial bones under ketogenic diet in rats. Exp. Ther. Med. 2019, 17, 2503–2510. [Google Scholar] [CrossRef]
- Wu, X.; Ding, J.; Xu, X.; Wang, X.; Liu, J.; Jiang, J.; Liu, Q.; Kong, G.; Huang, Z.; Yang, Z.; et al. Ketogenic diet compromises vertebral microstructure and biomechanical characteristics in mice. J. Bone Miner. Metab. 2019, 37, 957–966. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, X.; Yang, Z.; Liu, Y.; Wu, X.; Huang, Z.; Liu, J.; Huang, Z.; Kong, G.; Ding, J.; et al. Metformin Alleviates the Bone Loss Induced by Ketogenic Diet: An In Vivo Study in Mice. Calcif. Tissue Int. 2019, 104, 59–69. [Google Scholar] [CrossRef]
- Bielohuby, M.; Matsuura, M.; Herbach, N.; Kienzle, E.; Slawik, M.; Hoeflich, A.; Bidlingmaier, M. Short-term exposure to low-carbohydrate, high-fat diets induces low bone mineral density and reduces bone formation in rats. J. Bone Miner. Res. 2009, 25, 275–284. [Google Scholar] [CrossRef]
- Nishizawa, Y.; Koyama, H.; Shoji, T.; Tahara, H.; Hagiwara, S.; Aratani, H.; Nakatsuka, K.; Miki, T.; Morii, H. Altered calcium homeostasis accompanying changes of regional bone mineral during a very-low-calorie diet. Am. J. Clin. Nutr. 1992, 56, 265S–267S. [Google Scholar] [CrossRef]
- Davie, M.W.J.; Abraham, R.R.; Hewins, B.; Wynn, V. Changes in bone and muscle constituents during dieting for obesity. Clin. Sci. 1986, 70, 285–293. [Google Scholar] [CrossRef]
- Andersen, R.E.; Wadden, T.A.; Herzog, R.J. Changes in bone mineral content in obese dieting women. Metabolism 1997, 46, 857–861. [Google Scholar] [CrossRef]
- Howard, A.N.; Kreitzman, S.N. The Swansea Trial: Body Composition and Metabolic Studies with a Very-Low-Calorie Diet (VLCD); Kreitzman, S., Howard, A., Eds.; Smith Gordon: London, UK, 1993; p. 173. ISBN 1-85463-070-9. [Google Scholar]
- Bonjour, J.-P.; Theintz, G.; Buchs, B.; Slosman, D.; Rizzoli, R. Critical Years and Stages of Puberty for Spinal and Femoral Bone Mass Accumulation during Adolescence. J. Clin. Endocrinol. Metab. 1991, 73, 555–563. [Google Scholar] [CrossRef]
- Joseph^Melton, L.; Kan, S.H.; Wahner, H.W.; Riggs, B.L. Lifetime fracture risk: An approach to hip fracture risk assessment based on bone mineral density and age. J. Clin. Epidemiol. 1988, 41, 985–994. [Google Scholar] [CrossRef]
- Henderson, R.C.; Lark, R.K.; Gurka, M.J.; Worley, G.; Fung, E.B.; Conaway, M.; Stallings, V.A.; Stevenson, R.D. Bone density and metabolism in children and adolescents with moderate to severe cerebral palsy. Pediatrics 2002, 110, e5. [Google Scholar] [CrossRef]
- McNamara, N.A.; Romanowski, E.M.F.; Olson, D.P.; Shellhaas, R.A. Bone Health and Endocrine Comorbidities in Pediatric Epilepsy. Semin. Pediatr. Neurol. 2017, 24, 301–309. [Google Scholar] [CrossRef]
- Simm, P.J.; Bicknell-Royle, J.; Lawrie, J.; Nation, J.; Draffin, K.; Stewart, K.G.; Cameron, F.J.; Scheffer, I.E.; Mackay, M.T. The effect of the ketogenic diet on the developing skeleton. Epilepsy Res. 2017, 136, 62–66. [Google Scholar] [CrossRef]
- Peterson, S.J.; Tangney, C.C.; Pimentel-Zablah, E.M.; Hjelmgren, B.; Booth, G.; Berry-Kravis, E. Changes in Growth and Seizure Reduction in Children on the Ketogenic Diet as a Treatment for Intractable Epilepsy. J. Am. Diet. Assoc. 2005, 105, 718–724. [Google Scholar] [CrossRef]
- Armeno, M.; Verini, A.; Del Pino, M.; Araujo, M.B.; Mestre, G.; Reyes, J.G.; Caraballo, R.H. A Prospective Study on Changes in Nutritional Status and Growth Following Two Years of Ketogenic Diet (KD) Therapy in Children with Refractory Epilepsy. Nutrients 2019, 11, 1596. [Google Scholar] [CrossRef]
- Colica, C.; Merra, G.; Gasbarrini, A.; De Lorenzo, A.; Cioccoloni, G.; Gualtieri, P.; Perrone, M.A.; Bernardini, S.; Bernardo, V.; Di Renzo, L.; et al. Efficacy and safety of very-low-calorie ketogenic diet: A double blind randomized crossover study. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2274–2289. [Google Scholar]
- Kossoff, E.H.; McGrogan, J.R.; Bluml, R.M.; Pillas, D.J.; Rubenstein, J.E.; Vining, E.P. A Modified Atkins Diet Is Effective for the Treatment of Intractable Pediatric Epilepsy. Epilepsia 2006, 47, 421–424. [Google Scholar] [CrossRef]
- Chen, W.; Kossoff, E.H. Long-Term Follow-Up of Children Treated with the Modified Atkins Diet. J. Child Neurol. 2012, 27, 754–758. [Google Scholar] [CrossRef]
- Svedlund, A.; Hallböök, T.; Magnusson, P.; Dahlgren, J.; Swolin-Eide, D. Prospective study of growth and bone mass in Swedish children treated with the modified Atkins diet. Eur. J. Paediatr. Neurol. 2019, 23, 629–638. [Google Scholar] [CrossRef]
- Draaisma, J.M.; Hampsink, B.M.; Janssen, M.; Van Houdt, N.B.; Linders, E.T.; Willemsen, M.A. The Ketogenic Diet and Its Effect on Bone Mineral Density: A Retrospective Observational Cohort Study. Neuropediatrics 2019, 50, 353–358. [Google Scholar] [CrossRef]
- Aghajanian, P.; Hall, S.; Wongworawat, M.D.; Mohan, S. The Roles and Mechanisms of Actions of Vitamin C in Bone: New Developments. J. Bone Miner. Res. 2015, 30, 1945–1955. [Google Scholar] [CrossRef]
- Falchetti, A.; Cosso, R. The interaction between vitamin C and bone health: A narrative review. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 215–223. [Google Scholar] [CrossRef]
- Yaşar, E.; Adigüzel, E.; Arslan, M.; Matthews, D.J. Basics of bone metabolism and osteoporosis in common pediatric neuromuscular disabilities. Eur. J. Paediatr. Neurol. 2018, 22, 17–26. [Google Scholar] [CrossRef]
- Ko, A.; Kong, J.; Samadov, F.; Mukhamedov, A.; Kim, Y.M.; Lee, Y.-J.; Nam, S.O. Bone health in pediatric patients with neurological disorders. Ann. Pediatr. Endocrinol. Metab. 2020, 25, 15–23. [Google Scholar] [CrossRef]
- Hahn, T.J.; Halstead, L.R.; DeVivo, D.C. Disordered mineral metabolism produced by ketogenic diet therapy. Calcif. Tissue Int. 1979, 28, 17–22. [Google Scholar] [CrossRef]
- Scheller, E.L.; Khoury, B.; Moller, K.L.; Wee, N.K.Y.; Khandaker, S.; Kozloff, K.M.; Abrishami, S.H.; Zamarron, B.F.; Singer, K. Changes in Skeletal Integrity and Marrow Adiposity during High-Fat Diet and after Weight Loss. Front. Endocrinol. 2016, 7, 102. [Google Scholar] [CrossRef]
- Scott, M.C.; Fuller, S.E.; Watt, J.D.; Osborn, M.L.; Johannsen, N.M.; Irving, B.A.; Noland, R.C. Cortical and Trabecular Bone Morphology in Response to Exercise and a Ketogenic Diet. Med. Sci. Sports Exerc. 2019, 51, 755–756. [Google Scholar] [CrossRef]
- Sheth, R.D.; Wesolowski, C.A.; Jacob, J.; Penney, S.; Hobbs, G.R.; Riggs, J.E.; Bodensteiner, J.B. Effect of carbamazepine and valproate on bone mineral density. J. Pediatr. 1995, 127, 256–262. [Google Scholar] [CrossRef]
- Petty, S.; Paton, L.M.; O’Brien, T.J.; Makovey, J.; Erbas, B.; Sambrook, P.; Berkovic, S.F.; Wark, J.D. Effect of antiepileptic medication on bone mineral measures. Neurology 2005, 65, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Babayigit, A.; Dirik, E.; Bober, E.; Cakmakcı, H. Adverse Effects of Antiepileptic Drugs on Bone Mineral Density. Pediatr. Neurol. 2006, 35, 177–181. [Google Scholar] [CrossRef]
- Sheth, R.D.; Montouris, G. Chapter 19 Metabolic Effects of AEDs. Int. Rev. Neurobiol. 2008, 83, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Fuleihan, G.E.-H.; Dib, L.; Yamout, B.; Sawaya, R.; Mikati, M.A. Predictors of bone density in ambulatory patients on antiepileptic drugs. Bone 2008, 43, 149–155. [Google Scholar] [CrossRef]
- Keene, D.L. A Systematic Review of the Use of the Ketogenic Diet in Childhood Epilepsy. Pediatr. Neurol. 2006, 35, 1–5. [Google Scholar] [CrossRef]
- Hartman, A.L.; Stafstrom, C.E. Harnessing the power of metabolism for seizure prevention: Focus on dietary treatments. Epilepsy Behav. 2013, 26, 266–272. [Google Scholar] [CrossRef]
- Neal, E.G.; Chaffe, H.M.; Edwards, N.; Lawson, M.S.; Schwartz, R.H.; Cross, J.H. Growth of Children on Classical and Medium-Chain Triglyceride Ketogenic Diets. Pediatrics 2008, 122, e334–e340. [Google Scholar] [CrossRef]
- Sampath, A.; Kossoff, E.H.; Furth, S.L.; Pyzik, P.L.; Vining, E.P.G. Kidney Stones and the Ketogenic Diet: Risk Factors and Prevention. J. Child Neurol. 2007, 22, 375–378. [Google Scholar] [CrossRef]
- Carter, J.D.; Vasey, F.B.; Valeriano, J. The effect of a low-carbohydrate diet on bone turnover. Osteoporos. Int. 2006, 17, 1398–1403. [Google Scholar] [CrossRef]
- Bertoli, S.; Trentani, C.; Ferraris, C.; De Giorgis, V.; Veggiotti, P.; Tagliabue, A. Long-term effects of a ketogenic diet on body composition and bone mineralization in GLUT-1 deficiency syndrome: A case series. Nutrients 2014, 30, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, M.C.; Henry, B.J.; Felton, E.A.; Patton, K.; Kossoff, E.H. Establishing an Adult Epilepsy Diet Center: Experience, efficacy and challenges. Epilepsy Behav. 2016, 58, 61–68. [Google Scholar] [CrossRef]
- Husari, K.S.; Cervenka, M.C. The ketogenic diet all grown up—Ketogenic diet therapies for adults. Epilepsy Res. 2020, 162, 106319. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.P.R.; Sale, C.; Greeves, J.P.; Casey, A.; Dutton, J.; Fraser, W.D. Effect of fasting versus feeding on the bone metabolic response to running. Bone 2012, 51, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Sale, C.; Varley, I.; Jones, T.W.; James, R.M.; Tang, J.C.Y.; Fraser, W.D.; Greeves, J.P. Effect of carbohydrate feeding on the bone metabolic response to running. J. Appl. Physiol. 2015, 119, 824–830. [Google Scholar] [CrossRef]
- Townsend, R.; Elliott-Sale, K.J.; Currell, K.; Tang, J.; Fraser, W.D.; Sale, C. The effect of post exercise carbohydrate and protein ingestion on bone metabolism. Med. Sci. Sports Exerc. 2017, 49, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Hammond, K.M.; Sale, C.; Fraser, W.; Tang, J.; Shepherd, S.O.; Strauss, J.A.; Close, G.L.; Cocks, M.; Louis, J.; Pugh, J.N.; et al. Post-exercise carbohydrate and energy availability induce independent effects on skeletal muscle cell signalling and bone turnover: Implications for training adaptation. J. Physiol. 2019, 597, 4779–4796. [Google Scholar] [CrossRef]
- McKay, A.K.A.; Peeling, P.; Pyne, D.B.; Welvaert, M.; Tee, N.; Leckey, J.J.; Sharma, A.P.; Ross, M.L.R.; Garvican-Lewis, L.A.; Swinkels, D.W.; et al. Chronic Adherence to a Ketogenic Diet Modifies Iron Metabolism in Elite Athletes. Med. Sci. Sports Exerc. 2019, 51, 548–555. [Google Scholar] [CrossRef]
- Heikura, I.A.; Bowen, D.J.; Hawley, J.A.; Ross, M.L.; Garvican-Lewis, L.; Sharma, A.P.; McKay, A.K.A.; Leckey, J.J.; Welvaert, M.; McCall, L.; et al. A Short-Term Ketogenic Diet Impairs Markers of Bone Health in Response to Exercise. Front. Endocrinol. 2020, 10, 880. [Google Scholar] [CrossRef]
- Fenton, T.R.; Lyon, A.W.; Eliasziw, M.; Tough, S.C.; Hanley, D.A. Meta-Analysis of the Effect of the Acid-Ash Hypothesis of Osteoporosis on Calcium Balance. J. Bone Miner. Res. 2009, 24, 1835–1840. [Google Scholar] [CrossRef]
- Barzel, U.S.; Massey, L.K. Excess Dietary Protein Can Adversely Affect Bone. J. Nutr. 1998, 128, 1051–1053. [Google Scholar] [CrossRef]
- Palermo, A.; Naciu, A.M.; Tabacco, G.; Manfrini, S.; Trimboli, P.; Vescini, F.; Falchetti, A.; Falchetti, A. Calcium citrate: From biochemistry and physiology to clinical applications. Rev. Endocr. Metab. Disord. 2019, 20, 353–364. [Google Scholar] [CrossRef]
- Heaney, R.P. Bone mineral content, not bone mineral density, is the correct bone measure for growth studies. Am. J. Clin. Nutr. 2003, 78, 350–351. [Google Scholar] [CrossRef]
- Schafer, A.; Vittinghoff, E.; Ramachandran, R.; Mahmoudi, N.; Bauer, D.C. Laboratory reproducibility of biochemical markers of bone turnover in clinical practice. Osteoporos. Int. 2009, 21, 439–445. [Google Scholar] [CrossRef][Green Version]
- Prentice, A.; Parsons, T.J.; Cole, T.J. Uncritical use of bone mineral density in absorptiometry may lead to size-related artifacts in the identification of bone mineral determinants. Am. J. Clin. Nutr. 1994, 60, 837–842. [Google Scholar] [CrossRef]
- Ruosi, C.; Liccardo, S.; Rossi, D.; Colella, G.; Di Somma, C.; Colao, A. Importance of spinal deformity index in risk evaluation of VCF (vertebral compression fractures) in obese subjects: Prospective study. Eur. Spine J. 2013, 22, 945–949. [Google Scholar] [CrossRef]
- Eller-Vainicher, C.; Falchetti, A.; Gennari, L.; Cairoli, E.; Bertoldo, F.; Vescini, F.; Scillitani, A.; Chiodini, I. DIAGNOSIS OF ENDOCRINE DISEASE: Evaluation of bone fragility in endocrine disorders. Eur. J. Endocrinol. 2019, 180, R213–R232. [Google Scholar] [CrossRef]
- Naylor, K.; Jacques, R.M.; Paggiosi, M.; Gossiel, F.; Peel, N.F.A.; McCloskey, E.V.; Walsh, J.S.; Eastell, R. Response of bone turnover markers to three oral bisphosphonate therapies in postmenopausal osteoporosis: The TRIO study. Osteoporos. Int. 2016, 27, 21–31. [Google Scholar] [CrossRef]
- Diez-Perez, A.; Adherence Working Group of the International Osteoporosis Foundation and the European Calcified Tissue Society; Naylor, K.E.; Abrahamsen, B.; Agnusdei, D.; Brandi, M.L.; Cooper, C.; Dennison, E.; Eriksen, E.F.; Gold, D.T.; et al. International Osteoporosis Foundation and European Calcified Tissue Society Working Group. Recommendations for the screening of adherence to oral bisphosphonates. Osteoporos. Int. 2017, 28, 767–774. [Google Scholar] [CrossRef]
- Shepherd, J.A.; Schousboe, J.T.; Broy, S.B.; Engelke, K.; Leslie, W.D. Executive Summary of the 2015 ISCD Position Development Conference on Advanced Measures from DXA and QCT: Fracture Prediction Beyond BMD. J. Clin. Densitom. 2015, 18, 274–286. [Google Scholar] [CrossRef]
- Schett, G. Effects of inflammatory and anti-inflammatory cytokines on the bone. Eur. J. Clin. Investig. 2011, 41, 1361–1366. [Google Scholar] [CrossRef]
- Loi, F.; Córdova, L.A.; Pajarinen, J.; Lin, T.-H.; Yao, Z.; Goodman, S.B. Inflammation, fracture and bone repair. Bone 2016, 86, 119–130. [Google Scholar] [CrossRef]
- Almeida, M.; Martin-Millan, M.; Ambrogini, E.; Bradsher, R.; Han, L.; Chen, X.-D.; Roberson, P.K.; Weinstein, R.S.; O’Brien, C.A.; Jilka, R.L.; et al. Estrogens Attenuate Oxidative Stress and the Differentiation and Apoptosis of Osteoblasts by DNA Binding-Independent Actions of the ERα. J. Bone Miner. Res. 2009, 25, 769–781. [Google Scholar] [CrossRef]
- Pfeilschifter, J.; Köditz, R.; Pfohl, M.; Schatz, H. Changes in Proinflammatory Cytokine Activity after Menopause. Endocr. Rev. 2002, 23, 90–119. [Google Scholar] [CrossRef]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Manolagas, S.C.; Parfitt, A.M. What old means to bone. Trends Endocrinol. Metab. 2010, 21, 369–374. [Google Scholar] [CrossRef]
- Shivappa, N.; E Steck, S.; Hurley, T.G.; Hussey, J.R.; Hébert, J.R. Designing and developing a literature-derived, population-based dietary inflammatory index. Public Health Nutr. 2014, 17, 1689–1696. [Google Scholar] [CrossRef]
- Shivappa, N.; Hébert, J.R.; Karamati, M.; Shariati-Bafghi, S.-E.; Rashidkhani, B. Increased inflammatory potential of diet is associated with bone mineral density among postmenopausal women in Iran. Eur. J. Nutr. 2015, 55, 561–568. [Google Scholar] [CrossRef]
- Orchard, T.; Yildiz, V.; Steck, S.E.; Hébert, J.R.; Ma, Y.; Cauley, J.A.; Li, W.; Mossavar-Rahmani, Y.; Johnson, K.C.; Sattari, M.; et al. Dietary Inflammatory Index, Bone Mineral Density, and Risk of Fracture in Postmenopausal Women: Results from the Women’s Health Initiative. J. Bone Miner. Res. 2017, 32, 1136–1146. [Google Scholar] [CrossRef]
- Kim, H.S.; Sohn, C.; Kwon, M.; Na, W.; Shivappa, N.; Hébert, J.R.; Kim, M.K. Positive Association between Dietary Inflammatory Index and the Risk of Osteoporosis: Results from the KoGES_Health Examinee (HEXA) Cohort Study. Nutrients 2018, 10, 1999. [Google Scholar] [CrossRef]
- Correa-Rodríguez, M.; Ortego-Centeno, N.; González-Jiménez, E.; Correa-Bautista, J.E.; Ramírez-Vélez, R.; Schmidt-RioValle, J. Dietary inflammatory index, bone health and body composition in a population of young adults: A cross-sectional study. Int. J. Food Sci. Nutr. 2018, 69, 1013–1019. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic Analysis of the Human Distal Gut Microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Backhed, F. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Healey, G.; Murphy, R.; Brough, L.; A Butts, C.; Coad, J. Interindividual variability in gut microbiota and host response to dietary interventions. Nutr. Rev. 2017, 75, 1059–1080. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Jones, R.M.; Schett, G.; Pacifici, R. The gut-bone axis: How bacterial metabolites bridge the distance. J. Clin. Investig. 2019, 129, 3018–3028. [Google Scholar] [CrossRef]
- Tyagi, A.M.; Yu, M.; Darby, T.M.; Vaccaro, C.; Li, J.-Y.; Owens, J.A.; Hsu, E.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; et al. The Microbial Metabolite Butyrate Stimulates Bone Formation via T Regulatory Cell-Mediated Regulation of WNT10B Expression. Immunity 2018, 49, 1116–1131.e7. [Google Scholar] [CrossRef]
- Lucas, S.; Omata, Y.; Hofmann, J.; Böttcher, M.; Iljazovic, A.; Sarter, K.; Albrecht, O.; Schulz, O.; Krishnacoumar, B.; Krönke, G.; et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Malik, T.A.; Yu, M.; Pal, S.; Malik, T.A.; Dar, H.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; Pacifici, R. Parathyroid hormone–dependent bone formation requires butyrate production by intestinal microbiota. J. Clin. Investig. 2020, 130, 1767–1781. [Google Scholar] [CrossRef]
- Yu, M.; Tyagi, A.M.; Li, J.-Y.; Adams, J.; Denning, T.L.; Weitzmann, M.N.; Jones, R.M.; Pacifici, R. PTH induces bone loss via microbial-dependent expansion of intestinal TNF+ T cells and Th17 cells. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Bellissimo, M.P.; Ziegler, T.R.; Jones, D.P.; Liu, K.H.; Fernandes, J.; Roberts, J.L.; Weitzmann, M.N.; Pacifici, R.; Alvarez, J.A. Plasma high-resolution metabolomics identifies linoleic acid and linked metabolic pathways associated with bone mineral density. Clin. Nutr. 2020, S0261-5614(20)30277-6. [Google Scholar] [CrossRef]
- Etheridge, A.; Lee, I.; Hood, L.; Galas, D.; Wang, K. Extracellular microRNA: A new source of biomarkers. Mutat. Res. Mol. Mech. Mutagen. 2011, 717, 85–90. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, L.; Gao, X.; Hu, J.; Wang, J.; Dai, Z.; Wang, J.-F.; Zhang, Z.; Lu, S.; Huang, X.; et al. Plasma MicroRNA Panel to Diagnose Hepatitis B Virus–Related Hepatocellular Carcinoma. J. Clin. Oncol. 2011, 29, 4781–4788. [Google Scholar] [CrossRef]
- Schultz, N.A.; Dehlendorff, C.; Jensen, B.V.; Bjerregaard, J.K.; Nielsen, K.R.; Bojesen, S.E.; Calatayud, D.; Nielsen, S.E.; Yilmaz, M.; Holländer, N.H.; et al. MicroRNA Biomarkers in Whole Blood for Detection of Pancreatic Cancer. JAMA 2014, 311, 392–404. [Google Scholar] [CrossRef]
- Lin, X.-J.; Chong, Y.; Guo, Z.-W.; Xie, C.; Yang, X.-J.; Zhang, Q.; Li, S.-P.; Xiong, Y.; Yuan, Y.; Min, J.; et al. A serum microRNA classifier for early detection of hepatocellular carcinoma: A multicentre, retrospective, longitudinal biomarker identification study with a nested case-control study. Lancet Oncol. 2015, 16, 804–815. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, G.; Yang, C.; Zhou, K.; Shen, B.; Liang, H.; Jiang, X. The Role of Circulating MicroRNA-126 (miR-126): A Novel Biomarker for Screening Prediabetes and Newly Diagnosed Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2014, 15, 10567–10577. [Google Scholar] [CrossRef]
- Willeit, P.; Skroblin, P.; Moschen, A.R.; Yin, X.; Kaudewitz, D.; Zampetaki, A.; Barwari, T.; Whitehead, M.; Ramírez, C.M.; Goedeke, L.; et al. Circulating MicroRNA-122 Is Associated with the Risk of New-Onset Metabolic Syndrome and Type 2 Diabetes. Diabetes 2017, 66, 347–357. [Google Scholar] [CrossRef]
- Shi, Q.; Yang, X. Circulating MicroRNA and Long Noncoding RNA as Biomarkers of Cardiovascular Diseases. J. Cell. Physiol. 2015, 231, 751–755. [Google Scholar] [CrossRef]
- Kessler, T.; Erdmann, J.; Vilne, B.; Bruse, P.; Kurowski, V.; Diemert, P.; Schunkert, H.; Sager, H.B. Serum microRNA-1233 is a specific biomarker for diagnosing acute pulmonary embolism. J. Transl. Med. 2016, 14, 1–10. [Google Scholar] [CrossRef]
- Gui, J.; Tian, Y.; Wen, X.; Zhang, W.; Zhang, P.; Gao, J.; Run, W.; Tian, L.; Jia, X.; Gao, Y. Serum microRNA characterization identifies miR-885-5p as a potential marker for detecting liver pathologies. Clin. Sci. 2010, 120, 183–193. [Google Scholar] [CrossRef]
- Seeliger, C.; Karpinski, K.; Haug, A.T.; Vester, H.; Schmitt, A.; Bauer, J.S.; Van Griensven, M. Five Freely Circulating miRNAs and Bone Tissue miRNAs Are Associated With Osteoporotic Fractures. J. Bone Miner. Res. 2014, 29, 1718–1728. [Google Scholar] [CrossRef]
- Panach, L.; Mifsut, D.; Tarín, J.; Cano, A.; Garciaperez, M.A. Serum Circulating MicroRNAs as Biomarkers of Osteoporotic Fracture. Calcif. Tissue Int. 2015, 97, 495–505. [Google Scholar] [CrossRef]
- Hackl, M.; Heilmeier, U.; Weilner, S.; Grillari, J. Circulating microRNAs as novel biomarkers for bone diseases—Complex signatures for multifactorial diseases? Mol. Cell. Endocrinol. 2016, 432, 83–95. [Google Scholar] [CrossRef]
- Kocijan, R.; Muschitz, C.; Geiger, E.; Skalicky, S.; Baierl, A.; Dormann, R.; Plachel, F.; Feichtinger, X.; Heimel, P.; Fahrleitner-Pammer, A.; et al. Circulating microRNA Signatures in Patients with Idiopathic and Postmenopausal Osteoporosis and Fragility Fractures. J. Clin. Endocrinol. Metab. 2016, 101, 4125–4134. [Google Scholar] [CrossRef]
- Mandourah, A.Y.; Ranganath, L.; Barraclough, R.; Vinjamuri, S.; Hof, R.V.; Hamill, S.; Czanner, G.; Dera, A.A.; Wang, D.; Barraclough, D.L. Circulating microRNAs as potential diagnostic biomarkers for osteoporosis. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Ramírez-Salazar, E.G.; Carrillo-Patiño, S.; Hidalgo-Bravo, A.; Rivera-Paredez, B.; Quiterio, M.; Ramírez-Palacios, P.; Patiño, N.; Valdés-Flores, M.; Salmerón, J.; Velázquez-Cruz, R. Serum miRNAs miR-140-3p and miR-23b-3p as potential biomarkers for osteoporosis and osteoporotic fracture in postmenopausal Mexican-Mestizo women. Gene 2018, 679, 19–27. [Google Scholar] [CrossRef]
- Ladang, A.; Beaudart, C.; Locquet, M.; Reginster, J.-Y.; Bruyère, O.; Cavalier, E. Evaluation of a Panel of MicroRNAs that Predicts Fragility Fracture Risk: A Pilot Study. Calcif. Tissue Int. 2019, 106, 239–247. [Google Scholar] [CrossRef]
- Zarecki, P.; Hackl, M.; Grillari, J.; Debono, M.; Eastell, R. Serum microRNAs as novel biomarkers for osteoporotic vertebral fractures. Bone 2019, 130, 115105. [Google Scholar] [CrossRef]
- Shuai, Y.; Liao, L.; Su, X.; Sha, N.; Li, X.; Wu, Y.; Jing, H.; Kuang, H.; Deng, Z.; Li, Y.; et al. Circulating microRNAs in serum as novel biomarkers for osteoporosis: A case-control study. Ther. Adv. Musculoskelet. Dis. 2020, 12. [Google Scholar] [CrossRef]
- Canning, K.L.; Brown, R.E.; Wharton, S.; Sharma, A.M.; Kuk, J.L. Edmonton Obesity Staging System Prevalence and Association with Weight Loss in a Publicly Funded Referral-Based Obesity Clinic. J. Obes. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Cannataro, R.; Perri, M.; Gallelli, L.; Caroleo, M.C.; De Sarro, G.; Cione, E. Ketogenic Diet Acts on Body Remodeling and MicroRNAs Expression Profile. MicroRNA 2019, 8, 116–126. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P. Nutrients and Oxidative Stress: Friend or Foe? Oxidative Med. Cell. Longev. 2018, 2018, 1–24. [Google Scholar] [CrossRef]
- Malivindi, R.; Rago, V.; De Rose, D.; Gervasi, M.C.; Cione, E.; Russo, G.; Santoro, M.; Aquila, S. Influence of all- trans retinoic acid on sperm metabolism and oxidative stress: Its involvement in the physiopathology of varicocele-associated male infertility. J. Cell. Physiol. 2018, 233, 9526–9537. [Google Scholar] [CrossRef]
- Perri, M.; Pingitore, A.; Cione, E.; Vilardi, E.; Perrone, V.; Genchi, G. Proliferative and anti-proliferative effects of retinoic acid at doses similar to endogenous levels in Leydig MLTC-1/R2C/TM-3 cells. Biochim. Biophys. Acta (BBA) Gen. Subj. 2010, 1800, 993–1001. [Google Scholar] [CrossRef]
- Cione, E.; Pingitore, A.; Perri, M.; Genchi, G. Influence of all-trans-retinoic acid on oxoglutarate carrier via retinoylation reaction. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2009, 1791, 3–7. [Google Scholar] [CrossRef]
- Alsahli, A.; Kiefhaber, K.; Gold, T.; Muluke, M.; Jiang, H.; Cremers, S.; Schulzespate, U. Palmitic Acid Reduces Circulating Bone Formation Markers in Obese Animals and Impairs Osteoblast Activity via C16-Ceramide Accumulation. Calcif. Tissue Int. 2016, 98, 511–519. [Google Scholar] [CrossRef]
- Gillet, C.; Spruyt, D.; Rigutto, S.; Valle, A.D.; Berlier, J.; Louis, C.; Debier, C.; Gaspard, N.; Malaisse, W.J.; Gangji, V.; et al. Oleate Abrogates Palmitate-Induced Lipotoxicity and Proinflammatory Response in Human Bone Marrow-Derived Mesenchymal Stem Cells and Osteoblastic Cells. Endocrinology 2015, 156, 4081–4093. [Google Scholar] [CrossRef]
- Gunaratnam, K.; Vidal, C.; Gimble, J.M.; Duque, G. Mechanisms of Palmitate-Induced Lipotoxicity in Human Osteoblasts. Endocrinology 2014, 155, 108–116. [Google Scholar] [CrossRef]
- Elbaz, A.; Wu, X.; Rivas, D.; Gimble, J.M.; Duque, G. Inhibition of fatty acid biosynthesis prevents adipocyte lipotoxicity on human osteoblasts in vitro. J. Cell. Mol. Med. 2009, 14, 982–991. [Google Scholar] [CrossRef]
- Platt, I.D.; El-Sohemy, A. Regulation of osteoblast and adipocyte differentiation from human mesenchymal stem cells by conjugated linoleic acid. J. Nutr. Biochem. 2009, 20, 956–964. [Google Scholar] [CrossRef]
- Rahman, M.; Halade, G.V.; Williams, P.J.; Fernandes, G. t10c12-CLA maintains higher bone mineral density during aging by modulating osteoclastogenesis and bone marrow adiposity. J. Cell. Physiol. 2010, 226, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Van Heerden, B.; Kasonga, A.; Kruger, M.C.; Kasonga, A. Palmitoleic Acid Inhibits RANKL-Induced Osteoclastogenesis and Bone Resorption by Suppressing NF-κB and MAPK Signalling Pathways. Nutrients 2017, 9, 441. [Google Scholar] [CrossRef] [PubMed]
- Yeung, D.K.W.; Griffith, J.F.; Antonio, G.E.; Lee, F.K.; Woo, J.; Leung, P.C. Osteoporosis is associated with increased marrow fat content and decreased marrow fat unsaturation: A proton MR spectroscopy study. J. Magn. Reson. Imaging 2005, 22, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Patsch, J.M.; Li, X.; Baum, T.; Yap, S.P.; Karampinos, D.C.; Schwartz, A.V.; Link, T.M. Bone marrow fat composition as a novel imaging biomarker in postmenopausal women with prevalent fragility fractures. J. Bone Miner. Res. 2013, 28, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, G.; Capuani, S.; Manenti, G.; Vinicola, V.; Fusco, A.; Baldi, J.; Scimeca, M.; E Hagberg, G.; Bozzali, M.; Simonetti, G.; et al. Bone Marrow Lipid Profiles from Peripheral Skeleton as Potential Biomarkers for Osteoporosis: A 1H-MR Spectroscopy Study. Acad. Radiol. 2016, 23, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Whitney, D.G.; Alford, A.I.; Devlin, M.J.; Li, Y.; Caird, M.S. Intersite reliability of vertebral bone marrow lipidomics-derived lipid composition among children with varying degrees of bone fragility undergoing routine orthopedic surgery. Bone 2021, 143, 115633. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Effects on Insulin Metabolism | Effects on Energy Metabolism and Other Suggested Endocrine Effects |
|---|---|
| Increase of β-cells proliferation, synthesis, and secretion of insulin | Improvement of energy expenditure through multiple mechanisms [15]. |
| Improvement of insulin sensitivity through multiple mechanisms [15]. | Increased expression of adiponectin (ADPN) in white fat and reducing lipid accumulation (and inflammation in the steatosis liver). |
| Modulation of hepatic insulin sensitivity | Stimulation of energy expenditure by increasing mitochondrial biogenesis in muscle and regulating the expression of genes involved in energy consumption in brown adipose and musculoskeletal tissue. |
| Promotion of male fertility by stimulating the synthesis of testosterone in Leydig cells. |
| Low O2 Conditions | Normal O2 Conditions |
|---|---|
| Glucose is converted into pyruvate with ATP production, and pyruvate is converted into lactate. Then, 80% of glucose is converted into lactate, representing the “predominant destiny” of glucose in OBL | Pyruvate enters the mitochondria where it is oxidized to CO2 + H2O |
| IGF-1, 3βOHB, and Famine | IGF-1 and KDs |
|---|---|
| IGF-1 levels reduce into famine [87]. | IGF-1 levels may be reduced up to 46% after 7 days of KD [88]. |
| 3βOHB blood levels and IGF-1 related growth speed inversely correlated [89]. | KD-induced famine-like metabolic state may reduce either the IGF-1 levels or its bioavailability [89,90]. |
| Model | Evaluated Parameters | Findings/Conclusions |
|---|---|---|
| Forty C57BL/6J 8-week-old female mice randomly divided into SD + Sham, SD+OVX, KD + Sham KD + OVX groups; KD fed for 12 weeks [103] | Micro-CT scanning of distal femur trabecular bone and middle femur of cortical bone; tibial maximum bending force and stiffness; TRAP, collagen type I (ColI) expression, and OC staining | KD compromised cancellous and cortical bone of long bones. Combination of KD and OVX may lead to more bone loss. |
| Sprague-Dawley rats control, KD and EODKD groups, fed with standard diet, continuous and intermittent KD, respectively [106] | ketone levels | EODKD demonstrated higher ketone levels than KD, inhibition of either osteoclastic process or early osteogenic differentiation. |
| DXA-assessed BMD and body fat percentage; micro-CT analysis of bone microstructure and mechanical properties, three-point bending test; bone turnover markers (serum ALP and TRAP), plus osteogenic capabilities of BMSCs (ALP activities and alizarin red stain) in different osteogenic stage. | ||
| Sprague-Dawley male rats KD or a standard diet for 12 weeks, equally divided into two groups [107] | Cortical and trabecular bone micro-CT morphometric analyses; micro-finite element analysis to calculate compressive stiffness and strength of the skeletal areas. | KD leaded to bone loss and reduction of the biomechanical function more on appendicular bones than axial bones |
| 48-weeks-old female C57BL/6J mice Two main groups: A) SD; and B) KD; SD and KD were then subdivided into four groups: A1) SD + Sham (SD + ovary intact); and A2) SD + OVX (SD + ovariectomy); B1) KD + Sham (KD + ovary intact); and B2) KD + OVX (KD + ovariectomy) [108] | L4 and L5 vertebral compressive test and histological staining of these vertebrae | The compressive test decreased in the failure load in OVX + Sham and KD + OVX. |
| female C57BL/6J mice Randomly divided: Sham, OVX, OVX + Met (100 mg/kg/day), KD (3:1 ratio of fat to carbohydrate and protein), and KD + Met (100 mg/kg/day) groups [109] | After 12 weeks: bone mass and biomechanical parameters in distal cancellous bone and femoral mid-shaft cortical bone; biochemical parameters: serum ALP and TRAP, OC, and TRAP immunohistochemistry staining | Met associated with increased serum ALP in the KD group, decreased serum TRAP in OVX group, and up-regulation and down-regulation of OC and TRAP expression, respectively. |
| Twenty-four male Wistar rats fed for 4 weeks either normal chow (CH, 9% fat, 33% protein, and 58% carbohydrates), LC-HF-1 (66% fat, 33% protein, and 1% carbohydrates), or LC-HF-2 (94.5% fat, 4.2% protein, and 1.3% carbohydrates) [110] | pQCT, µCT analysis; three-point bending test; histology of humeri, bone marrow quantitative real-time PCR; P1NP, OC, PINP1, CTX, IGF-1, 25OHD, GH, leptin, IGF-1, Insulin-like growth factor binding protein 2-3, rat intact PTH, serum calcium and phosphate | LC-HF diets associated with more visceral and bone marrow fat, increased leptin but decreased IGF-1 and body length; LC-HF-2 reduced humerus, tibia, and femur lengths; pQCT and µCT revealed significant reductions in tibial BMD in both groups; in LC-HF groups, tibial maximum load was impaired and serum PINP1 reduced; in LC-HF, real-time PCR showed bone marrow reduced expression (70 to 80%) of Runx2, osterix, and C/EBPβ. |
| Population | Type of Study | Outcome Considered and Methods to Evaluate Diet and/or Bone Health Parameters | Findings/Conclusions |
|---|---|---|---|
| Twenty-five US children with intractable epilepsy (IE), on a 4:1 KD (by weight fat:CHO and protein) [5] | 2-phase, 15-mo longitudinal study of growth and nutritional effects of KD | Effect of KD on bone health, growth, nutritional status, and seizures; DXA-assessed whole-body and LS-BMC z score; evaluation/measurements of demographics, anthropometry, serum 25OHD, intact PTH, electrolytes, and dietary intake. All at baseline and at 3, 6, 12, and 15 mo.; evaluation of BMC change over time. | Bone health was poor, particularly for younger nonambulatory children with low BMI status. KD resulted in progressive loss of BMC |
| Eight UK obese subjects on therapy with low energy (2741–3301 kJ/day), high calcium (28.9–35.1 mmol/day) diets [112] | Observational | Effects of dietary CHO content and of tri-iodothyronine administration on calcium, zinc, and phosphate balances and on urinary hydroxyproline output | Calcium balances generally positive at the high levels of intake, but significantly more calcium was retained when dietary CHO intake increased |
| Twenty-one US healthy obese women aged 38 +/− 9 years, randomly on either diet alone or diet plus resistance training. Both groups with a 925-kcal/d portion-controlled diet for the first 16 of 17 weeks and a 1000 to 1500-kcal/d balanced deficit diet thereafter [113] | RCT | DXA-BMC, -BMD, fat-free mass (FFM), and DXA-fat mass before and after 24 weeks of dieting | Diet and resistance training not associated with a better outcome vs. diet alone. Increasing the energy content of very-low-calorie diets to 925 kcal/d may prevent the loss of total BMD, but not the loss from the FN and GT |
| Two hundred and seven healthy Caucasian, aged 9–18 yrs. [115] | Cross-sectional study | Rate of skeletal growth at LS-BMD-BMC and FN-BMD-BMC sites, in relation to age and pubertal stages in both sexes | Marked age-related delay in L2-L4-BMD or -BMC increase in males vs. females, unrelated to pubertal stages; higher mean values in males for BMD and BMC, at the end of the rapid growth spurt; dramatic reduction (2–4 years after menarche), in bone mass growth in females >15 yrs. of age |
| One hundred and seventeen US subjects, from 3-county areas, age range 2–19 years with moderate to severe cerebral palsy (CP) according to the Gross Motor Functional Classification scale. Population-based sampling from both hospital- and school-based sources [117] | Observational study | DXA assessed LS- and distal femur-BMD; anthropometric assessment of growth and evaluation of nutritional status; Child Health Status Questionnaire; serum calcium, phosphate, ALP (total and bone-specific), OC, NTx, total protein, albumin, prealbumin, 25OHD | Femur osteopenia in 77% of the cohort and 97% of all study participants. Fractures in 26% of the children older than 10 years. Severity of neurologic impairment, increasing difficulty feeding, use of anticonvulsants, and lower triceps skinfold z scores, independently contribute to lower femoral BMD z scores. Low BMD prevailed in moderate-severe CP and associated with significant fracture risk |
| Fifty-seven US subjects, age range 1-26 years, on KD for seizures management and one on KD for GLUT1 deficiency. Fourteen children had CP other than seizure disorder [120] | Retrospective study analysis | 3-day food diary nutrient analysis. Height or length and weight at 0, 6 mo., and 12 mo. follow-up visits. | KD for 12 months significantly decreased height z scores. Daily urinary ketone levels (reagent strip) categorized into two groups: <80 mg/dL (moderate ketosis) and 80–120 mg/dL (high ketosis). High ketosis significantly decreased height z scores. Subjects on the KD showed a delay in growth |
| Fourthy-five Argentinian children, 23 boys, on KD for at least two years, between 0.8 and 17.3 years of age (mean 6.6) [121] | Prospective cohort | Growth assessment (weight, height, and BMI). Standard deviation scores for all measurements calculated at KD initiation and at follow-up | Height growth < percentile third for 2 boys and 2 girls |
| Sixty-three Australian patients on the KD, 29 patients on the KD for a minimum of 6 months (range 0.5–6.5 years, mean 2.1 years) [109] | Prospective, longitudinal study | LS-, right TH-DXA z-scores at baseline and 6 mo. intervals; serum calcium, phosphate, 25OHD, PTH, ALP, OC, urine calcium/creatinine ratio at baseline and 3 mo. intervals; gross motor functional classification system (GMFCS) | Trend in reduction of LS-BMD z-score/year; 68% had lower BMD z-score at the end of treatment. Lower baseline z-scores in less mobile patients, but the rate of bone loss on the diet was greater in the more mobile patients. Only 2 patients fractured. Elevated mean urinary calcium-creatinine ratios; 1 patient developed renal calculi |
| Seventy-five UK children providing growth data on 1 of 2 KDs at baseline and after 3, 6, and 12 months, if continued [141] | Prospective, longitudinal study | Weight, height, and BMI z scores at baseline and 3, 6, 12 mo.; evaluation of growth trend. | Height z scores decreased at 6 and 12 mo., particularly in younger and ambulatory children; weight z scores decrease in the MCT group only at 3 and 6 mo. and in both groups at 12 mo.; forty children completed the study with no differences in growth trend between classical and MCT diets for weight, height, or BMI; the MCT group had significantly higher protein intake |
| One hundred ninety-five US children on the KD for intractable epilepsy from 2000 to 2005 [142] | Cohort study kidney stones formers vs. not kidney stones formers | Demographics, urine laboratory markers, and intervention with urine alkalization (potassium citrate) | Thirteen children developed kidney stones; oral potassium citrate significantly decreased the prevalence of stones and increased the mean time on the KD before a stone was first noted; the prevalence of kidney stones trended toward higher correlation with hypercalciuria (92% vs. 71%). No child stopped the diet due to stones; kidney stones continue to occur in approximately 1 in 20 children on the KD |
| Thirty US patients (15 study subjects and 15 controls). The 15 patients on diet to consume less than 20 g of CHO per day for the 1st month and then less than 40 g per day for months 2 and 3. Control subjects without restrictions on diet [45] | 3-month study | Primary end point: urinary UNTx at 3 mo. Secondary end points: UNTx at 1 mo., BSAP at 1 mo., bone turnover ratio (BSAP/UNTx) at 1 mo., and weight loss | Patients on low-CHO diet lost significantly more weight than controls, the diet not increased bone turnover markers vs. controls. No significant change in the bone turnover ratio compared with controls |
| Thirty-eight Swedish patients, mean age (SD) 6.1 years (4.8 years), 21 girls, with intractable epilepsy, glucose transporter type 1 deficiency syndrome, or pyruvate dehydrogenase complex deficiency [125] | Prospective longitudinal cohort study | Assessment of growth, body composition, and bone mass in children on MAD for 24 months. Body weight, height, BMI, DXA-BMD, serum calcium, phosphorus, magnesium, ALP, cholesterol, 25OHD, IGF-1 and IGFBP3 at baseline and 24 mo. of MAD | Approximately, 50% of the patients responded with more than 50% seizure reduction. Weight and height standard deviation score stabilized over 24 months; median BMI SDS significantly increased. MAD efficiently reduced seizures. No negative effect observed on longitudinal growth or bone mass after 24 months MAD treatment. |
| Sixty-eight Dutch children on KDT for more than 6 months and who had at least two DXA scans [126] | Retrospective, observational cohort study | Changes in DXA-BMD in children with KDT and evaluation of efficacy of i.v. bisphosphonate therapy | In 50% of patients, ≥ 1 LS-DXA scans performed. 8.8% got a fracture during KDT, and also 8.8% got kidney stones. Not significant BMD decrease. BMD significantly increased in the five patients treated with i.v. bisphosphonate therapy, vs. not treated. |
| Three adult Italian patients with GLUT-1 deficiency syndrome on KD for > 5 years. Normocaloric KD on a 3:1 ketogenic ratio [129] | Case series report | Long-term effects of KD on body composition and bone mineral status in GLUT-1 deficiency syndrome; Anthropometric and body composition measurements (weight, height, BMI, waist circumference, abdominal circumference, skinfold-thickness measurement, arm muscle circumference, DXA-assessed body fat mass, lean body mass (LBM), BMC, and BMD. | No appreciable changes in weight and body composition of adults with GLUT-1 deficiency. No evidence of potential adverse effects of KD on bone health |
| Ten healthy, physically active UK men, age 24 ± 3 yrs., nonsmokers, not suffered a bone fracture or injury of any type in the previous 12 mo., free from musculoskeletal injury, not taking any medication, and not suffering from any condition known to affect bone metabolism. Two randomized, repeated-measures, counterbalanced 7-day experimental trials, involving either placebo (PBO) or CHO ingestion during 120 min of treadmill running [149] | Clinical trial | Immediate and short-term bone metabolic responses to CHO feeding during treadmill running; β-CTX, P1NP, OPG, OC, PTH, leptin, GLP-2, and IL-6, cortisol, insulin, serum calcium, albumin, and phosphate. | CHO feeding during exercise attenuated the β-CTX and P1NP responses in the hours following exercise, indicating an acute effect of CHO feeding on bone turnover. |
| Ten physically active UK men with at least one bout of endurance running per week, free from fracture in the previous 12 months and any condition known to affect bone metabolism. Free from musculoskeletal injury [149] | Clinical trial | Effect of an overnight fast vs. feeding of a single mixed meal, on the bone metabolic response to an acute bout of treadmill running; three-day food diary, β-CTX, P1NP, OC, OPG, cortisol, bone ALP, PTH, albumin-adjusted calcium, phosphate, leptin, and ghrelin | Bone markers not significantly differ from baseline on follow up 1–4. Fasting had a minor effect on the bone metabolic response to subsequent acute, endurance exercise, reducing the duration of the increase in β-CTX during early recovery, but no effect on changes in bone formation markers. Reduction of duration of the β-CTX response with fasting not fully explained by changes in PTH, OPG, leptin or ghrelin |
| Nine UK male runners, age 21 ± 1.9 years, completing a morning and afternoon high-intensity interval running protocol (interspersed by 3.5 h) under dietary conditions: high CHO availability, reduced CHO but high fat availability, or reduced CHO and reduced energy availability [150] | Clinical trial | Effects of post-exercise CHO and caloric restriction on the modulation of skeletal muscle cell signaling pathways as well as indicators of bone metabolism. | Post-exercise circulating βCTX was significantly lower in high CHO compared to low CHO-high fat and low CHO and reduced energy availability |
| Twenty-five males, and 5 females Australian World-class race walkers completing 3.5-weeks of energy-matched high CHO or ketogenic low-carbohydrate, high-fat diet followed by acute CHO restoration [152] | Clinical trial | Diet-exercise interactions related to bone markers in elite endurance athletes after a 3.5-week ketogenic low-CHO, high-fat diet and subsequent restoration of CHO feeding; serum CTX, P1NP, OC assessed at rest (fasting and 2 h post meal) and after exercise (0 and 3 h) at baseline, after the 3.5-week intervention (Adaptation) and after acute CHO feeding (Restoration) | Markers of bone modeling/remodeling were impaired after short-term LCHF diet, and only a marker of resorption recovered after acute CHO restoration. |
| Clinical Parameters | Biochemical Parameters | Instrumental Parameters |
|---|---|---|
| Baseline Familial clinical history, physiological and pathological personal history (especially regarding clinical fracture risk factors); calcium/phosphate daily intake questionnaire; sun exposure questionnaire; drug history; presence/absence and type of physical activity; smoke and alcohol habitus. Tanner stage or bone age results should be applied in the pediatric age. Baseline, 6, 12, 18, and 24 mos. Height Weight BMI Waist circumference Clinical evaluation | Baseline, 6 mo., 12 mo., 18 mo., and 24 mo. Serum: 25OHD, PTH, calcium, phosphate, BTMs *°, total proteins, fractioned proteins, insulin, glucose, creatinine. Standard urine test and 24 hrs. urinary collection for: calcium, phosphate, and citrates To assess and confirm a stable ketosis state: early morning blood β–OHbutyrate and post-dinner urine acetoacetate. Alternatively, urinary stick tests for ketone bodies. * also, serum OPG, sclerostin, and IL-6 could be considered in the biochemical assessment. ° Follow-up BTMs testing should be done when its expected change equals or exceeds the least significant change (LSC) [157]. | Baseline, 12 mo., 24 mo. DXA for LS, FN/Total hip, Nondominant BMC/BMD (when the population includes females prior to menopause and males younger than age 50 use Z-score; this is particularly important in children whose adjustments have to be made for growth, and interpretation) [163]. Follow-up BMD testing should be done when its expected change equals or exceeds the least significant change (LSC) [156]. One year after initiation or change of therapy is appropriate, with longer intervals once therapeutic effect is established [163] DXA Fracture Risk Assessment when subjects at high fracture risk are included [163] Baseline, 6, 12, 18, and 24 mos. Bio-impedancemetry |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merlotti, D.; Cosso, R.; Eller-Vainicher, C.; Vescini, F.; Chiodini, I.; Gennari, L.; Falchetti, A. Energy Metabolism and Ketogenic Diets: What about the Skeletal Health? A Narrative Review and a Prospective Vision for Planning Clinical Trials on this Issue. Int. J. Mol. Sci. 2021, 22, 435. https://doi.org/10.3390/ijms22010435
Merlotti D, Cosso R, Eller-Vainicher C, Vescini F, Chiodini I, Gennari L, Falchetti A. Energy Metabolism and Ketogenic Diets: What about the Skeletal Health? A Narrative Review and a Prospective Vision for Planning Clinical Trials on this Issue. International Journal of Molecular Sciences. 2021; 22(1):435. https://doi.org/10.3390/ijms22010435
Chicago/Turabian StyleMerlotti, Daniela, Roberta Cosso, Cristina Eller-Vainicher, Fabio Vescini, Iacopo Chiodini, Luigi Gennari, and Alberto Falchetti. 2021. "Energy Metabolism and Ketogenic Diets: What about the Skeletal Health? A Narrative Review and a Prospective Vision for Planning Clinical Trials on this Issue" International Journal of Molecular Sciences 22, no. 1: 435. https://doi.org/10.3390/ijms22010435
APA StyleMerlotti, D., Cosso, R., Eller-Vainicher, C., Vescini, F., Chiodini, I., Gennari, L., & Falchetti, A. (2021). Energy Metabolism and Ketogenic Diets: What about the Skeletal Health? A Narrative Review and a Prospective Vision for Planning Clinical Trials on this Issue. International Journal of Molecular Sciences, 22(1), 435. https://doi.org/10.3390/ijms22010435