Immunity as Cornerstone of Non-Alcoholic Fatty Liver Disease: The Contribution of Oxidative Stress in the Disease Progression

 , ,
, ,  , , and
, , and 
Abstract
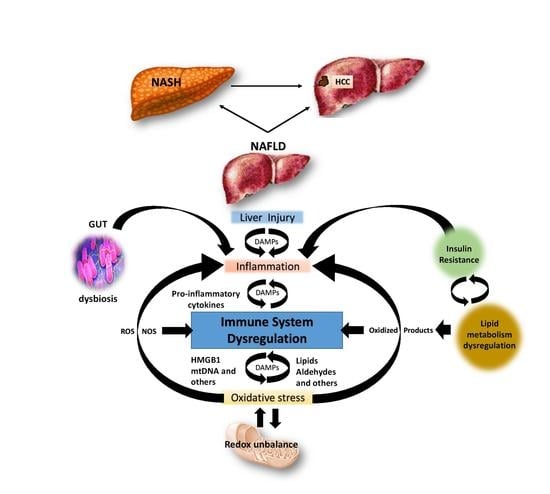
1. Introduction
1.1. Overview on Molecular Mechanisms of Oxidative Stress in NAFLD
1.1.1. Oxidative Stress Derived from Mitochondrial Dysfunction
1.1.2. ER Contribution to Oxidative Stress
2. Redox Biology and Immune Regulation in NAFLD Progression
2.1. The Role of PRRs
2.2. The Role of OSEs
3. The Immune Response in HCC Development
4. Future Perspectives: The Role of Trained Immunity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | Adenosine monophosphate-activated protein kinase |
| AP-1 | Activator protein-1 |
| APCs | Antigen Presenting Cells |
| ASK 1 | Apoptosis signal-regulating kinase 1 |
| CAFs | Cancer-associated fibroblasts |
| CCL-2 | Chemokine (C-C motif) ligand 2 |
| CHOP | C/EBP Homologous Protein |
| COX | Cytochrome c oxidase |
| COX-2 | Cyclooxygenase 2 |
| CTLA | CD14+ cytotoxic T-lymphocyte-associated protein |
| CXCL12 | Chemokine C-X-C motif ligand 12 |
| CYP | Cytochrome P450 |
| DAMPs | Damage-associated molecular patterns |
| DC | Dendritic cell |
| EGF | Epidermal growth factor |
| eIF2a | RNA-dependent protein kinase-like ER eukaryotic initiation factor-2 α kinase |
| ER | Endoplasmic Reticulum |
| ETC | Electron transport chain |
| FAO | Fatty acid oxidation |
| FFAs | Free fatty acids |
| FGF | Fibroblast growth factor |
| FoxP3 | Forkhead box P3 |
| HCC | Hepatocellular Carcinoma |
| HFD | High fat diet |
| HGF | Hepatocyte growth factor |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HLA | Human leukocyte antigen |
| HMGB1 | high mobility group box 1 protein |
| HNE | Dydroxinonenal |
| HO-1 | Hemeoxygenase-1 |
| HSCs | Hepatic stellate cells |
| ICI | Immune checkpoint inhibitors |
| IFN-γ | Interferon γ |
| IKK | Inhibitor of nuclear factor kappa-B kinase |
| IR | Insulin Resistance |
| IRG1 | Immune responsive gene 1 |
| JMJ | Jumonji domain-containing |
| JNK | c-Jun-N-terminal kinase |
| KCs | Kupffer cells |
| KEAP1 | Kelch-like ECH associated protein 1 |
| LPS | Lipopolysaccharides |
| MAA | Malondialdehyde-acetaldehyde |
| MAFLD | Metabolic (dysfunction) associated fatty liver disease |
| MAP3K | Mitogen-activated protein kinase kinase kinase |
| MAPK | Mitogen activated protein kinase. |
| MCSF | Macrophage colony-stimulating factor |
| MDA | Malondialdehyde |
| MDSCs | Myeloid-derived suppressor cells |
| MMP | Matrix metalloproteases |
| MPT | Mitochondrial permeability transition |
| MS | Metabolic Syndrome |
| mTOR | Mammalian target of rapamycin |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural Killer |
| NLRP | NLR family pyrin domain containing |
| NLRS | Nucleotide-binding oligomerization domain-like receptors |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| OLRs | Oligoadenylate synthase (OAS)-like receptors |
| OSEs | Oxidation-specific epitopes |
| OXPHOS | Oxidative phosphorylation |
| PAMPs | Pathogen Associated Molecular Patterns |
| PDGF | Platelet-Derived Growth Factor |
| PPARs | Peroxisome proliferator-activated receptor |
| PD1 | Programmed cell Death 1 |
| PRRs | Pattern recognition receptors |
| PUFAs | Polyunsaturated fatty acids |
| RAGEs | Receptors for advanced glycation end products |
| ROS | Reactive oxygen species |
| SDH | Succinate dehydrogenase |
| SNPs | Single Nucleotide Polymorphisms |
| SOD | Superoxide dismutase |
| SPARC | Secreted protein acidic rich in cysteine |
| TAK 1 | TGF-β-activated kinase 1 |
| TAMs | Tumour-associated macrophages |
| TET | Ten eleven translocation |
| TGF-β | Transforming Growth Factor-β |
| TI | Trained Immunity |
| TLRs | Toll-like receptors |
| TNF-α | Tumor Necrosis Factor-α |
| TRAIL | Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) |
| Tregs | Regulatory T cells |
| VEGF | Vascular endothelial growth factor |
References
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver international. Off. J. Int. Assoc. Study Liver 2017, 37 (Suppl. 1), 81–84. [Google Scholar]
- Lindenmeyer, C.C.; McCullough, A. The Natural History of Nonalcoholic Fatty Liver Disease—An Evolving View. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Boccuto, L.; Federico, A.; Dallio, M.; Loguercio, C.; Di Renzo, L.; De Lorenzo, A. Diet and Non-Alcoholic Fatty Liver Disease: The Mediterranean Way. Int. J. Environ. Res. Public Heal. 2019, 16, 3011. [Google Scholar] [CrossRef] [PubMed]
- Wijarnpreecha, K.; Aby, E.S.; Ahmed, A.; Kim, D. Evaluation and Management of Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Clin. Mol. Hepatol. 2020, 14, 2, 168–178. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Masarone, M.; Persico, M.; Loguercio, C. The epidemiology of non-alcoholic fatty liver disease and its connection with cardiovascular disease: Role of endothelial dysfunction. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4731–4741. [Google Scholar]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 1–8. [Google Scholar] [CrossRef]
- Masarone, M.; Federico, A.; Abenavoli, L.; Loguercio, C.; Persico, M. Non Alcoholic Fatty Liver: Epidemiology and Natural History. Rev. Recent Clin. Trials 2015, 9, 126–133. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Sherlock, S. Sherlock’s Diseases of the Liver and Biliary System, 13th ed.; Dooley, J.S., Garcia-Tsao, G., Pinzani, M., Eds.; John Wiley & Sons Ltd: Hoboken, NJ, USA, 2018; Volume 28, pp. 540–560. [Google Scholar] [CrossRef]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment. Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxidative Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2019, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, X.-J.; Li, H. Role of Innate Immune Signaling in Non-Alcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2018, 29, 712–722. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Devkar, R.; Nammi, S. Oxidative Stress in Liver Diseases: Pathogenesis, Prevention, and Therapeutics. Oxidative Med. Cell. Longev. 2017, 2017, 1–2. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metab. Clin. Exp. 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free. Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Sanchez-Jimenez, F. Antioxidant enzymes and their implications in pathophysiologic processes. Frontiers in bioscience. J. Virtual Libr. 1999, 4, D339–D345. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Villani, R.; Facciorusso, A.; Vendemiale, G.; Serviddio, G. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free. Radic. Biol. Med. 2017, 111, 173–185. [Google Scholar] [CrossRef]
- Bellanti, F.; Villani, R.; Tamborra, R.; Blonda, M.; Iannelli, G.; Di Bello, G.; Facciorusso, A.; Poli, G.; Iuliano, L.; Avolio, C.; et al. Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol. 2017, 15, 86–96. [Google Scholar] [CrossRef]
- Sangineto, M.; Villani, R.; Cavallone, F.; Romano, A.D.; Loizzi, D.; Serviddio, G. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers 2020, 12, 1419. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Vendemiale, G.; Altomare, E. Mitochondrial dysfunction in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 233–244. [Google Scholar] [CrossRef]
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of Hepatic Carnitine Palmitoyl Transferase-I (CPT-I) Impairs Fatty Acid Beta-Oxidation in Rats Fed a Methionine-Choline Deficient Diet. PLoS ONE 2011, 6, e24084. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, Y.; Cai, J.; Li, H. Emerging Molecular Targets for Treatment of Nonalcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2019, 30, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Simões, I.C.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in non-alcoholic fatty liver disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Rodríguez-Juan, C.; Díaz-Sanjuan, T.; Del Hoyo, P.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006, 44, 581–591. [Google Scholar] [CrossRef]
- Garcia-Ruiz, I.; Rodriguez-Juan, C.; Diaz-Sanjuan, T.; Martinez, M.A.; Munoz-Yague, T.; Solis-Herruzo, J.A. Ef-fects of rosiglitazone on the liver histology and mitochondrial function in ob/ob mice. Hepatology 2007, 46, 414–423. [Google Scholar] [CrossRef]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; DeSimone, C.; Song, X.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef]
- Rizki, G.; Arnaboldi, L.; Gabrielli, B.; Yan, J.; Lee, G.S.; Ng, R.K.; Turner, S.M.; Badger, T.M.; Pitas, R.E.; Maher, J.J. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J. Lipid Res. 2006, 47, 2280–2290. [Google Scholar] [CrossRef]
- Romestaing, C.; Piquet, M.A.; Letexier, D.; Rey, B.; Mourier, A.; Servais, S.; Belouze, M.; Rouleau, V.; Dautresme, M.; Ollivier, I.; et al. Mitochondrial adaptations to steatohepatitis in-duced by a methionine- and choline-deficient diet. American journal of physiology. Endocrinol. Metab. 2008, 294, E110–E119. [Google Scholar]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen-Kirberg, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Gorski, J.C.; Asghar, M.S.; Asghar, A.; Foresman, B.; Hall, S.D.; Crabb, D.W. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003, 37, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, S.; Kasumov, T.; Edmison, J.M.; Gruca, L.L.; Bennett, C.; Duenas, C.; Marczewski, S.; McCullough, A.J.; Hanson, R.W.; Kalhan, S. Glycine and urea kinetics in nonalcoholic steatohepatitis in human: Effect of intralipid infusion. Am. J. Physiol. Liver Physiol. 2009, 297, G567–G575. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, S.; Yang, Y.; McCullough, A.J.; Marczewski, S.; Bennett, C.; Kalhan, S.C. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2011, 23, 382–388. [Google Scholar] [CrossRef]
- Hodson, L.; McQuaid, S.E.; Humphreys, S.M.; Milne, R.; Fielding, B.A.; Frayn, K.N.; Karpe, F. Greater dietary fat oxidation in obese compared with lean men: An adaptive mechanism to prevent liver fat accumulation? Am. J. Physiol. Metab. 2010, 299, E584–E592. [Google Scholar] [CrossRef]
- Miele, L.; Grieco, A.; Armuzzi, A.; Candelli, M.; Forgione, A.; Gasbarrini, A.; Gasbarrini, G. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am. J. Gastroenterol. 2003, 98, 2335–2336. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Schneider, A.R.J.; Kraut, C.; Lindenthal, B.; Braden, B.; Caspary, W.F.; Stein, J. Total body metabolism of 13C-octanoic acid is preserved in patients with non-alcoholic steatohepatitis, but differs between women and men. Eur. J. Gastroenterol. Hepatol. 2005, 17, 1181–1184. [Google Scholar] [CrossRef]
- Mawatari, H.; Inamori, M.; Fujita, K.; Yoneda, M.; Iida, H.; Endo, H.; Hosono, K.; Nozaki, Y.; Yoneda, K.; Akiyama, T.; et al. The continuous real-time 13C-octanoate breath test for patients with nonalcoholic steatohepatitis using the BreathID system. Hepatogastroenterology 2009, 56, 1436–1438. [Google Scholar]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.E.; Dufour, S.; Rothman, D.L.; Shulman, G.I. Assessment of Hepatic Mitochondrial Oxidation and Pyruvate Cycling in NAFLD by 13 C Magnetic Resonance Spectroscopy. Cell Metab. 2016, 24, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, P.A.; Chausse, B.; Caldeira da Silva, C.C.; Donato Junior, J.; Kowaltowski, A.J. Resilient hepatic mi-tochondrial function and lack of iNOS dependence in diet-induced insulin resistance. PLoS ONE 2019, 14, e0211733. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Liddle, C. Nonalcoholic fatty liver disease: Pathogenesis and potential for nuclear receptors as thera-peutic targets. Mol. Pharm. 2008, 5, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Cocco, T.; Di Paola, M.; Papa, P.; Lorusso, M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free. Radic. Biol. Med. 1999, 27, 51–59. [Google Scholar] [CrossRef]
- Loskovich, M.V.; Grivennikova, V.G.; Cecchini, G.; Vinogradov, A.D. Inhibitory effect of palmitate on the mi-tochondrial NADH:ubiquinone oxidoreductase (complex I) as related to the active-de-active enzyme transition. Biochem. J. 2005, 387, 677–683. [Google Scholar] [CrossRef]
- Schönfeld, P.; Reiser, G. Rotenone-like Action of the Branched-chain Phytanic Acid Induces Oxidative Stress in Mitochondria. J. Biol. Chem. 2006, 281, 7136–7142. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Fatty acids decrease mitochondrial generation of reactive oxygen species at the reverse electron transport but increase it at the forward transport. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1032–1040. [Google Scholar] [CrossRef]
- Gille, L.; Nohl, H. The Ubiquinol/bc1 Redox Couple Regulates Mitochondrial Oxygen Radical Formation. Arch. Biochem. Biophys. 2001, 388, 34–38. [Google Scholar] [CrossRef]
- Schönfeld, P.; Struy, H. Refsum disease diagnostic marker phytanic acid alters the physical state of membrane proteins of liver mitochondria. FEBS Lett. 1999, 457, 179–183. [Google Scholar] [CrossRef]
- Serviddio, G.; Blonda, M.; Bellanti, F.; Villani, R.; Iuliano, L.; Vendemiale, G. Oxysterols and redox signaling in the pathogenesis of non-alcoholic fatty liver disease. Free. Radic. Res. 2013, 47, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Aoun, M.; Fouret, G.; Michel, F.; Bonafos, B.; Ramos, J.; Cristol, J.P.; Carbonneau, M.-A.; Coudray, C.; Feillet-Coudray, C. Dietary fatty acids modulate liver mitochondrial cardiolipin content and its fatty acid composition in rats with non alcoholic fatty liver disease. J. Bioenerg. Biomembr. 2012, 44, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Arvier, M.; Gallois, Y.; Simard, G.; Malthièry, Y.; Ritz, P.; Ducluzeau-Fieloux, P.-H. Fatty liver and insulin resistance in obese Zucker rats: No role for mitochondrial dysfunction. Biochimie 2008, 90, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Rieusset, J.; Vidal, H.; Simard, G.; Malthièry, Y.; Fromenty, B.; Ducluzeau, P.-H. Regulation of hepatic mitochondrial metabolism in response to a high fat diet: A longitudinal study in rats. J. Physiol. Biochem. 2012, 68, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Casellas, A.; Amengual-Cladera, E.; Proenza, A.M.; Lladó, I.; Gianotti, M. Long-term High-fat-diet Feeding Impairs Mitochondrial Biogenesis in Liver of Male and Female Rats. Cell. Physiol. Biochem. 2010, 26, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Dubouchaud, H.; Couturier, K.; Cottet-Rousselle, C.; Taleux, N.; Athias, A.; Galinier, A.; Casteilla, L.; Leverve, X.M. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J. Hepatol. 2011, 54, 348–356. [Google Scholar] [CrossRef]
- Wardlaw, G.M.; Kaplan, M.L. Oxygen Consumption and Oxidative Capacity of Hepatocytes from Young Male Obese and Nonobese Zucker Rats. Exp. Biol. Med. 1986, 183, 199–206. [Google Scholar] [CrossRef]
- Poussin, C.; Ibberson, M.; Hall, D.; Ding, J.; Soto, J.; Abel, E.D.; Thorens, B. Oxidative Phosphorylation Flexibility in the Liver of Mice Resistant to High-Fat Diet-Induced Hepatic Steatosis. Diabetes 2011, 60, 2216–2224. [Google Scholar] [CrossRef]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef]
- Valdecantos, M.P.; Pérez-Matute, P.; González-Muniesa, P.; Prieto-Hontoria, P.L.; Moreno-Aliaga, M.J.; Martínez, J.A. Lipoic acid administration prevents nonalcoholic steatosis linked to long-term high-fat feeding by modulating mitochondrial function. J. Nutr. Biochem. 2012, 23, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2004, 19, 136–138. [Google Scholar] [CrossRef]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: A pilot study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhao, M.; An, W. Increased hepatic apoptosis in high-fat diet-induced NASH in rats may be associated with downregulation of hepatic stimulator substance. J. Mol. Med. 2011, 89, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Mingorance, C.; Duluc, L.; Chalopin, M.; Simard, G.; Ducluzeau, P.-H.; Herrera, M.D.; De Sotomayor, M.A.; Andriantsitohaina, R. Propionyl-L-carnitine Corrects Metabolic and Cardiovascular Alterations in Diet-Induced Obese Mice and Improves Liver Respiratory Chain Activity. PLoS ONE 2012, 7, e34268. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.D.; Claudel, T.; Scharnagl, H.; Stojakovic, T.; Trauner, M. FXR controls CHOP expression in steatohepatitis. FEBS Lett. 2017, 591, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free. Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent Activation of Nrf2 Contributes to Redox Homeostasis and Cell Survival following Endoplasmic Reticulum Stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive Oxygen Species in the Immune System. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, M.; Tan, H.-Y.; Wang, N.; Feng, Y. Insights into the Role and Interdependence of Oxidative Stress and Inflammation in Liver Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 1–21. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Bubici, C.; Papa, S.; Pham, C.G.; Zazzeroni, F.; Franzoso, G. The NF-kappaB-mediated control of ROS and JNK signaling. Histol. Histopathol. 2006, 21, 69–80. [Google Scholar]
- Zhu, L.H.; Wang, A.; Luo, P.; Wang, X.; Jiang, D.-S.; Deng, W.; Zhang, X.; Wang, T.; Liu, Y.; Gao, L.; et al. Mindin/Spondin 2 inhibits hepatic steatosis, insulin resistance, and obesity via interaction with peroxisome proliferator-activated receptor α in mice. J. Hepatol. 2014, 60, 1046–1054. [Google Scholar] [CrossRef]
- Wang, X.-A.; Deng, S.; Jiang, D.; Zhang, R.; Zhang, S.; Zhong, J.; Yang, L.; Wang, T.; Hong, S.; Guo, S.; et al. CARD3 Deficiency Exacerbates Diet-Induced Obesity, Hepatosteatosis, and Insulin Resistance in Male Mice. Endocrinology 2013, 154, 685–697. [Google Scholar] [CrossRef]
- Tong, J.; Han, C.J.; Zhang, J.Z.; He, W.Z.; Zhao, G.J.; Cheng, X.; Zhang, L.; Deng, K.Q.; Liu, Y.; Fan, H.F.; et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor gamma in Mice. Hepatology 2019, 69, 2471–2488. [Google Scholar]
- Wang, P.-X.; Zhang, R.; Huang, L.; Zhu, L.-H.; Jiang, D.-S.; Chen, H.-Z.; Zhang, Y.; Tian, S.; Zhang, X.-F.; Liu, D.; et al. Interferon regulatory factor 9 is a key mediator of hepatic ischemia/reperfusion injury. J. Hepatol. 2015, 62, 111–120. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, X.-H.; Zhang, X.-J.; Wang, P.-X.; Zhang, R.; Zhang, P.; Zhao, G.-N.; Gao, L.; Tian, S.; Li, H. Targeting TRAF3 signaling protects against hepatic ischemia/reperfusions injury. J. Hepatol. 2016, 64, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, J.; Ji, Y.X.; Mei, F.; Wang, P.X.; Deng, K.Q.; Jiang, X.; Ma, G.; Li, H. Tripartite Motif 8 Contributes to Pathological Cardiac Hypertrophy Through Enhancing Transforming Growth Factor beta-Activated Kinase 1-Dependent Signaling Pathways. Hypertension 2017, 69, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-N.; Zhang, P.; Gong, J.; Zhang, X.-J.; Wang, P.-X.; Yin, M.; Jiang, Z.; Shen, L.-J.; Ji, Y.-X.; Tong, J.; et al. Tmbim1 is a multivesicular body regulator that protects against non-alcoholic fatty liver disease in mice and monkeys by targeting the lysosomal degradation of Tlr4. Nat. Med. 2017, 23, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Huang, Y.; Wang, X.; Wang, H.; Ren, F.; Tian, R.; Cheng, X.; Cai, J.; Zhang, Y.; Zhu, X.; et al. Integrated Omics Reveals Tollip as an Regulator and Therapeutic Target for Hepatic Ischemia-Reperfusion Injury in Mice. Hepatology 2019, 70, 1750–1769. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Xu, M.; Zhang, X.; Li, H. Innate Immune Signaling in Nonalcoholic Fatty Liver Disease and Cardiovascular Diseases. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 153–184. [Google Scholar] [CrossRef]
- Federico, A.; Caprio, G.G.; Ormando, V.M.; Loguercio, C. Gut Microbiota and Gastrointestinal Tract, Liver and Pancreas: From Physiology to Pathology. Minerva Gastroenterol. Dietol. 2017, 63, 385–398. [Google Scholar]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nature reviews. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar]
- Li, L.; Gao, J.; Li, J. Emerging role of HMGB 1 in fibrotic diseases. J. Cell. Mol. Med. 2014, 18, 2331–2339. [Google Scholar] [CrossRef]
- Al-Khafaji, A.B.; Tohme, S.; Yazdani, H.O.; Miller, D.; Huang, H.; Tsung, A. Superoxide induces Neutrophil Extracellular Trap Formation in a TLR-4 and NOX-Dependent Mechanism. Mol. Med. 2015, 22, 621–631. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cai, J.; She, Z.; Li, H. Insights into the Epidemiology, Pathogenesis, and Therapeutics of Nonalcoholic Fatty Liver Diseases. Adv. Sci. 2019, 6, 1801585. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Doridot, L.; Jeljeli, M.; Chêne, C.; Batteux, F. Implication of oxidative stress in the pathogenesis of systemic sclerosis via inflammation, autoimmunity and fibrosis. Redox Biol. 2019, 25, 101122. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Li, G.; Gehr, T.W.B.; Boini, K.M.; Li, P.-L. Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free. Radic. Biol. Med. 2014, 67, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.Y.; Park, H.H. Crystal Structure of NALP3 Protein Pyrin Domain (PYD) and Its Implications in Inflammasome Assembly. J. Biol. Chem. 2011, 286, 39528–39536. [Google Scholar] [CrossRef]
- Yan, F.-J.; Zhang, X.-J.; Wang, W.-X.; Ji, Y.-X.; Wang, P.-X.; Yang, Y.; Gong, J.; Shen, L.-J.; Zhu, X.-Y.; Huang, Z.; et al. The E3 ligase tripartite motif 8 targets TAK1 to promote insulin resistance and steatohepatitis. Hepatology 2017, 65, 1492–1511. [Google Scholar] [CrossRef]
- Xiang, M.; Wang, P.-X.; Wang, A.-B.; Zhang, X.-J.; Zhang, Y.; Zhang, P.; Mei, F.-H.; Chen, M.-H.; Li, H. Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. J. Hepatol. 2016, 64, 1365–1377. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, H.; Fu, J.; Cai, L.; Li, P.; Zhao, C.; Dong, Z.; Ma, J.P.; Wang, X.; Tian, H.; et al. Hepatocyte TNF Receptor–Associated Factor 6 Aggravates Hepatic Inflammation and Fibrosis by Promoting Lysine 6–Linked Polyubiquitination of Apoptosis Signal-Regulating Kinase 1. Hepatology 2020, 71, 93–111. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Fernández-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-N.; Jiang, D.-S.; Li, H. Interferon regulatory factors: At the crossroads of immunity, metabolism, and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.-J.; Wang, P.-X.; Zhang, P.; Li, H. Reprogramming Innate Immune Signaling in Cardiometabolic Disease. Hypertension 2017, 69, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Li, H. Targeting Interferon Regulatory Factor for Cardiometabolic Diseases: Opportunities and Challenges. Curr. Drug Targets 2017, 18, 1754–1778. [Google Scholar] [CrossRef]
- Wang, X.A.; Zhang, R.; She, Z.G.; Zhang, X.F.; Jiang, D.S.; Wang, T.; Gao, L.; Deng, W.; Zhang, S.M.; Zhu, L.H.; et al. Interferon regulatory factor 3 constrains IKKbeta/NF-kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 2014, 59, 870–885. [Google Scholar] [CrossRef]
- Yadav, U.C.; Ramana, K.V. Regulation of NF-kappaB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxidative Med. Cell. Longev. 2013, 2013, 690545. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Furnkranz, A.; Leitinger, N. Regulation of inflammatory responses by oxidized phospholipids: Structure-function relationships. Curr. Pharm. Des. 2004, 10, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, X.; Li, H. Liver capsule: IRFs in hepatocytes: Pathophysiology. Hepatology 2016, 63, 1706. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Wang, P.X.; Li, Z.Z.; Zhang, X.J.; Jiang, X.; Gong, J.; Qin, J.J.; Guo, J.; Zhu, X.; Yang, S.; et al. Hepatic Oncostatin M Receptor beta Regulates Obesity-Induced Steatosis and Insulin Resistance. Am. J. Pathol. 2016, 186, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Li, H. Innate immune regulatory networks in hepatic lipid metabolism. J. Mol. Med. 2019, 97, 593–604. [Google Scholar] [CrossRef]
- Azzimato, V.; Jager, J.; Chen, P.; Morgantini, C.; Levi, L.; Barreby, E.; Sulen, A.; Oses, C.; Willerbrords, J.; Xu, C.; et al. Liver macrophages inhibit the endogenous antioxidant response in obesity-associated insulin resistance. Sci. Transl. Med. 2020, 12, eaaw9709. [Google Scholar] [CrossRef]
- Milicevic, N.P.; Busch, C.J.-L.; Binder, C.J. Malondialdehyde Epitopes as Targets of Immunity and the Implications for Atherosclerosis. Dev. Funct. Myeloid Subsets 2016, 131, 1–59. [Google Scholar] [CrossRef]
- Nobili, V.; Parola, M.; Alisi, A.; Marra, F.; Piemonte, F.; Mombello, C.; Sutti, S.; Povero, D.; Maina, V.; Novo, E.; et al. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int. J. Mol. Med. 2010, 26, 471–476. [Google Scholar] [CrossRef]
- Chou, M.-Y.; Fogelstrand, L.; Hartvigsen, K.; Hansen, L.F.; Woelkers, D.; Shaw, P.X.; Choi, J.; Perkmann, T.; Bäckhed, F.; Miller, Y.I.; et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J. Clin. Investig. 2009, 119, 1335–1349. [Google Scholar] [CrossRef]
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free. Radic. Biol. Med. 2018, 125, 3–14. [Google Scholar] [CrossRef]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef]
- Albano, E.; Mottaran, E.; Vidali, M.; Reale, E.; Saksena, S.; Occhino, G.; Burt, A.D.; Day, C.P. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut 2005, 54, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Bruzzì, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free. Radic. Biol. Med. 2018, 124, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Baumgardner, J.N.; Shankar, K.; Hennings, L.; Albano, E.; Badger, T.M.; Ronis, M.J. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. J. Nutr. 2008, 138, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Rolla, S.; Alchera, E.; Imarisio, C.; Bardina, V.; Valente, G.; Cappello, P.; Mombello, C.; Follenzi, A.; Novelli, F.; Carini, R. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin. Sci. 2015, 130, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Rau, M.; Schilling, A.-K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2015, 196, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Inzaugarat, M.E.; Solari, N.E.F.; Billordo, L.A.; Abecasis, R.; Gadano, A.C.; Cherñavsky, A.C. Altered Phenotype and Functionality of Circulating Immune Cells Characterize Adult Patients with Nonalcoholic Steatohepatitis. J. Clin. Immunol. 2011, 31, 1120–1130. [Google Scholar] [CrossRef]
- Solari, N.E.F.; Inzaugarat, M.E.; Baz, P.; De Matteo, E.; Lezama, C.; Galoppo, M.; Galoppo, C.; Cherñavsky, A.C. The Role of Innate Cells Is Coupled to a Th1-Polarized Immune Response in Pediatric Nonalcoholic Steatohepatitis. J. Clin. Immunol. 2012, 32, 611–621. [Google Scholar] [CrossRef]
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.A.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, P.J.D.F.; Terabe, M.; Kapoor, V.; Elgindi, M.; et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nat. Cell Biol. 2016, 531, 253–257. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Weyler, J.; Kwanten, W.J.; Dirinck, E.L.; De Winter, B.Y.; Francque, S.M.; Vonghia, L. The Differential Roles of T Cells in Non-alcoholic Fatty Liver Disease and Obesity. Front. Immunol. 2019, 10, 82. [Google Scholar] [CrossRef]
- Laurent, S.; Carrega, P.; Saverino, D.; Piccioli, P.; Camoriano, M.; Morabito, A.; Dozin, B.; Fontana, V.; Simone, R.; Mortara, L.; et al. CTLA-4 is expressed by human monocyte-derived dendritic cells and regulates their functions. Hum. Immunol. 2010, 71, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Watzenböck, M.L.; Walenbergh, S.M.A.; Amir, S.; Gruber, S.; Kozma, M.O.; Grabsch, H.I.; Koek, G.H.; Pierik, M.J.; Staufer, K.; et al. Low levels of IgM antibodies recognizing oxidation-specific epitopes are associated with human non-alcoholic fatty liver disease. BMC Med. 2016, 14, 107. [Google Scholar] [CrossRef] [PubMed]
- Bieghs, V.; Van Gorp, P.J.J.; Walenbergh, S.M.; Gijbels, M.J.; Verheyen, F.; Buurman, W.A.; Briles, D.E.; Hofker, M.H.; Binder, C.J.; Shiri-Sverdlov, R. Specific immunization strategies against oxidized low-density lipoprotein: A novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology 2012, 56, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491.e1. [Google Scholar] [CrossRef] [PubMed]
- Margini, C.; Dufour, J.-F. The story of HCC in NAFLD: From epidemiology, across pathogenesis, to prevention and treatment. Liver Int. 2015, 36, 317–324. [Google Scholar] [CrossRef]
- Sachdeva, M.; Chawla, Y.K.; Arora, S.K. Immunology of hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2080–2090. [Google Scholar] [CrossRef]
- Fu, Y.; Chung, F.-L. Oxidative stress and hepatocarcinogenesis. Hepatoma Res. 2018, 4, 4. [Google Scholar] [CrossRef]
- Yu, L.-X.; Ling, Y.; Wang, H. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. npj Precis. Oncol. 2018, 2, 1–10. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- El–Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Sahasrabuddhe, V.V.; Gunja, M.Z.; Graubard, B.I.; Trabert, B.; Schwartz, L.M.; Park, Y.; Hollenbeck, A.R.; Freedman, N.D.; McGlynn, K.A. Nonsteroidal Anti-inflammatory Drug Use, Chronic Liver Disease, and Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2012, 104, 1808–1814. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.C.; Lin, J.T.; Jeng, L.B.; Ho, H.J.; Yang, H.R.; Wu, M.S.; Kuo, K.N.; Wu, C.Y. Nonsteroidal anti-inflammatory drugs are associated with reduced risk of early hepatocellular carcinoma recurrence after curative liver resection: A nationwide cohort study. Ann. Surg. 2015, 261, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Zappavigna, S.; Vanacore, D.; Lama, S.; Potenza, N.; Russo, A.; Ferranti, P.; Dallio, M.; Federico, A.; Loguercio, C.; Sperlongano, P.; et al. Silybin-Induced Apoptosis Occurs in Parallel to the Increase of Ceramides Synthesis and miRNAs Secretion in Human Hepatocarcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2190. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The Danger Model: A Renewed Sense of Self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.S.D.; Kang, M.-J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, X.; Hu, Q.; Wu, J.; Wang, G.; Hong, Z.; Ren, J.-A.; Infections, L.F.T.A.S. Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci. 2019, 236, 116464. [Google Scholar] [CrossRef]
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Bao, D.; Zhao, J.; Zhou, X.; Yang, Q.; Chen, Y.; Zhu, J.; Yuan, P.; Yang, J.; Qin, T.; Wan, S.; et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene 2019, 38, 5007–5020. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Tracey, K.J. HMGB1 Is a Therapeutic Target for Sterile Inflammation and Infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Kazama, H.; Ricci, J.-E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of Immunological Tolerance by Apoptotic Cells Requires Caspase-Dependent Oxidation of High-Mobility Group Box-1 Protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, W.; Wang, L.; Xiao, C.; Gong, Y.; Huang, D.; Guo, B.; Li, Q.; Xiang, Y.; Nan, Y. Co-culture of dendritic cells and cytokine-induced killer cells effectively suppresses liver cancer stem cell growth by inhibiting pathways in the immune system. BMC Cancer 2018, 18, 984. [Google Scholar] [CrossRef]
- Dou, L.; Ono, Y.; Chen, Y.-F.; Chen, X.-P.; Thomson, A.W. Hepatic Dendritic Cells, the Tolerogenic Liver Environment, and Liver Disease. Semin. Liver Dis. 2018, 38, 170–180. [Google Scholar] [CrossRef]
- Streba, L.A.M.; Streba, L.A.M.; Sandulescu, D.L.; Vere, C.C.; Mitrut, P.; Cotoi, B.V.; Popescu, L.N.; Ion, D.A. Dendritic cells and hepatocellular carcinoma. Romanian J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2014, 55, 1287–1293. [Google Scholar]
- Chen, S.; Akbar, S.M.; Tanimoto, K.; Ninomiya, T.; Iuchi, H.; Michitaka, K.; Horiike, N.; Onji, M. Absence of CD83-positive mature and activated dendritic cells at cancer nodules from patients with hepatocellular carcinoma: Relevance to hepatocarcinogenesis. Cancer Lett. 2000, 148, 49–57. [Google Scholar] [CrossRef]
- Matsui, M.; Machida, S.; Itani-Yohda, T.; Akatsuka, T. Downregulation of the proteasome subunits, transporter, and antigen presentation in hepatocellular carcinoma, and their restoration by interferon-gamma. J. Gastroenterol. Hepatol. 2002, 17, 897–907. [Google Scholar] [CrossRef]
- Roliński, J.; Hus, I. Breaking immunotolerance of tumors: A new perspective for dendritic cell therapy. J. Immunotoxicol. 2014, 11, 311–318. [Google Scholar] [CrossRef]
- Onishi, Y.; Fehervari, Z.; Yamaguchi, T.; Sakaguchi, S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. USA 2008, 105, 10113–10118. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Zeh, H.J.; Lotze, M.T. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid. Redox Signal. 2011, 15, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Szlosarek, P.W.; Balkwill, F.R. Tumour necrosis factor alpha: A potential target for the therapy of solid tumours. Lancet Oncol. 2003, 4, 565–573. [Google Scholar] [CrossRef]
- Schrader, J.; Iredale, J.P. The inflammatory microenvironment of HCC-the plot becomes complex. J. Hepatol. 2010, 54, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-D.; Ma, Y.-S.; Fang, Y.; Liu, L.-L.; Fu, D.; Shen, X. Role of the microenvironment in hepatocellular carcinoma development and progression. Cancer Treat. Rev. 2012, 38, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S. NF-κB, JNK, and TLR Signaling Pathways in Hepatocarcinogenesis. Gastroenterol. Res. Pr. 2010, 2010, 1–10. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumor-Associated Macrophages. BioMed Res. Int. 2012, 2013, 1–15. [Google Scholar] [CrossRef]
- Komohara, Y.; Takeya, M. CAFs and TAMs: Maestros of the tumour microenvironment. J. Pathol. 2016, 241, 313–315. [Google Scholar] [CrossRef]
- Oršolić, N.; Kunštić, M.; Kukolj, M.; Gračan, R.; Nemrava, J. Oxidative stress, polarization of macrophages and tumour angiogenesis: Efficacy of caffeic acid. Chem. Biol. Interact. 2016, 256, 111–124. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.L.; Linnoila, I.; Liu, Z.-G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef]
- Zhu, X.-D.; Zhang, J.-B.; Zhuang, P.-Y.; Zhu, H.-G.; Zhang, W.; Xiong, Y.-Q.; Wu, W.-Z.; Wang, L.; Tang, Z.-Y.; Sun, H.-C. High Expression of Macrophage Colony-Stimulating Factor in Peritumoral Liver Tissue Is Associated With Poor Survival After Curative Resection of Hepatocellular Carcinoma. J. Clin. Oncol. 2008, 26, 2707–2716. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Forgues, M.; Ye, Q.-H.; Jia, H.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.; Tang, Z.-Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Immuno-Oncology in Hepatocellular Carcinoma: 2017 Update. Oncology 2017, 93, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Attallah, A.M.; Tabll, A.A.; El-Sadany, M.; Ibrahim, T.A.; El-Dosoky, I. Dysregulation of blood lymphocyte subsets and natural killer cells in schistosomal liver cirrhosis and hepatocellular carcinoma. Z. Die Gesamte Exp. Med. 2003, 3, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. NAFLD Related-HCC: The Relationship with Metabolic Disorders. Adv. Exp. Med. Biol. 2018, 1061, 55–62. [Google Scholar] [CrossRef]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased Populations of Regulatory T Cells in Peripheral Blood of Patients with Hepatocellular Carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef]
- Greten, T.F.; Ormandy, L.A.; Fikuart, A.; Höchst, B.; Henschen, S.; Hörning, M.; Manns, M.P.; Korangy, F. Low-dose Cyclophosphamide Treatment Impairs Regulatory T Cells and Unmasks AFP-specific CD4+ T-cell Responses in Patients with Advanced HCC. J. Immunother. 2010, 33, 211–218. [Google Scholar] [CrossRef]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, B.; Du, Z.; Bai, T.; Gao, Y.T.; Wang, Y.J.; Lou, C.; Wang, F.M.; Bai, Y. Aberrant methylation of SPARC in human hepatocellular carcinoma and its clinical implication. World J. Gastroenterol. 2012, 18, 2043–2052. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology 2002, 35, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.E.; Baldeosingh, R.; Lamm, R.; Patel, K.; Zhang, K.; Dominguez, D.A.; Kirton, K.J.; Shah, A.P.; Dang, H. Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell Dev. Biol. 2020, 8, 709. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Vermijlen, D.; Luo, D.; Froelich, C.J.; Medema, J.P.; Kummer, J.A.; Willems, E.; Braet, F.; Wisse, E. Hepatic natural killer cells exclusively kill splenic/blood natural killer-resistant tumor cells by the perforin/granzyme pathway. J. Leukoc. Biol. 2002, 72, 668–676. [Google Scholar]
- Li, T.; Yang, Y.; Song, H.; Li, H.; Cui, A.; Liu, Y.; Su, L.; Crispe, I.N.; Tu, Z. Activated NK cells kill hepatic stellate cells via p38/PI3K signaling in a TRAIL-involved degranulation manner. J. Leukoc. Biol. 2019, 105, 695–704. [Google Scholar] [CrossRef]
- Singh, H.D.; Otano, I.; Rombouts, K.; Singh, K.P.; Peppa, D.; Gill, U.S.; Böttcher, K.; Kennedy, P.T.F.; Oben, J.; Pinzani, M.; et al. TRAIL regulatory receptors constrain human hepatic stellate cell apoptosis. Sci. Rep. 2017, 7, 5514. [Google Scholar] [CrossRef]
- Radaeva, S.; Wang, L.; Radaev, S.; Jeong, W.-I.; Park, O.; Gao, B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am. J. Physiol. Liver Physiol. 2007, 293, G809–G816. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, L.; Xu, Y.; Zhang, Z.; Ren, G.; Tang, K.; Kuang, P.; Zhao, B.; Yin, Z.; Wang, X. Hepatic stellate cells promote tumor progression by enhancement of immunosuppressive cells in an orthotopic liver tumor mouse model. Lab. Investig. 2014, 94, 182–191. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased Regulatory T Cells Correlate With CD8 T-Cell Impairment and Poor Survival in Hepatocellular Carcinoma Patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Ju, M.-J.; Qiu, S.-J.; Gao, Q.; Fan, J.; Cai, M.-Y.; Li, Y.-W.; Tang, Z.-Y. Combination of peritumoral mast cells and T-regulatory cells predicts prognosis of hepatocellular carcinoma. Cancer Sci. 2009, 100, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Schmidt, C.S.; Mondino, A.; Lins, D.C.; Kedl, R.M.; Jenkins, M.K.; Mescher, M.F. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol. 1999, 162, 3256–3262. [Google Scholar]
- Lever, M.; Maini, P.K.; Van Der Merwe, P.A.; Dushek, O. Phenotypic models of T cell activation. Nat. Rev. Immunol. 2014, 14, 619–629. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- El Dika, I.; Khalil, D.N.; Abou-Alfa, G.K. Immune checkpoint inhibitors for hepatocellular carcinoma. Cancer 2019, 125, 3312–3319. [Google Scholar] [CrossRef]
- Ritz, T.; Krenkel, O.; Tacke, F. Dynamic plasticity of macrophage functions in diseased liver. Cell. Immunol. 2018, 330, 175–182. [Google Scholar] [CrossRef]
- Devisscher, L.; Verhelst, X.; Colle, I.; Van Vlierberghe, H.; Geerts, A. The role of macrophages in obesity-driven chronic liver disease. J. Leukoc. Biol. 2016, 99, 693–698. [Google Scholar] [CrossRef]
- Bekkering, S.; Blok, B.A.; Joosten, L.A.; Riksen, N.P.; van Crevel, R.; Netea, M.G. In Vitro Experimental Model of Trained Innate Immunity in Human Primary Monocytes. Clin. Vaccine Immunol. 2016, 23, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Mourits, V.P.; Arts, R.J.; Novakovic, B.; Matzaraki, V.; De Bree, L.C.J.; Koeken, V.A.; Moorlag, S.J.; Van Puffelen, J.H.; Groh, L.; Van Der Heijden, C.D.; et al. The role of Toll-like receptor 10 in modulation of trained immunity. Immunology 2019, 159, 289–297. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar] [CrossRef] [PubMed]
- Quintin, J.; Saeed, S.; Martens, J.H.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.-J.; Wijmenga, C.; et al. Candida albicans Infection Affords Protection against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.B.; Ifrim, D.C.; Saeed, S.; Jacobs, C.; Van Loenhout, J.; De Jong, D.; Stunnenberg, H.G.; et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 17537–17542. [Google Scholar] [CrossRef]
- Morgillo, F.; Dallio, M.; Morgillo, F.; Gravina, A.G.; Viscardi, G.; Loguercio, C.; Ciardiello, F.; Federico, A. Carcinogenesis as a Result of Multiple Inflammatory and Oxidative Hits: A Comprehensive Review from Tumor Microenvironment to Gut Microbiota. Neoplasia 2018, 20, 721–733. [Google Scholar] [CrossRef]
- Ganeshan, K.; Chawla, A. Metabolic Regulation of Immune Responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef]
- Sangineto, M.; Grabherr, F.; Adolph, T.E.; Grander, C.; Reider, S.; Jaschke, N.; Mayr, L.; Schwärzler, J.; Dallio, M.; Moschen, A.R.; et al. Dimethyl fumarate ameliorates hepatic inflammation in alcohol related liver disease. Liver Int. 2020, 40, 1610–1619. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef]
- Dominguez-Andres, J.; Novakovic, B.; Li, Y.; Scicluna, B.P.; Gresnigt, M.S.; Arts, R.J.; Oosting, M.; Moorlag, S.J.; Groh, L.A.; Zwaag, J.; et al. The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab. 2019, 29, 211–220.e5. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, L.; Xiao, R.; Wu, M.; Jinquan, T.; He, Y. Expression profile of human immune-responsive gene 1 and generation and characterization of polyclonal antiserum. Mol. Cell. Biochem. 2011, 353, 177–187. [Google Scholar] [CrossRef]
- Ganta, V.C.; Choi, M.H.; Kutateladze, A.; Fox, T.E.; Farber, C.R.; Annex, B.H. A MicroRNA93–Interferon Regulatory Factor-9–Immunoresponsive Gene-1–Itaconic Acid Pathway Modulates M2-Like Macrophage Polarization to Revascularize Ischemic Muscle. Circulation 2017, 135, 2403–2425. [Google Scholar] [CrossRef]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.H.; et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef]
- Costa, A.S.H.; Higgins, M.; Hams, E.; Szpyt, J.; Runtsch, M.C.; King, M.S.; McGouran, J.F.; Fischer, R.; Kessler, B.M.; McGettrick, A.F.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar]
- Cordes, T.; Lucas, A.; Divakaruni, A.S.; Murphy, A.N.; Cabrales, P.; Metallo, C.M. Itaconate modulates tricarboxylic acid and redox metabolism to mitigate reperfusion injury. Mol. Metab. 2020, 32, 122–135. [Google Scholar] [CrossRef]
- Yi, Z.; Deng, M.; Scott, M.J.; Fu, G.; Loughran, P.A.; Lei, Z.; Li, S.; Sun, P.; Yang, C.; Li, W.; et al. IRG1/Itaconate Activates Nrf2 in Hepatocytes to Protect Against Liver Ischemia-Reperfusion Injury. Hepatology 2020, 72, 1394–1411. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Di Sarno, R.; Giorgio, V.; Miele, L. Gut microbiota, obesity and metabolic disorders. Minerva Gastroenterol. Dietol. 2017, 63, 337–344. [Google Scholar] [PubMed]



{kind=link}
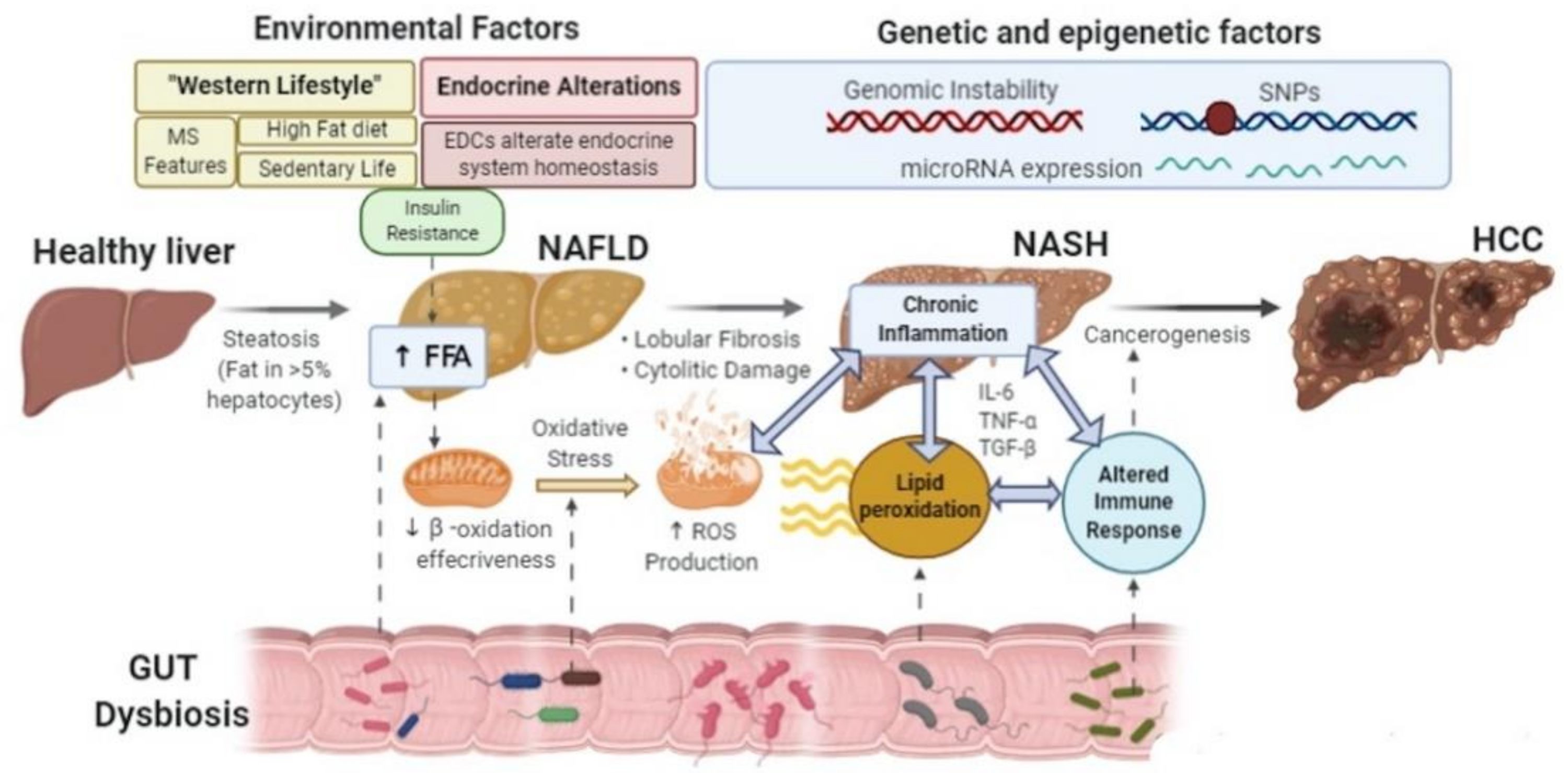{kind=link}
{kind=link}
| Main Mechanisms and Activities | Main Effects and Epiphenomena |
|---|---|
| Redox unbalance: ROS 11 production overcomes the scavenging capacity so that superoxide anion radicals (O2·−) and hydrogen peroxide (H2O2) are continuously formed. As a consequence: | Perpetuation of chronic inflammation Creation of a vicious circle where the chronic inflammation and the oxidative stress fuelling each other, involving adaptive immune cell mechanisms, contribute to the disease progression. Increased insulin resistance Chronic inflammation worsens insulin resistance, FFAs 1 accumulation and thus NAFLD evolution. |
| |
| |
| |
| OSEs 3: oxidative stress promotes the production of oxidised phospholipids and aldehydes generated by lipid peroxidation such as MAA 4 and 4-HNE 5-protein adducts. As a consequence: | Inflammation and Liver Damage Pro-inflammatory cytokine release and cell death promote the worsening of liver histological damage |
| |
| PAMPs 13 and DAMPs 14 hepatic invasion:13 PAMPs (e.g., LPS 15), 14 DAMPs (e.g., HMGB1, mtDNA, lipotoxic products, and other metabolites) continuously invade the liver in NAFLD 16. As a consequence: | Inflammation and Liver Damage Pro-inflammatory cytokine release promotes the worsening of liver histological damage. Fibrosis By HSCs fibrogenic response. |
| |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dallio, M.; Sangineto, M.; Romeo, M.; Villani, R.; Romano, A.D.; Loguercio, C.; Serviddio, G.; Federico, A. Immunity as Cornerstone of Non-Alcoholic Fatty Liver Disease: The Contribution of Oxidative Stress in the Disease Progression. Int. J. Mol. Sci. 2021, 22, 436. https://doi.org/10.3390/ijms22010436
Dallio M, Sangineto M, Romeo M, Villani R, Romano AD, Loguercio C, Serviddio G, Federico A. Immunity as Cornerstone of Non-Alcoholic Fatty Liver Disease: The Contribution of Oxidative Stress in the Disease Progression. International Journal of Molecular Sciences. 2021; 22(1):436. https://doi.org/10.3390/ijms22010436
Chicago/Turabian StyleDallio, Marcello, Moris Sangineto, Mario Romeo, Rosanna Villani, Antonino Davide Romano, Carmelina Loguercio, Gaetano Serviddio, and Alessandro Federico. 2021. "Immunity as Cornerstone of Non-Alcoholic Fatty Liver Disease: The Contribution of Oxidative Stress in the Disease Progression" International Journal of Molecular Sciences 22, no. 1: 436. https://doi.org/10.3390/ijms22010436
APA StyleDallio, M., Sangineto, M., Romeo, M., Villani, R., Romano, A. D., Loguercio, C., Serviddio, G., & Federico, A. (2021). Immunity as Cornerstone of Non-Alcoholic Fatty Liver Disease: The Contribution of Oxidative Stress in the Disease Progression. International Journal of Molecular Sciences, 22(1), 436. https://doi.org/10.3390/ijms22010436