Intracellular and Extracellular Markers of Lethality in Osteogenesis Imperfecta: A Quantitative Proteomic Approach

 , , , ,
, , , , 
Abstract
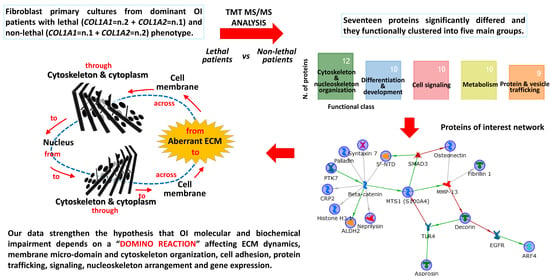
1. Introduction
2. Results and Discussion
2.1. Fibrillin-1 Higher Abundance in Lethal Fibroblasts: Signaling and Metabolism as Possible Players for OI Phenotype
2.2. Decorin Reduction May Modulate OI Severity Acting on Cell Signaling, Intracellular Trafficking, Cytoskeletal Organization, and Collagen Assembly
2.3. SPARC Abundance Negatively Affects OI Outcome
2.4. Chondroitin Sulfate Proteoglycan 4: A Sprout Protein between Extra and Intracellular Compartment
2.5. Tyrosine-Protein Kinase 7 (PTK7) Points to the Wnt Pathway as Modulator of OI Outcome
2.6. Altered Cytoskeleton Modulates OI Phenotype
3. Materials and Methods
3.1. Patients
3.2. Tissue Culture and Lysate Preparation
3.3. Sample Preparation for Tandem Mass Tag Labeling and LC-MS/MS
3.4. Protein Identification and Quantification
3.5. Gene Ontology (GO) Clustering
3.6. MetaCore Functional Analysis
3.7. Western Blotting
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forlino, A.; Cabral, W.A.; Barnes, A.M.; Marini, J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011, 7, 540–557. [Google Scholar] [CrossRef]
- Marini, J.C.; Forlino, A.; Cabral, W.A.; Barnes, A.M.; Antonio, J.D.S.; Milgrom, S.; Hyland, J.C.; Körkkö, J.; Prockop, D.J.; De Paepe, A.; et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007, 28, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.M.; Skovby, F.; Schwartz, M. Serine for glycine substitutions in the C-terminal third of the alpha 1(I) chain of collagen I in five patients with nonlethal osteogenesis imperfecta. Hum. Mutat. 1997, 9, 378–382. [Google Scholar] [CrossRef]
- Kang, H.; AC, S.A.; Marini, J.C.; Aryal, A.S. Osteogenesis imperfecta: New genes reveal novel mechanisms in bone dysplasia. Transl. Res. 2017, 181, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, I.M.; Glorieux, F.H.; Rauch, F. Genotype-Phenotype Correlations in Autosomal Dominant Osteogenesis Imperfecta. J. Osteoporos. 2011, 2011, 540178. [Google Scholar] [CrossRef] [PubMed]
- Rauch, F.; Moffatt, P.; Cheung, M.; Roughley, P.J.; Lalic, L.; Lund, A.M.; Ramirez, N.; Fahiminiya, S.; Majewski, J.; Glorieux, F.H. Osteogenesis imperfecta type V: Marked phenotypic variability despite the presence of theIFITM5c.−14C>T mutation in all patients. J. Med. Genet. 2013, 50, 21–24. [Google Scholar] [CrossRef]
- Bodian, D.L.; Chan, T.F.; Poon, A.; Schwarze, U.; Yang, K.; Byers, P.H.; Kwok, P.-Y.; Klein, T.E. Mutation and polymorphism spectrum in osteogenesis imperfecta type II: Implications for genotype-phenotype relationships. Hum. Mol. Genet. 2009, 18, 463–471. [Google Scholar] [CrossRef]
- Forlino, A.; Tani, C.; Rossi, A.; Lupi, A.; Campari, E.; Gualeni, B.; Bianchi, L.; Armini, A.; Cetta, G.; Bini, L.; et al. Differential expression of both extracellular and intracellular proteins is involved in the lethal or nonlethal phenotypic variation of BrtlIV, a murine model for osteogenesis imperfecta. Proteomics 2007, 7, 1877–1891. [Google Scholar] [CrossRef]
- Bianchi, L.; Gagliardi, A.; Maruelli, S.; Besio, R.; Landi, C.; Gioia, R.; Kozloff, K.M.; Khoury, B.M.; Coucke, P.J.; Symoens, S.; et al. Altered cytoskeletal organization characterized lethal but not surviving Brtl+/- mice: Insight on phenotypic variability in osteogenesis imperfecta. Hum. Mol. Genet. 2015, 24, 6118–6133. [Google Scholar] [CrossRef]
- Gagliardi, A.; Besio, R.; Carnemolla, C.; Landi, C.; Armini, A.; Aglan, M.; Otaify, G.; Temtamy, S.A.; Forlino, A.; Bini, L.; et al. Cytoskeleton and nuclear lamina affection in recessive osteogenesis imperfecta: A functional proteomics perspective. J. Proteom. 2017, 167, 46–59. [Google Scholar] [CrossRef]
- Marom, R.; Rabenhorst, B.M.; Morello, R. Osteogenesis imperfecta: An update on clinical features and therapies. Eur. J. Endocrinol. 2020, 183, R95–R106. [Google Scholar] [CrossRef] [PubMed]
- Besio, R.; Iula, G.; Garibaldi, N.; Cipolla, L.; Sabbioneda, S.; Biggiogera, M.; Marini, J.C.; Rossi, A.; Forlino, A. 4-PBA ameliorates cellular homeostasis in fibroblasts from osteogenesis imperfecta patients by enhancing autophagy and stimulating protein secretion. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Forlino, A.; Kuznetsova, N.V.; Marini, J.C.; Leikin, S. Selective retention and degradation of molecules with a single mutant α1(I) chain in the Brtl IV mouse model of OI. Matrix Biol. 2007, 26, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Folkestad, L.; Hald, J.; Gram, J.; Langdahl, B.; Hermann, A.P.; Diederichsen, A.C.P.; Abrahamsen, B.; Brixen, K. Cardiovascular disease in patients with osteogenesis imperfecta—A nationwide, register-based cohort study. Int. J. Cardiol. 2016, 225, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Folkestad, L.; Hald, J.D.; Canudas-Romo, V.; Gram, J.; Hermann, A.P.; Langdahl, B.; Abrahamsen, B.; Brixen, K. Mortality and Causes of Death in Patients With Osteogenesis Imperfecta: A Register-Based Nationwide Cohort Study. J. Bone Miner. Res. 2016, 31, 2159–2166. [Google Scholar] [CrossRef]
- DiGirolamo, D.J.; Singhal, V.; Chang, X.; Lee, S.-J.; Germain-Lee, E.L. Administration of soluble activin receptor 2B increases bone and muscle mass in a mouse model of osteogenesis imperfecta. Bone Res. 2015, 3, 14042. [Google Scholar] [CrossRef]
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657–1671. [Google Scholar] [CrossRef]
- Bianchi, L.; Gagliardi, A.; Gioia, R.; Besio, R.; Tani, C.; Landi, C.; Cipriano, M.; Gimigliano, A.; Rossi, A.; Marini, J.C.; et al. Differential response to intracellular stress in the skin from osteogenesis imperfecta Brtl mice with lethal and non lethal phenotype: A proteomic approach. J. Proteom. 2012, 75, 4717–4733. [Google Scholar] [CrossRef]
- Senarath-Yapa, K.; Meyer, N.; Li, S.; Longaker, M.T.; Quarto, N. Abstract 54: TGF Beta and BMP Signaling Pathways Influence Regenerative Capacity of Calvarial Bones via Cross-Talk and Modulation of Apoptosis: The Potential Therapeutic Role of Small Molecule Inhibitors of TGF Beta Signaling. Plast. Reconstr. Surg. 2014, 133, 65–66. [Google Scholar] [CrossRef]
- Wagner, I.; Wang, H.; Weissert, P.M.; Straube, W.L.; Shevchenko, A.; Gentzel, M.; Brito, G.; Tazaki, A.; Oliveira, C.; Sugiura, T.; et al. Serum Proteases Potentiate BMP-Induced Cell Cycle Re-entry of Dedifferentiating Muscle Cells during Newt Limb Regeneration. Dev. Cell 2017, 40, 608–617.e6. [Google Scholar] [CrossRef]
- Filvaroff, E.; Erlebacher, A.; Ye, J.; Gitelman, S.E.; Lotz, J.; Heillman, M.; Derynck, R. Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development 1999, 126, 4267–4279. [Google Scholar]
- Zieba, J.; Munivez, E.; Castellon, A.; Jiang, M.; Dawson, B.; Ambrose, C.G.; Lee, B.H.L. Fracture Healing in Collagen-Related Preclinical Models of Osteogenesis Imperfecta. J. Bone Miner. Res. 2020, 35, 1132–1148. [Google Scholar] [CrossRef]
- Jann, J.; Gascon, S.; Roux, S.; Faucheux, N. Influence of the TGF-β Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions. Int. J. Mol. Sci. 2020, 21, 7597. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Deng, C.; Li, Y.-P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Grafe, I.; Bae, Y.; Chen, S.; Chen, Y.; Bertin, T.K.; Jiang, M.-M.; Ambrose, C.G.; Lee, B. E-selectin ligand 1 regulates bone remodeling by limiting bioactive TGF-β in the bone microenvironment. Proc. Natl. Acad. Sci. USA 2013, 110, 7336–7341. [Google Scholar] [CrossRef]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone Morphogenetic Proteins: A critical review. Cell. Signal. 2011, 23, 609–620. [Google Scholar] [CrossRef]
- Nistala, H.; Lee-Arteaga, S.; Smaldone, S.; Siciliano, G.; Carta, L.; Ono, R.N.; Sengle, G.; Arteaga-Solis, E.; Levasseur, R.; Ducy, P.; et al. Fibrillin-1 and -2 differentially modulate endogenous TGF-β and BMP bioavailability during bone formation. J. Cell Biol. 2010, 190, 1107–1121. [Google Scholar] [CrossRef]
- Vehviläinen, P.; Hyytiäinen, M.; Keski-Oja, J. Matrix association of latent TGF-beta binding protein-2 (LTBP-2) is dependent on fibrillin-1. J. Cell. Physiol. 2009, 221, 586–593. [Google Scholar] [CrossRef]
- Muthu, M.L.; Reinhardt, D.P. Fibrillin-1 and fibrillin-1-derived asprosin in adipose tissue function and metabolic disorders. J. Cell Commun. Signal. 2020, 14, 159–173. [Google Scholar] [CrossRef]
- Grafe, I.; Yang, T.; Alexander, S.; Homan, E.P.; Lietman, C.; Jiang, M.M.; Bertin, T.K.; Munivez, E.; Chen, Y.; Dawson, B.; et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 2014, 20, 670–675. [Google Scholar] [CrossRef]
- Tauer, J.T.; Abdullah, S.; Rauch, F. Effect of Anti-TGF-β Treatment in a Mouse Model of Severe Osteogenesis Imperfecta. J. Bone Miner. Res. 2019, 34, 207–214. [Google Scholar] [CrossRef]
- Boraschi-Diaz, I.; Tauer, J.T.; El Rifai, O.; Guillemette, D.; Lefebvre, G.; Rauch, F.; Ferron, M.; Komarova, S.V. Metabolic phenotype in the mouse model of osteogenesis imperfecta. J. Endocrinol. 2017, 234, 279–289. [Google Scholar] [CrossRef]
- Etich, J.; Rehberg, M.; Eckes, B.; Sengle, G.; Semler, O.; Zaucke, F. Signaling pathways affected by mutations causing osteogenesis imperfecta. Cell. Signal. 2020, 76, 109789. [Google Scholar] [CrossRef] [PubMed]
- Chafey, P.; Finzi, L.; Boisgard, R.; Caüzac, M.; Clary, G.; Broussard, C.; Pégorier, J.-P.; Guillonneau, F.; Mayeux, P.; Camoin, L.; et al. Proteomic analysis of beta-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics 2009, 9, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Rainero, E.; Howe, J.D.; Caswell, P.T.; Jamieson, N.B.; Anderson, K.; Critchley, D.R.; Machesky, L.; Norman, J.C. Ligand-Occupied Integrin Internalization Links Nutrient Signaling to Invasive Migration. Cell Rep. 2015, 10, 398–413. [Google Scholar] [CrossRef] [PubMed]
- Dornier, E.; Norman, J.C. Tensin links energy metabolism to extracellular matrix assembly. J. Cell Biol. 2017, 216, 867–869. [Google Scholar] [CrossRef]
- Dietz, H.C.; Cutting, C.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef]
- Robinson, P.N.; Arteaga-Solis, E.; Baldock, C.; Collod-Béroud, G.; Booms, P.; De Paepe, A.; Dietz, H.C.; Guo, G.; Handford, P.A.; Judge, D.P.; et al. The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet. 2006, 43, 769–787. [Google Scholar] [CrossRef]
- Maroteaux, P.; Stanescu, R.; Stanescu, V.; Rappaport, R.; Reynolds, J.F. Acromicric dysplasia. Am. J. Med Genet. 1986, 24, 447–459. [Google Scholar] [CrossRef]
- Le Goff, C.; Mahaut, C.; Wang, L.W.; Allali, S.; Abhyankar, A.; Jensen, S.; Zylberberg, L.; Collod-Beroud, G.; Bonnet, D.; Alanay, Y.; et al. Mutations in the TGFβ Binding-Protein-Like Domain 5 of FBN1 Are Responsible for Acromicric and Geleophysic Dysplasias. Am. J. Hum. Genet. 2011, 89, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Schrenk, S.; Cenzi, C.; Bertalot, T.; Conconi, M.T.; Di Liddo, R. Structural and functional failure of fibrillin‑1 in human diseases (Review). Int. J. Mol. Med. 2017, 41, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Fedarko, N.S.; Robey, P.; Vetter, U.K. Extracellular matrix stoichiometry in osteoblasts from patients with osteogenesis imperfecta. J. Bone Miner. Res. 1995, 10, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, M.K.; Otero-Viñas, M.; Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, X.J.; Liu, Y.; Zhang, X.; Li, Y.Y.; Xu, W.S. Recombinant human decorin inhibits cell proliferation and downregulates TGF-beta1 production in hypertrophic scar fibroblasts. Burns 2007, 33, 634–641. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin causes autophagy in endothelial cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Instructive Roles of Extracellular Matrix on Autophagy. Am. J. Pathol. 2014, 184, 2146–2153. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Yamamoto, A.; Kitamura, A.; Lamandé, S.R.; Yoshimori, T.; Bateman, J.F.; Kubota, H.; Nagata, K. Autophagic Elimination of Misfolded Procollagen Aggregates in the Endoplasmic Reticulum as a Means of Cell Protection. Mol. Biol. Cell 2009, 20, 2744–2754. [Google Scholar] [CrossRef]
- Mirigian, L.S.; Makareeva, E.; Mertz, E.L.; Omari, S.; Roberts-Pilgrim, A.M.; Oestreich, A.K.; Phillips, C.L.; Leikin, S. Osteoblast Malfunction Caused by Cell Stress Response to Procollagen Misfolding in α2(I)-G610C Mouse Model of Osteogenesis Imperfecta. J. Bone Miner. Res. 2016, 31, 1608–1616. [Google Scholar] [CrossRef]
- Kim, S.-W.; Hayashi, M.; Lo, J.-F.; Yang, Y.; Yoo, J.-S.; Lee, J.-D. ADP-ribosylation Factor 4 Small GTPase Mediates Epidermal Growth Factor Receptor-dependent Phospholipase D2 Activation. J. Biol. Chem. 2003, 278, 2661–2668. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Jackson, C.L. ARF family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Nakai, W.; Kondo, Y.; Saitoh, A.; Naito, T.; Nakayama, K.; Shin, H.-W. ARF1 and ARF4 regulate recycling endosomal morphology and retrograde transport from endosomes to the Golgi apparatus. Mol. Biol. Cell 2013, 24, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chia, J.; Canning, C.A.; Jones, C.M.; Bard, F.; Virshup, D.M. WLS Retrograde Transport to the Endoplasmic Reticulum during Wnt Secretion. Dev. Cell 2014, 29, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.-X.; Calafat, J.; Janssen, H.; De La Iglesia-Vicente, J.; Mollinedo, F. Intracellular location of syntaxin 7 in human neutrophils. Immunol. Lett. 2010, 129, 72–77. [Google Scholar] [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Oncosuppressive functions of decorin. Mol. Cell. Oncol. 2015, 2, e975645. [Google Scholar] [CrossRef]
- Levy, O.; Ruvinov, E.; Reem, T.; Granot, Y.; Cohen, S. Highly efficient osteogenic differentiation of human mesenchymal stem cells by eradication of STAT3 signaling. Int. J. Biochem. Cell Biol. 2010, 42, 1823–1830. [Google Scholar] [CrossRef]
- Ahmed, Z.; Berry, M.; Esmaeili, M.; Logan, A. Decorin treatment of spinal cord injury. Neural Regen. Res. 2014, 9, 1653–1656. [Google Scholar] [CrossRef]
- Jungmann, O.; Nikolovska, K.; Stock, C.; Schulz, J.-N.; Eckes, B.; Riethmüller, C.; Owens, R.T.; Iozzo, R.V.; Seidler, D.G. The Dermatan Sulfate Proteoglycan Decorin Modulates α2β1 Integrin and the Vimentin Intermediate Filament System during Collagen Synthesis. PLoS ONE 2012, 7, e50809. [Google Scholar] [CrossRef]
- Goldman, R.D.; Khuon, S.; Chou, Y.H.; Opal, P.; Steinert, P.M. The function of intermediate filaments in cell shape and cytoskeletal integrity. J. Cell Biol. 1996, 134, 971–983. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef]
- Ivaska, J.; Pallari, H.-M.; Nevo, J.; Eriksson, J.E. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 2007, 313, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the Vimentin Network under Control: Cell–Matrix Adhesion–associated Plectin 1f Affects Cell Shape and Polarity of Fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.A.; Wong, P. Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nature 2004, 6, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.A.; Sun, M.; Barnum, C.E.; Weiss, S.N.; Huegel, J.; Shetye, S.S.; Lin, L.; Saez, D.; Adams, S.M.; Iozzo, R.V.; et al. Decorin and biglycan are necessary for maintaining collagen fibril structure, fiber realignment, and mechanical properties of mature tendons. Matrix Biol. 2017, 64, 81–93. [Google Scholar] [CrossRef]
- Danielson, K.G.; Baribault, H.; Holmes, D.F.; Graham, H.; Kadler, K.E.; Iozzo, R.V. Targeted Disruption of Decorin Leads to Abnormal Collagen Fibril Morphology and Skin Fragility. J. Cell Biol. 1997, 136, 729–743. [Google Scholar] [CrossRef]
- Gordon, J.; Freedman, B.; Zuskov, A.; Iozzo, R.V.; Birk, D.; Soslowsky, L. Achilles tendons from decorin- and biglycan-null mouse models have inferior mechanical and structural properties predicted by an image-based empirical damage model. J. Biomech. 2015, 48, 2110–2115. [Google Scholar] [CrossRef]
- Jarvelainen, H.; Puolakkainen, P.; Pakkanen, S.; Brown, E.L.; Hook, M.; Iozzo, R.V.; Sage, E.; Wight, T.N. A role for decorin in cutaneous wound healing and angiogenesis. Wound Repair Regen. 2006, 14, 443–452. [Google Scholar] [CrossRef]
- Bi, Y.; Stuelten, C.H.; Kilts, T.; Wadhwa, S.; Iozzo, R.V.; Robey, P.G.; Chen, X.-D.; Young, M.F. Extracellular Matrix Proteoglycans Control the Fate of Bone Marrow Stromal Cells. J. Biol. Chem. 2005, 280, 30481–30489. [Google Scholar] [CrossRef]
- Morello, R. Osteogenesis imperfecta and therapeutics. Matrix Biol. 2018, 71–72, 294–312. [Google Scholar] [CrossRef]
- Jeong, Y.; Daghlas, S.A.; Xie, Y.; Hulbert, M.A.; Pfeiffer, F.M.; Dallas, M.R.; Omosule, C.L.; Pearsall, R.S.; Dallas, S.L.; Phillips, C.L. Skeletal Response to Soluble Activin Receptor Type IIB in Mouse Models of Osteogenesis Imperfecta. J. Bone Miner. Res. 2018, 33, 1760–1772. [Google Scholar] [CrossRef]
- Carmen, L.; Maria, V.; Morales-Medina, J.C.; Vallelunga, A.; Palmieri, B.; Iannitti, T. Role of proteoglycans and glycosaminoglycans in Duchenne muscular dystrophy. Glycobiology 2019, 29, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Riley, H.J.; Bradshaw, A.D. The Influence of the Extracellular Matrix in Inflammation: Findings from the SPARC-Null Mouse. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2020, 303, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.D. The role of SPARC in extracellular matrix assembly. J. Cell Commun. Signal. 2009, 3, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Londono, R.; Fahiminiya, S.; Majewski, J.; Tétreault, M.; Nadaf, J.; Kannu, P.; Sochett, E.; Howard, A.W.; Stimec, J.; Dupuis, L.; et al. Recessive Osteogenesis Imperfecta Caused by Missense Mutations in SPARC. Am. J. Hum. Genet. 2015, 96, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.D.; Baicu, C.F.; Rentz, T.J.; van Laer, A.O.; Boggs, J.; Lacy, J.M.; Zile, M.R. Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: Role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation 2009, 119, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.A.; Tillet, E.; Timpl, R.; Stallcup, W.B. Binding of the NG2 proteoglycan to type VI collagen and other extracellular matrix molecules. J. Biol. Chem. 1996, 271, 26110–26116. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Kawakami, K.; Huangfu, W.-C.; Liu, I.-H. Chondroitin sulfate proteoglycan 4 regulates zebrafish body axis organization via Wnt/planar cell polarity pathway. PLoS ONE 2020, 15, e0230943. [Google Scholar] [CrossRef]
- Iida, J.; Pei, D.; Kang, T.; Simpson, M.A.; Herlyn, M.; Furcht, L.T.; McCarthy, J.B. Melanoma Chondroitin Sulfate Proteoglycan Regulates Matrix Metalloproteinase-dependent Human Melanoma Invasion into Type I Collagen. J. Biol. Chem. 2001, 276, 18786–18794. [Google Scholar] [CrossRef]
- Iieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2017, 8, 1911. [Google Scholar]
- Peradziryi, H.; Tolwinski, N.S.; Borchers, A. The many roles of PTK7: A versatile regulator of cell–cell communication. Arch. Biochem. Biophys. 2012, 524, 71–76. [Google Scholar] [CrossRef]
- Puppo, F.; Thomé, V.; Lhoumeau, A.-C.; Cibois, M.; Gangar, A.; Lembo, F.; Belotti, E.; Marchetto, S.; Lecine, P.; Prebet, T.; et al. Protein tyrosine kinase 7 has a conserved role in Wnt/β-catenin canonical signalling. EMBO Rep. 2011, 12, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Keupp, K.; Beleggia, F.; Kayserili, H.; Barnes, A.M.; Steiner, M.; Semler, O.; Fischer, B.; Yigit, G.; Janda, C.Y.; Becker, J.; et al. Mutations in WNT1 Cause Different Forms of Bone Fragility. Am. J. Hum. Genet. 2013, 92, 565–574. [Google Scholar] [CrossRef]
- Pyott, S.M.; Tran, T.T.; Leistritz, D.F.; Pepin, M.G.; Mendelsohn, N.J.; Temme, R.T.; Fernandez, B.A.; Elsayed, S.M.; Elsobky, E.; Verma, I.; et al. WNT1 Mutations in Families Affected by Moderately Severe and Progressive Recessive Osteogenesis Imperfecta. Am. J. Hum. Genet. 2013, 92, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Kihara, T.; Shinohara, S.; Fujikawa, R.; Sugimoto, Y.; Murata, M.; Miyake, J. Regulation of cysteine-rich protein 2 localization by the development of actin fibers during smooth muscle cell differentiation. Biochem. Biophys. Res. Commun. 2011, 411, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Midorikawa, Y.; Tsutsumi, S.; Taniguchi, H.; Ishii, M.; Kobune, Y.; Kodama, T.; Makuuchi, M.; Aburatani, H. Identification of genes associated with dedifferentiation of hepatocellular carcinoma with expression profiling analysis. Jpn. J. Cancer Res. 2002, 93, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Mao, X.; Dieterle, M.; Moreau, F.; Al Absi, A.; Steinmetz, A.; Oudin, A.; Berchem, G.; Janji, B.; Thomas, C. CRP2, a new invadopodia actin bundling factor critically promotes breast cancer cell invasion and metastasis. Oncotarget 2016, 7, 13688–13705. [Google Scholar] [CrossRef]
- Bodakuntla, S.; Jijumon, A.; Villablanca, C.; González-Billault, C.; Janke, C. Microtubule-Associated Proteins: Structuring the Cytoskeleton. Trends Cell Biol. 2019, 29, 804–819. [Google Scholar] [CrossRef]
- Eckes, B.; Colucci-Guyon, E.; Smola, H.; Nodder, S.; Babinet, C.; Krieg, T.; Martin, P. Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell Sci. 2000, 113 Pt 13, 2455–2462. [Google Scholar]
- Charbaut, E.; Curmi, P.A.; Ozon, S.; Lachkar, S.; Redeker, V.; Sobel, A. Stathmin Family Proteins Display Specific Molecular and Tubulin Binding Properties. J. Biol. Chem. 2001, 276, 16146–16154. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, R.; Ko, S.-Y.; Oyajobi, B.O.; Papasian, C.J.; Deng, H.-W.; Zhang, S.; Zhao, M. Microtubule assembly affects bone mass by regulating both osteoblast and osteoclast functions: Stathmin deficiency produces an osteopenic phenotype in mice. J. Bone Miner. Res. 2011, 26, 2052–2067. [Google Scholar] [CrossRef]
- Bamburg, J.R. Proteins of the ADF/Cofilin Family: Essential Regulators of Actin Dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.; Parmar, H.; Roelandse, M.; Bornmann, C.; Matus, A. Cytoskeletal microdifferentiation: A mechanism for organizing morphological plasticity in dendrites. Proc. Natl. Acad. Sci. USA 2001, 98, 7086–7092. [Google Scholar] [CrossRef] [PubMed]
- Hehnly, H.; Stamnes, M. Regulating cytoskeleton-based vesicle motility. FEBS Lett. 2007, 581, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Parast, M.M.; Otey, C.A. Characterization of Palladin, a Novel Protein Localized to Stress Fibers and Cell Adhesions. J. Cell Biol. 2000, 150, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Gurung, R.; Yadav, R.; Brungardt, J.G.; Orlova, A.; Egelman, E.H.; Beck, M.R. Actin polymerization is stimulated by actin cross-linking protein palladin. Biochem. J. 2016, 473, 383–396. [Google Scholar] [CrossRef]
- Kim, H.; Lee, Y.D.; Kim, M.K.; Kwon, J.O.; Song, M.K.; Lee, Z.H.; Kim, H.H. Extracellular S100A4 negatively regulates osteoblast function by activating the NF-κB pathway. BMB Rep. 2017, 50, 97–102. [Google Scholar] [CrossRef]
- Ambartsumian, N.; Klingelhöfer, J.; Grigorian, M. The Multifaceted S100A4 Protein in Cancer and Inflammation. Methods Mol. Biol. 2019, 1929, 339–365. [Google Scholar]
- Berge, G.; Pettersen, S.; Grotterød, I.; Bettum, I.J.; Boye, K.; Mælandsmo, G.M. Osteopontin-An important downstream effector of S100A4-mediated invasion and metastasis. Int. J. Cancer 2011, 129, 780–790. [Google Scholar] [CrossRef]
- Mirastschijski, U.; Lupse, B.; Maedler, K.; Sarma, B.; Radtke, A.; Belge, G.; Dorsch, M.M.; Wedekind, D.; McCawley, L.J.; Böhm, G.; et al. Matrix Metalloproteinase-3 is Key Effector of TNF-α-Induced Collagen Degradation in Skin. Int. J. Mol. Sci. 2019, 20, 5234. [Google Scholar] [CrossRef]
- Zhang, X.; Peterson, K.A.; Liu, X.S.; McMahon, A.P.; Ohba, S. Gene Regulatory Networks Mediating Canonical Wnt Signal-Directed Control of Pluripotency and Differentiation in Embryo Stem Cells. Stem Cells 2013, 31, 2667–2679. [Google Scholar] [CrossRef]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Terawaki, S.-I.; Kitano, K.; Hakoshima, T.; Palamakumbura, A.H.; Jeay, S.; Guo, Y.; Pischon, N.; Sommer, P.; Sonenshein, G.E.; Trackman, P.C. Structural Basis for Type II Membrane Protein Binding by ERM Proteins Revealed by the Radixin-neutral Endopeptidase 24.11 (NEP) Complex. J. Biol. Chem. 2007, 282, 19854–19862. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, M.; Kumar, S.; Mayer, G.; Walter, J. Casein Kinase 2 Dependent Phosphorylation of Neprilysin Regulates Receptor Tyrosine Kinase Signaling to Akt. PLoS ONE 2010, 5, e13134. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, J.; Tycksen, E.; Nunley, R.M.; McAlinden, A. MicroRNA-181a/b-1 over-expression enhances osteogenesis by modulating PTEN/PI3K/AKT signaling and mitochondrial metabolism. Bone 2019, 123, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Ruchon, A.F.; Marcinkiewicz, M.; Ellefsen, K.; Basak, A.; Aubin, J.E.; Crine, P.; Boileau, G. Cellular Localization of Neprilysin in Mouse Bone Tissue and Putative Role in Hydrolysis of Osteogenic Peptides. J. Bone Miner. Res. 2000, 15, 1266–1274. [Google Scholar] [CrossRef]
- Ganju, R.K.; Shpektor, R.G.; Brenner, D.G.; Shipp, M.A. CD10/neutral endopeptidase 24.11 is phosphorylated by casein kinase II and coassociates with other phosphoproteins including the lyn src-related kinase. Blood 1996, 88, 4159–4165. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Zhuravin, I.; Turner, A.J. Neprilysin expression and functions in development, ageing and disease. Mech. Ageing Dev. 2020, 192, 111363. [Google Scholar] [CrossRef] [PubMed]
- Wiese, C.; Rolletschek, A.; Kania, G.; Blyszczuk, P.; Tarasov, K.V.; Tarasova, Y.; Wersto, R.P.; Boheler, K.R.; Wobus, A.M. Nestin expression—A property of multi-lineage progenitor cells? Cell. Mol. Life Sci. 2004, 61, 2510–2522. [Google Scholar] [CrossRef]
- Bernal, A.; Arranz, L. Nestin-expressing progenitor cells: Function, identity and therapeutic implications. Cell. Mol. Life Sci. 2018, 75, 2177–2195. [Google Scholar] [CrossRef] [PubMed]
- Twomey, J.D.; Thakore, P.I.; Hartman, D.A.; Myers, E.G.H.; Hsieh, A.H. Roles of type VI collagen and decorin in human mesenchymal stem cell biophysics during chondrogenic differentiation. Eur. Cell Mater. 2014, 27, 237–250; discussion 249–250. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Bruzzese, F.; Leone, A.; Gagliardi, A.; Puglia, M.; Di Gennaro, E.; Rocco, M.; Gimigliano, A.; Pucci, B.; Armini, A.; et al. Proteomic analysis identifies differentially expressed proteins after HDAC vorinostat and EGFR inhibitor gefitinib treatments in Hep-2 cancer cells. Proteomics 2011, 11, 3725–3742. [Google Scholar] [CrossRef] [PubMed]
- Alcorta-Sevillano, N.; Macías, I.; Rodríguez, C.I.; Infante, A. Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone. Cells 2020, 9, 1330. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.M.; Dimori, M.; Heard-Lipsmeyer, M.E.; Morello, R. The Osteocyte Transcriptome Is Extensively Dysregulated in Mouse Models of Osteogenesis Imperfecta. JBMR Plus 2019, 3, e10171. [Google Scholar] [CrossRef] [PubMed]
- Mottes, M.; Lira, M.G.; Zolezzi, F.; Valli, M.; Lisi, V.; Freising, P. Four new cases of lethal osteogenesis imperfecta due to glycine substitutions in COL1A1 and genes. Hum. Mutat. 1998, 12, 71–72. [Google Scholar] [CrossRef]
- Gomez-Lira, M.; Sangalli, A.; Pignatti, P.F.; Digilio, M.C.; Giannotti, A.; Carnevale, E.; Mottes, M. Determination of a new collagen type I alpha 2 gene point mutation which causes a Gly640 Cys substitution in osteogenesis imperfecta and prenatal diagnosis by DNA hybridisation. J. Med. Genet. 1994, 31, 965–968. [Google Scholar] [CrossRef]
- Venturi, G.; Tedeschi, E.; Mottes, M.; Valli, M.; Camilot, M.; Viglio, S.; Antoniazzi, F.; Tatò, L. Osteogenesis imperfecta: Clinical, biochemical and molecular findings. Clin. Genet. 2006, 70, 131–139. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Müller, A.; Dayon, L.; Köcher, T.; Hainard, A.; Pichler, P.; Schmidt-Erfurth, U.; Superti-Furga, G.; Sanchez, J.-C.; Mechtler, K.; et al. General Statistical Modeling of Data from Protein Relative Expression Isobaric Tags. J. Proteome Res. 2011, 10, 2758–2766. [Google Scholar] [CrossRef]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef]
- Huntley, R.P.; Sawford, T.; Mutowo-Meullenet, P.; Shypitsyna, A.; Bonilla, C.; Martin, M.J.; O’Donovan, C. The GOA database: Gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015, 43, D1057–D1063. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Landi, C.; Luddi, A.; Bianchi, L.; Pannuzzo, G.; Pavone, V.; Piomboni, P.; Bini, L. Proteostasis network alteration in lysosomal storage disorders: Insights from the mouse model of Krabbe disease. J. Neurosci. Res. 2019, 98, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Puglia, M.; Landi, C.; Gagliardi, A.; Breslin, L.; Armini, A.; Brunetti, J.; Pini, A.; Bianchi, L.; Bini, L. The proteome speciation of an immortalized cystic fibrosis cell line: New perspectives on the pathophysiology of the disease. J. Proteom. 2018, 170, 28–42. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein Defect $ | OI Type # | Gender | Age § | PPERK/PERK | LC3II+ Chloroquine | CleavedCasp3 |
|---|---|---|---|---|---|---|---|
| COL1A1 | α1(I)-G226S | III | M | 8 m | ↑ | ↑ | ↑ |
| COL1A1 | α 1(I)-G478S | II | M | 0 m | ↑ | ↑ | ↑ |
| COL1A1 | α 1(I)-G994D | II | M | f | ↑ | ↑ | ↑ |
| COL1A2 | α 2(I)-G640C | II/III * | M | 6 m | ~ | ~ | ↑ |
| COL1A2 | α 2(I)-G697C | II | F | 1 m | ↑ | ↑ | ↑ |
| COL1A2 | α 2(I)-G745C | III | M | 3 y | ↑ | ~ | ↑ |
| Swiss-Prot Accession Number | Swiss-Prot Identifier | Protein Description | Dead p. vs. CTRL | Dead p. vs. Survived p. ¥ | Survived p. vs. CTRL |
|---|---|---|---|---|---|
| P08473 | NEP_HUMAN | Neprilysin | 0.55 | 0.50 | NS |
| P26447 | S10A4_HUMAN | Protein S100-A4 | 0.26 | 0.63 | 0.42 |
| P21589 | 5NTD_HUMAN | 5’-nucleotidase | 0.40 | 0.64 | 0.69 |
| P07585 | PGS2_HUMAN | Decorin | 0.27 | 0.64 | 0.43 |
| P68431 | H31_HUMAN | Histone H3.1 | 0.57 | 0.66 | NS |
| P18085 | ARF4_HUMAN | ADP-ribosylation factor 4 | NS | 0.66 | NS |
| Q8WX93 | PALLD_HUMAN | Palladin | 2.06 | 1.51 | 1.35 |
| P35555 | FBN1_HUMAN | Fibrillin-1 | NS | 1.58 | NS |
| O15400 | STX7_HUMAN | Syntaxin-7 | 1.52 | 1.62 | NS |
| P05091 | ALDH2_HUMAN | Aldehyde dehydrogenase, mitochondrial | NS | 1.63 | NS |
| P78559 | MAP1A_HUMAN | Microtubule-associated protein 1A | NS | 1.68 | NS |
| Q13308 | PTK7_HUMAN | Inactive tyrosine-protein kinase 7 | 1.5 | 1.68 | NS |
| P09486 | SPRC_HUMAN | SPARC | 2.91 | 1.69 | 1.66 |
| P11216 | PYGB_HUMAN | Glycogen phosphorylase, brain form | NS | 1.70 | 0.6 |
| Q6UVK1 | CSPG4_HUMAN | Chondroitin sulfate proteoglycan 4 | 1.98 | 1.80 | NS |
| Q16527 | CSRP2_HUMAN | Cysteine and glycine-rich protein 2 | 2.44 | 1.88 | NS |
| P48681 | NEST_HUMAN | Nestin | 2.11 | 2.33 | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bini, L.; Schvartz, D.; Carnemolla, C.; Besio, R.; Garibaldi, N.; Sanchez, J.-C.; Forlino, A.; Bianchi, L. Intracellular and Extracellular Markers of Lethality in Osteogenesis Imperfecta: A Quantitative Proteomic Approach. Int. J. Mol. Sci. 2021, 22, 429. https://doi.org/10.3390/ijms22010429
Bini L, Schvartz D, Carnemolla C, Besio R, Garibaldi N, Sanchez J-C, Forlino A, Bianchi L. Intracellular and Extracellular Markers of Lethality in Osteogenesis Imperfecta: A Quantitative Proteomic Approach. International Journal of Molecular Sciences. 2021; 22(1):429. https://doi.org/10.3390/ijms22010429
Chicago/Turabian StyleBini, Luca, Domitille Schvartz, Chiara Carnemolla, Roberta Besio, Nadia Garibaldi, Jean-Charles Sanchez, Antonella Forlino, and Laura Bianchi. 2021. "Intracellular and Extracellular Markers of Lethality in Osteogenesis Imperfecta: A Quantitative Proteomic Approach" International Journal of Molecular Sciences 22, no. 1: 429. https://doi.org/10.3390/ijms22010429
APA StyleBini, L., Schvartz, D., Carnemolla, C., Besio, R., Garibaldi, N., Sanchez, J.-C., Forlino, A., & Bianchi, L. (2021). Intracellular and Extracellular Markers of Lethality in Osteogenesis Imperfecta: A Quantitative Proteomic Approach. International Journal of Molecular Sciences, 22(1), 429. https://doi.org/10.3390/ijms22010429