Role of IL-36 Cytokines in the Regulation of Angiogenesis Potential of Trophoblast Cells

 , , , , and
, , , , and 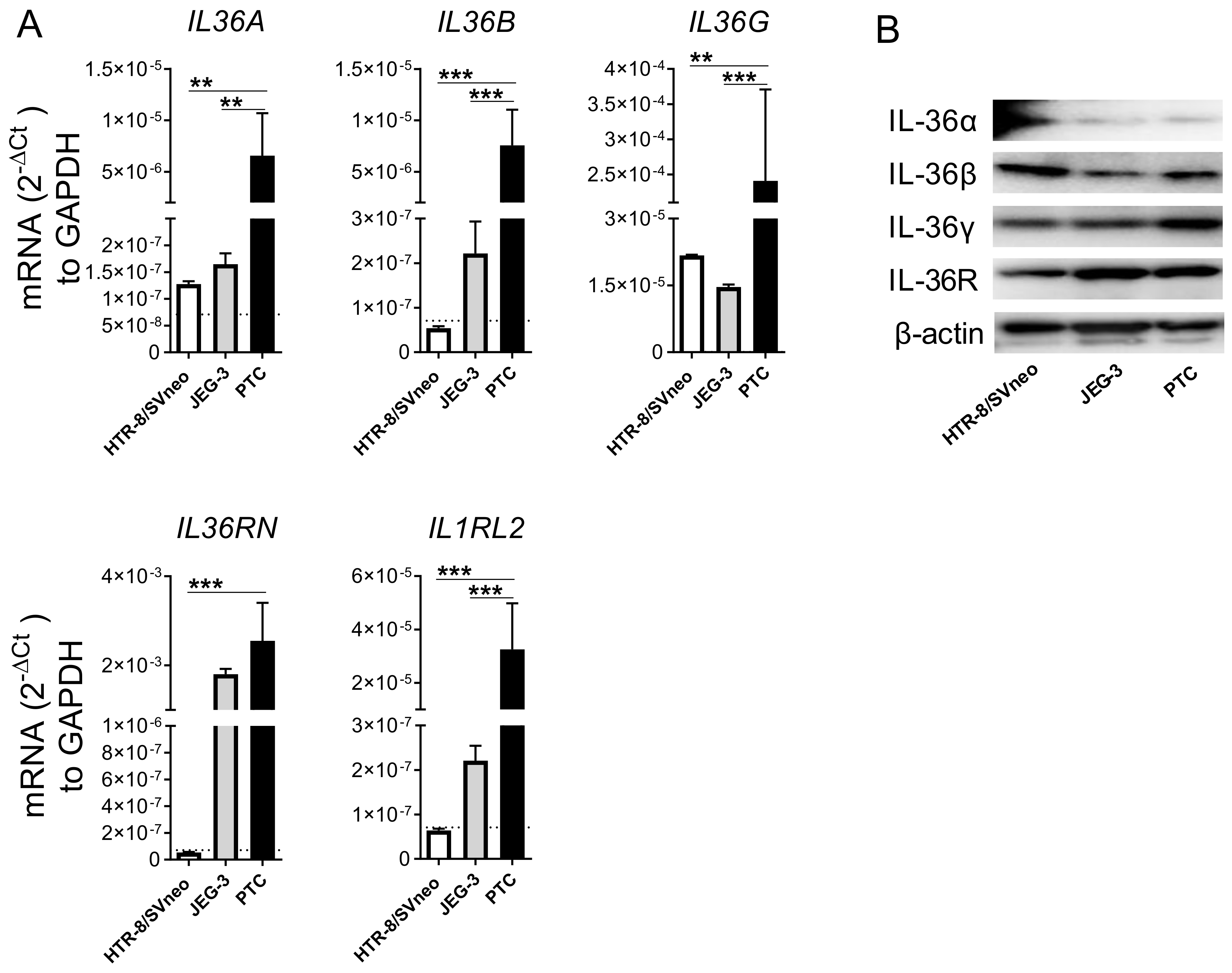{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Primary Trophoblast Cells but Not Their Cell Line Counterparts Express all Members of the IL-36 Family
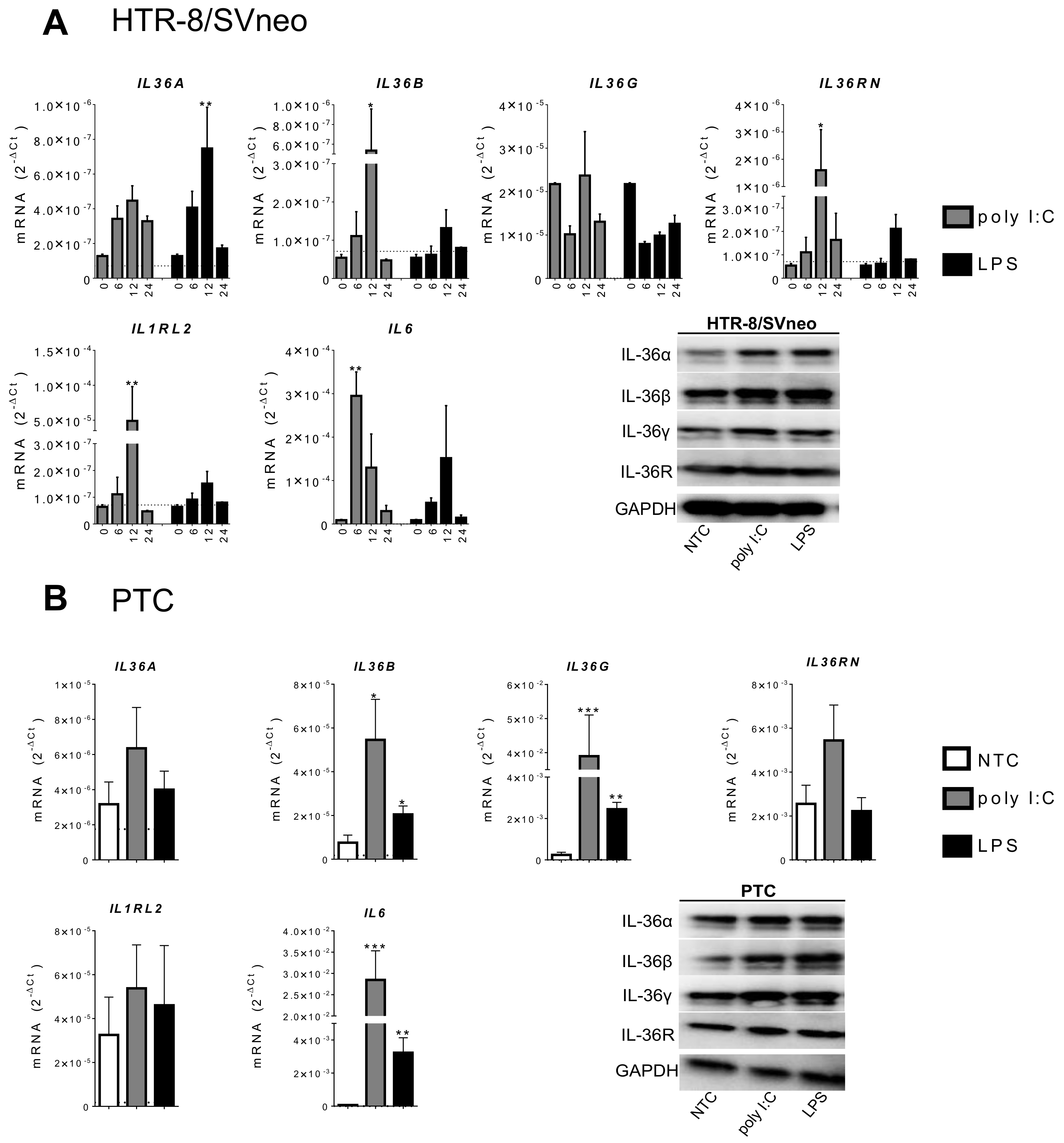
2.2. IL-36 Expression Induced by Poly I:C and LPS Stimulation in Trophoblast Cells
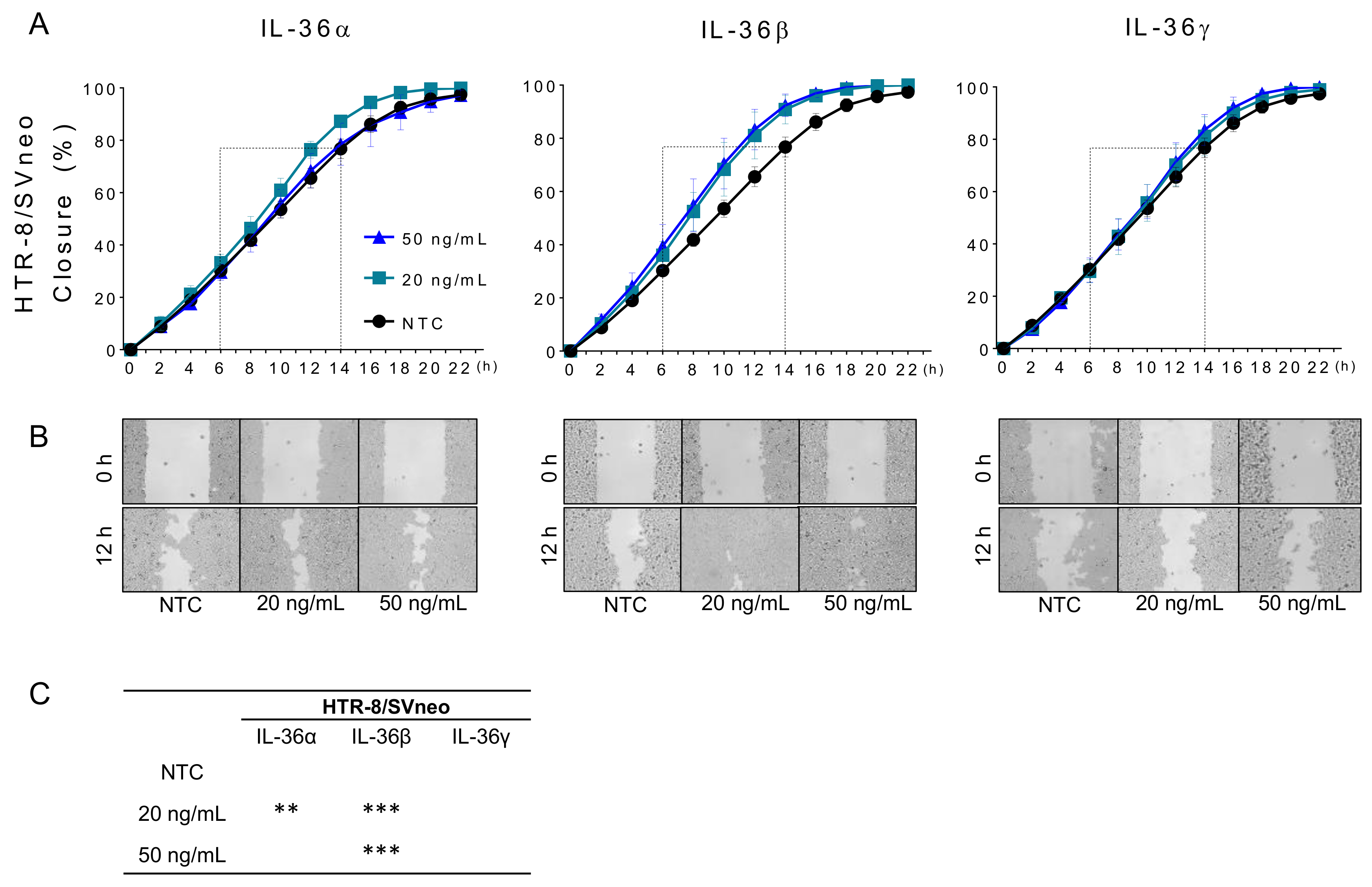
2.3. IL-36 (α, β, γ) Promote Migration of Trophoblastic Cells
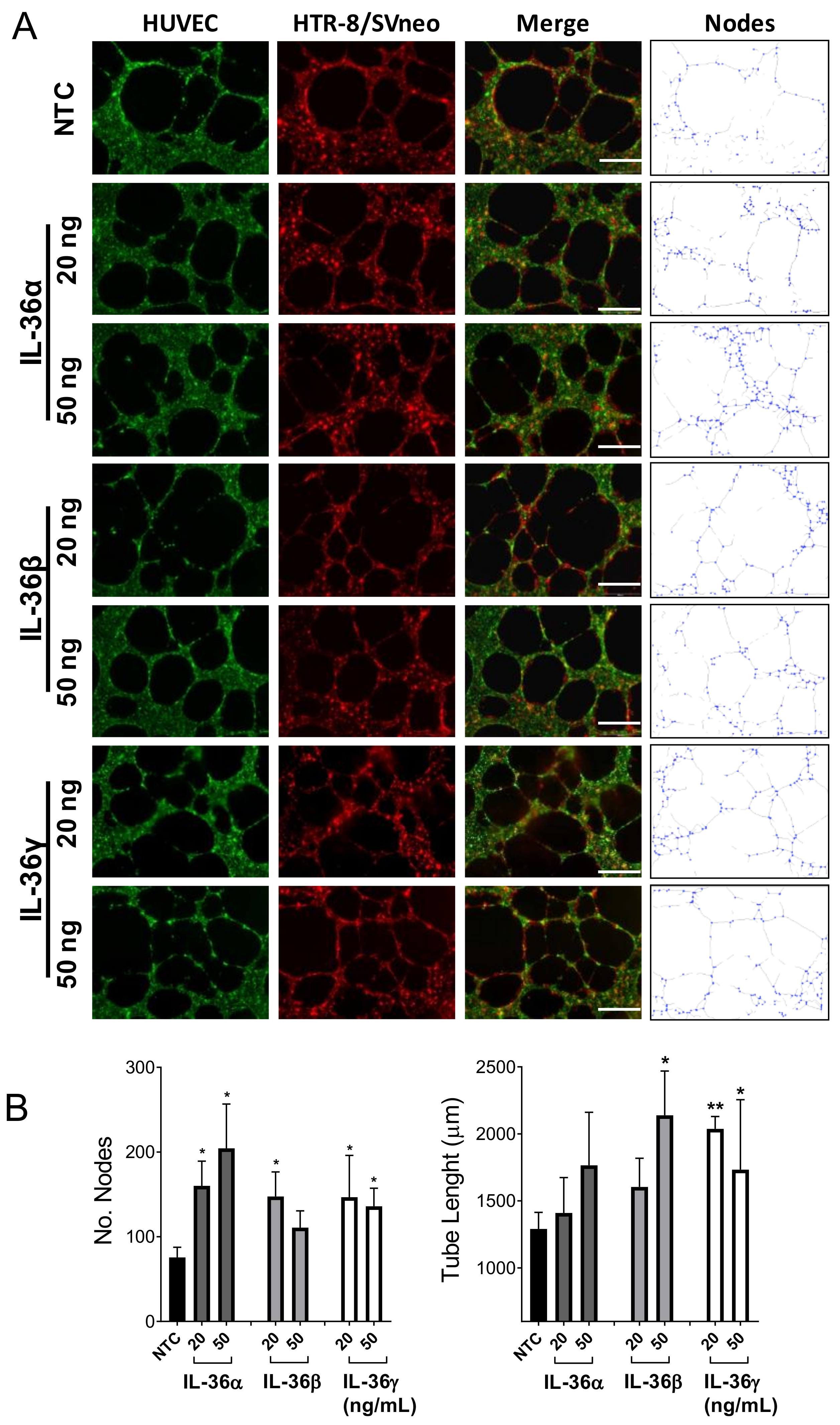
2.4. Effect of IL-36 (α, β, γ) in the Interaction between Trophoblast and Endothelial Cells
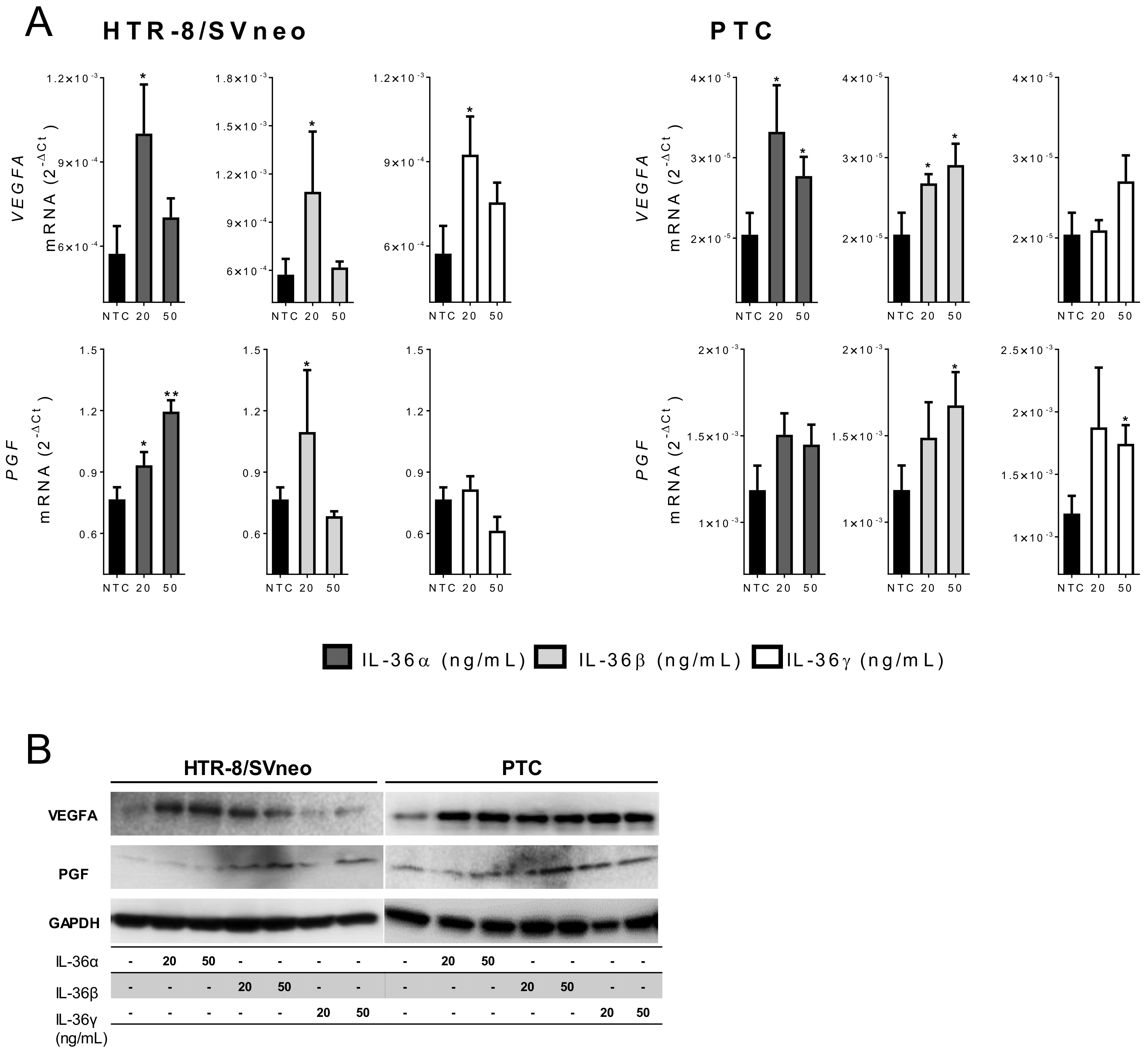
2.5. IL-36 (α, β, and γ) Induce Expression of Angiogenic Factors in Trophoblast Cells
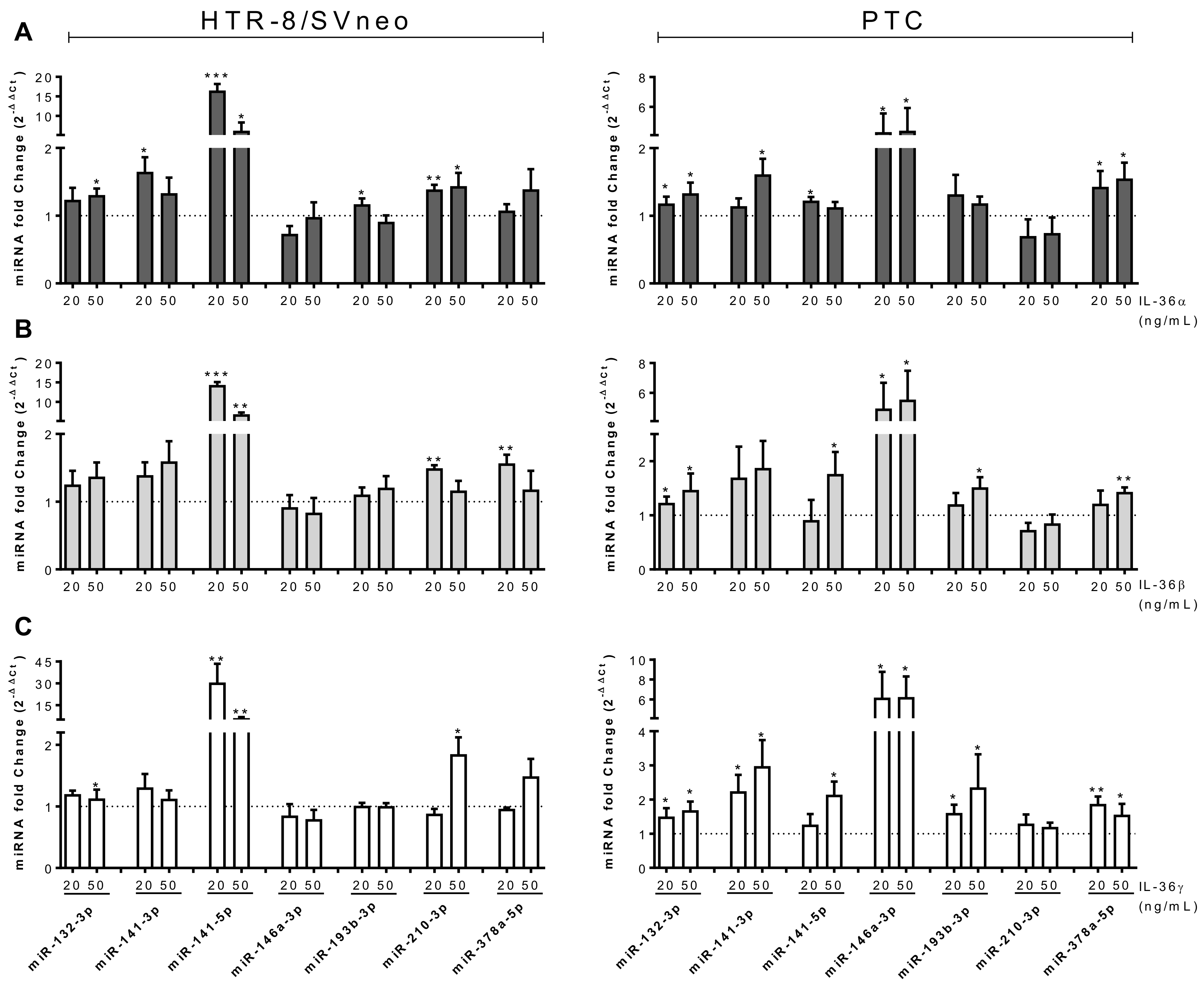
2.6. microRNA Expression is Regulated by IL-36 Cytokines in Trophoblastic Cells
3. Discussion
4. Materials and Methods
4.1. Isolation of PTC
4.2. Cell Culture
4.3. Cell Stimulation
4.4. RNA Isolation and qPCR
4.5. Western Blotting Analysis
4.6. “Wound Healing” Assay (Cell Migration)
4.7. Tube Formation Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AECs | Alveolar epithelial cells |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| ECGM | Endothelial Cell Growth Medium |
| ECM | Extracellular matrix |
| FRT | Female reproductive tract |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| HBSS | Hanks’ Balanced Salt Solution |
| HSV-2 | Herpes-simplex-Virus 2 |
| HUVEC | Human umbilical vein endothelial cells |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| IL-1RAcP | Interleukin-1 receptor accessory protein |
| IL1RL2 | Transcript for IL-36R protein |
| IL36A | Transcript for IL-36α protein |
| IL36B | Transcript for IL-36β protein |
| IL36G | Transcript for IL-36γ protein |
| IL36RN | Transcript for IL-36Ra protein |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinases |
| MyD88 | Myeloid differentiation primary response 88 |
| NF-kB | Nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells |
| NTC | Non-treated cells |
| PCR | Polymerase chain reaction |
| PGF | Placental growth factor |
| PTC | Primary trophoblast cells |
| RPMI | Roswell Park Memorial Institute medium |
| VCAM-1 | Vascular cell adhesion protein 1 |
| VEGFA | Vascular endothelial growth factor A |
References
- Mor, G.; Cardenas, I.; Abrahams, V.; Guller, S. Inflammation and pregnancy: The role of the immune system at the implantation site. Ann. N. Y. Acad. Sci. 2011, 1221, 80–87. [Google Scholar] [CrossRef]
- Simon, C.; Valbuena, D.; Krussel, J.; Bernal, A.; Murphy, C.R.; Shaw, T.; Pellicer, A.; Polan, M.L. Interleukin–1 receptor antagonist prevents embryonic implantation by a direct effect on the endometrial epithelium. Fertil. Steril. 1998, 70, 896–906. [Google Scholar] [CrossRef]
- Espinoza, J.; Romero, R.; Mee Kim, Y.; Kusanovic, J.P.; Hassan, S.; Erez, O.; Gotsch, F.; Than, N.G.; Papp, Z.; Jai Kim, C. Normal and abnormal transformation of the spiral arteries during pregnancy. J Perinat. Med. 2006, 34, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Weckman, A.M.; Ngai, M.; Wright, J.; McDonald, C.R.; Kain, K.C. The Impact of Infection in Pregnancy on Placental Vascular Development and Adverse Birth Outcomes. Front. Microbiol. 2019, 10, 1924. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin–36 (IL–36) ligands require processing for full agonist (IL–36alpha, IL–36beta, and IL–36gamma) or antagonist (IL–36Ra) activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef] [PubMed]
- Dunn, E.; Sims, J.E.; Nicklin, M.J.; O’Neill, L.A. Annotating genes with potential roles in the immune system: Six new members of the IL–1 family. Trends Immunol. 2001, 22, 533–536. [Google Scholar] [CrossRef]
- Ding, L.; Wang, X.; Hong, X.; Lu, L.; Liu, D. IL–36 cytokines in autoimmunity and inflammatory disease. Oncotarget 2018, 9, 2895–2901. [Google Scholar] [CrossRef]
- Gresnigt, M.S.; van de Veerdonk, F.L. Biology of IL–36 cytokines and their role in disease. Semin. Immunol. 2013, 25, 458–565. [Google Scholar] [CrossRef]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)–1F6, IL–1F8, and IL–1F9 signal through IL–1Rrp2 and IL–1RAcP to activate the pathway leading to NF–kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef]
- Nishida, A.; Hidaka, K.; Kanda, T.; Imaeda, H.; Shioya, M.; Inatomi, O.; Bamba, S.; Kitoh, K.; Sugimoto, M.; Andoh, A. Increased Expression of Interleukin–36, a Member of the Interleukin–1 Cytokine Family, in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 303–314. [Google Scholar] [CrossRef]
- Murrieta-Coxca, J.M.; Gomez-Chavez, F.; Baeza-Martinez, D.A.; Cancino-Diaz, M.E.; Cancino-Diaz, J.C.; Perez-Tapia, S.M.; Reyes-Maldonado, E.; Rodriguez-Martinez, S. Estrous Cycle and Gestational Age-Dependent Expression of Members of the Interleukin–36 Subfamily in a Semi-Allogeneic Model of Infected and Non-Infected Murine Pregnancy. Front. Immunol. 2016, 7, 376. [Google Scholar] [CrossRef] [PubMed]
- Coxca, J.; Chavez, F.G.; Diaz, M.E.C.; Diaz, J.C.C.; Martinez, S.R. L. monocytogenes induces overexpression of proinflammatory IL–36 cytokines in a murine model of early pregnancy. J. Reprod. Immunol. 2016, 115, 55. [Google Scholar] [CrossRef]
- Southcombe, J.H.; Redman, C.W.; Sargent, I.L.; Granne, I. Interleukin–1 family cytokines and their regulatory proteins in normal pregnancy and pre–eclampsia. Clin. Exp. Immunol. 2015, 181, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Blankenship, T.N.; Enders, A.C.; King, B.F. Trophoblastic invasion and modification of uterine veins during placental development in macaques. Cell Tissue Res. 1993, 274, 135–144. [Google Scholar] [CrossRef]
- Thaler, I.; Manor, D.; Itskovitz, J.; Rottem, S.; Levit, N.; Timor-Tritsch, I.; Brandes, J.M. Changes in uterine blood flow during human pregnancy. Am. J. Obstet. Gynecol. 1990, 162, 121–125. [Google Scholar] [CrossRef]
- Arechavaleta-Velasco, F.; Koi, H.; Strauss, J.F.; Parry, S. Viral infection of the trophoblast: Time to take a serious look at its role in abnormal implantation and placentation? J. Reprod. Immunol. 2002, 55, 113–121. [Google Scholar] [CrossRef]
- Gomez-Chavez, F.; Lopez-Portales, O.H.; Baeza-Martinez, D.A.; Cancino-Diaz, J.C.; Murrieta-Coxca, J.M.; Cancino-Diaz, M.E.; Perez-Tapia, S.M.; Rodriguez-Martinez, S. IkappaBNS and IL–6 expression is differentially established in the uterus of pregnant healthy and infected mice. Heliyon 2020, 6, e04122. [Google Scholar] [CrossRef]
- Aldo, P.B.; Krikun, G.; Visintin, I.; Lockwood, C.; Romero, R.; Mor, G. A novel three-dimensional in vitro system to study trophoblast–endothelium cell interactions. Am. J. Reprod. Immunol. 2007, 58, 98–110. [Google Scholar] [CrossRef]
- Morales-Prieto, D.M.; Barth, E.; Murrieta-Coxca, J.M.; Favaro, R.R.; Gutiérrez-Samudio, R.N.; Chaiwangyen, W.; Ospina-Prieto, S.; Gruhn, B.; Schleußner, E.; Marz, M.; et al. Identification of miRNAs and associated pathways regulated by Leukemia Inhibitory Factor in trophoblastic cell lines. Placenta 2019, 88, 20–27. [Google Scholar] [CrossRef]
- Dubinsky, V.; Poehlmann, T.G.; Suman, P.; Gentile, T.; Markert, U.R.; Gutierrez, G. Role of regulatory and angiogenic cytokines in invasion of trophoblastic cells. Am. J. Reprod. Immunol. 2010, 63, 193–199. [Google Scholar] [CrossRef]
- Gabay, C.; Towne, J.E. Regulation and function of interleukin–36 cytokines in homeostasis and pathological conditions. J. Leukoc. Biol. 2015, 97, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Torales-Cardena, A.; Martinez-Torres, I.; Rodriguez-Martinez, S.; Gomez-Chavez, F.; Cancino-Diaz, J.C.; Vazquez-Sanchez, E.A.; Cancino-Diaz, M.E. Cross Talk between Proliferative, Angiogenic, and Cellular Mechanisms Orchestred by HIF–1alpha in Psoriasis. Mediat. Inflamm. 2015, 2015, 607363. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Sims, J.E. IL–36 in psoriasis. Curr. Opin. Pharmacol. 2012, 12, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Morales-Prieto, D.M.; Chaiwangyen, W.; Ospina-Prieto, S.; Schneider, U.; Herrmann, J.; Gruhn, B.; Markert, U.R. MicroRNA expression profiles of trophoblastic cells. Placenta 2012, 33, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Bilban, M.; Tauber, S.; Haslinger, P.; Pollheimer, J.; Saleh, L.; Pehamberger, H.; Wagner, O.; Knofler, M. Trophoblast invasion: Assessment of cellular models using gene expression signatures. Placenta 2010, 31, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Q.; Gardner, L.; Turco, M.; Zhao, N.; Murray, M.J.; Coleman, N.; Rossant, J.; Hemberger, M.; Moffett, A. What Is Trophoblast? A Combination of Criteria Define Human First–Trimester Trophoblast. Stem Cell Rep. 2016, 6, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Lieblich, J.M.; Weintraub, B.D.; Krauth, G.H.; Kohler, P.O.; Rabson, A.S.; Rosen, S.W. Ectopic and eutopic secretion of chorionic gonadotropin and its sub–nits in vitro: Comparison of clonal strains from carcinomas of lung and placenta. J. Nat. Cancer Inst. 1976, 56, 911–917. [Google Scholar] [CrossRef]
- Graham, C.H.; Hawley, T.S.; Hawley, R.G.; MacDougall, J.R.; Kerbel, R.S.; Khoo, N.; Lala, P.K. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp. Cell Res. 1993, 206, 204–211. [Google Scholar] [CrossRef]
- Heine, R.P.; Ness, R.B.; Roberts, J.M. Seroprevalence of antibodies to Chlamydia pneumoniae in women with preeclampsia. Obstet. Gynecol. 2003, 101, 221–226. [Google Scholar]
- Xie, F.; Hu, Y.; Magee, L.A.; Money, D.M.; Patrick, D.M.; Krajden, M.; Thomas, E.; von Dadelszen, P.; Toxemia Study, G. An association between cytomegalovirus infection and pre–eclampsia: A case–control study and data synthesis. Acta Obstet. Gynecol. Scand. 2010, 89, 1162–1167. [Google Scholar] [CrossRef]
- Nourollahpour Shiadeh, M.; Behboodi Moghadam, Z.; Adam, I.; Saber, V.; Bagheri, M.; Rostami, A. Human infectious diseases and risk of preeclampsia: An updated review of the literature. Infection 2017, 45, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.A.; Bart, G.; Penhoat, M.; Amiaud, J.; Brulin, B.; Charrier, C.; Morel, F.; Lecron, J.C.; Rolli-Derkinderen, M.; Bourreille, A.; et al. Distinct expression of interleukin (IL)–36alpha, beta and gamma, their antagonist IL–36Ra and IL–38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 2016, 184, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Newstead, M.W.; Zeng, X.; Kunkel, S.L.; Kaku, M.; Standiford, T.J. IL–36 receptor deletion attenuates lung injury and decreases mortality in murine influenza pneumonia. Mucosal Immunol. 2017, 10, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Chustz, R.T.; Nagarkar, D.R.; Poposki, J.A.; Favoreto, S., Jr.; Avila, P.C.; Schleimer, R.P.; Kato, A. Regulation and function of the IL–1 family cytokine IL–1F9 in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011, 45, 145–153. [Google Scholar] [CrossRef]
- Rana, A.A.; Lucs, A.V.; DeVoti, J.; Blanc, L.; Papoin, J.; Wu, R.; Papayannakos, C.J.; Abramson, A.; Bonagura, V.R.; Steinberg, B.M. Poly(I:C) induces controlled release of IL–36gamma from keratinocytes in the absence of cell death. Immunol. Res. 2015, 63, 228–235. [Google Scholar] [CrossRef]
- Winkle, S.M.; Throop, A.L.; Herbst-Kralovetz, M.M. IL–36gamma Augments Host Defense and Immune Responses in Human Female Reproductive Tract Epithelial Cells. Front. Microbiol. 2016, 7, 955. [Google Scholar] [CrossRef]
- Gardner, J.K.; Herbst-Kralovetz, M.M. IL–36gamma induces a transient HSV–2 resistant environment that protects against genital disease and pathogenesis. Cytokine 2018, 111, 63–71. [Google Scholar] [CrossRef]
- Anton, L.; Brown, A.G.; Parry, S.; Elovitz, M.A. Lipopolysaccharide induces cytokine production and decreases extravillous trophoblast invasion through a mitogen-activated protein kinase-mediated pathway: Possible mechanisms of first trimester placental dysfunction. Hum. Reprod. 2012, 27, 61–72. [Google Scholar] [CrossRef]
- Abrahams, V.M.; Visintin, I.; Aldo, P.B.; Guller, S.; Romero, R.; Mor, G. A role for TLRs in the regulation of immune cell migration by first trimester trophoblast cells. J. Immunol. 2005, 175, 8096–8104. [Google Scholar] [CrossRef]
- Sokolov, D.I.; Lvova, T.Y.; Okorokova, L.S.; Belyakova, K.L.; Sheveleva, A.R.; Stepanova, O.I.; Mikhailova, V.A.; Sel’kov, S.A. Effect of Cytokines on the Formation Tube-Like Structures by Endothelial Cells in the Presence of Trophoblast Cells. Bull. Exp. Biol. Med. 2017, 163, 148–158. [Google Scholar] [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for Image—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef] [PubMed]
- Bridgewood, C.; Fearnley, G.W.; Berekmeri, A.; Laws, P.; Macleod, T.; Ponnambalam, S.; Stacey, M.; Graham, A.; Wittmann, M. IL–36gamma Is a Strong Inducer of IL–23 in Psoriatic Cells and Activates Angiogenesis. Front. Immunol. 2018, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Shore, V.H.; Wang, T.H.; Wang, C.L.; Torry, R.J.; Caudle, M.R.; Torry, D.S. Vascular endothelial growth factor, placenta growth factor and their receptors in isolated human trophoblast. Placenta 1997, 18, 657–665. [Google Scholar] [CrossRef]
- Andraweera, P.H.; Dekker, G.A.; Laurence, J.A.; Roberts, C.T. Placental expression of VEGF family mRNA in adverse pregnancy outcomes. Placenta 2012, 33, 467–742. [Google Scholar] [CrossRef]
- Chung, J.Y.; Song, Y.; Wang, Y.; Magness, R.R.; Zheng, J. Differential expression of vascular endothelial growth factor (VEGF), endocrine gland derived–VEGF, and VEGF receptors in human placentas from normal and preeclamptic pregnancies. J. Clin. Endocrinol. Metab 2004, 89, 2484–2490. [Google Scholar] [CrossRef]
- Cooper, J.C.; Sharkey, A.M.; Charnock-Jones, D.S.; Palmer, C.R.; Smith, S.K. VEGF mRNA levels in placentae from pregnancies complicated by pre-eclampsia. Br. J. Obstet. Gynaecol 1996, 103, 1191–1196. [Google Scholar] [CrossRef]
- Ranheim, T.; Staff, A.C.; Henriksen, T. VEGF mRNA is unaltered in decidual and placental tissues in preeclampsia at delivery. Acta Obstet. Gynecol. Scand. 2001, 80, 93–98. [Google Scholar] [CrossRef]
- Gobble, R.M.; Groesch, K.A.; Chang, M.; Torry, R.J.; Torry, D.S. Differential regulation of human PlGF gene expression in trophoblast and nontrophoblast cells by oxygen tension. Placenta 2009, 30, 869–875. [Google Scholar] [CrossRef]
- Hoeller, A.; Ehrlich, L.; Golic, M.; Herse, F.; Perschel, F.H.; Siwetz, M.; Henrich, W.; Dechend, R.; Huppertz, B.; Verlohren, S. Placental expression of sFlt–1 and PlGF in early preeclampsia vs. early IUGR vs. age-matched healthy pregnancies. Hypertens. Pregnancy 2017, 36, 151–160. [Google Scholar] [CrossRef]
- Hayder, H.; O’Brien, J.; Nadeem, U.; Peng, C. MicroRNAs: Crucial regulators of placental development. Reproduction 2018, 155, R259–R271. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Pan, Q.; Zhang, X.; Kong, L.Q.; Fan, J.; Dai, Z.; Wang, L.; Yang, X.R.; Hu, J.; Wan, J.-L.; et al. MiR–146a enhances angiogenic activity of endothelial cells in hepatocellular carcinoma by promoting PDGFRA expression. Carcinogenesis 2013, 34, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Ospina-Prieto, S.; Chaiwangyen, W.; Herrmann, J.; Groten, T.; Schleussner, E.; Markert, U.R.; Morales-Prieto, D.M. MicroRNA–141 is upregulated in preeclamptic placentae and regulates trophoblast invasion and intercellular communication. Transl. Res. J. Lab. Clin. Med. 2016, 172, 61–72. [Google Scholar]
- Masoumi-Dehghi, S.; Babashah, S.; Sadeghizadeh, M. MicroRNA–141–3p–containing small extracellular vesicles derived from epithelial ovarian cancer cells promote endothelial cell angiogenesis through activating the JAK/STAT3 and NF-κB signaling pathways. J. Cell Commun. Signal. 2020, 14, 233–244. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murrieta-Coxca, J.M.; Gutiérrez-Samudio, R.N.; El-Shorafa, H.M.; Groten, T.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E.; Cancino-Diaz, J.C.; Favaro, R.R.; Markert, U.R.; Morales-Prieto, D.M. Role of IL-36 Cytokines in the Regulation of Angiogenesis Potential of Trophoblast Cells. Int. J. Mol. Sci. 2021, 22, 285. https://doi.org/10.3390/ijms22010285
Murrieta-Coxca JM, Gutiérrez-Samudio RN, El-Shorafa HM, Groten T, Rodríguez-Martínez S, Cancino-Diaz ME, Cancino-Diaz JC, Favaro RR, Markert UR, Morales-Prieto DM. Role of IL-36 Cytokines in the Regulation of Angiogenesis Potential of Trophoblast Cells. International Journal of Molecular Sciences. 2021; 22(1):285. https://doi.org/10.3390/ijms22010285
Chicago/Turabian StyleMurrieta-Coxca, José M., Ruby N. Gutiérrez-Samudio, Heba M. El-Shorafa, Tanja Groten, Sandra Rodríguez-Martínez, Mario E. Cancino-Diaz, Juan C. Cancino-Diaz, Rodolfo R. Favaro, Udo R. Markert, and Diana M. Morales-Prieto. 2021. "Role of IL-36 Cytokines in the Regulation of Angiogenesis Potential of Trophoblast Cells" International Journal of Molecular Sciences 22, no. 1: 285. https://doi.org/10.3390/ijms22010285
APA StyleMurrieta-Coxca, J. M., Gutiérrez-Samudio, R. N., El-Shorafa, H. M., Groten, T., Rodríguez-Martínez, S., Cancino-Diaz, M. E., Cancino-Diaz, J. C., Favaro, R. R., Markert, U. R., & Morales-Prieto, D. M. (2021). Role of IL-36 Cytokines in the Regulation of Angiogenesis Potential of Trophoblast Cells. International Journal of Molecular Sciences, 22(1), 285. https://doi.org/10.3390/ijms22010285